

Recycling of Commercially Available Biobased Thermoset Polyurethane Using Covalent Adaptable Network Mechanisms

 , ,
, ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. PU Formulation
2.3. Reprcessing
2.4. Characterizaion
3. Results and Discussion
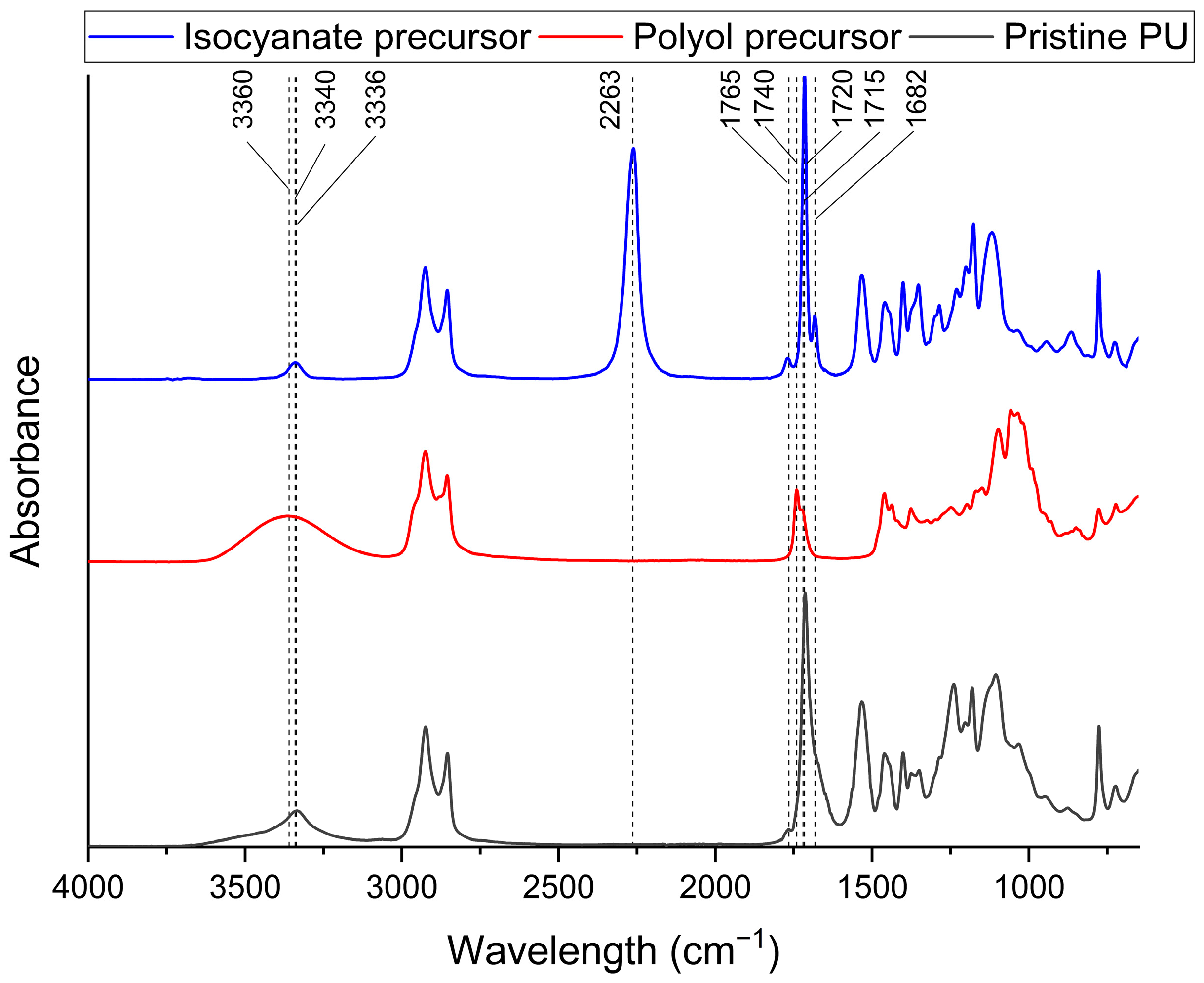
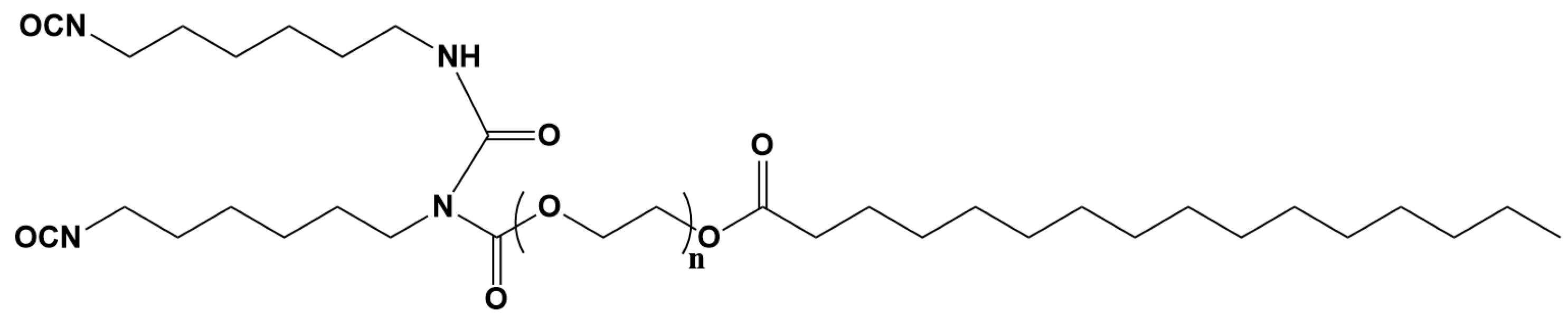
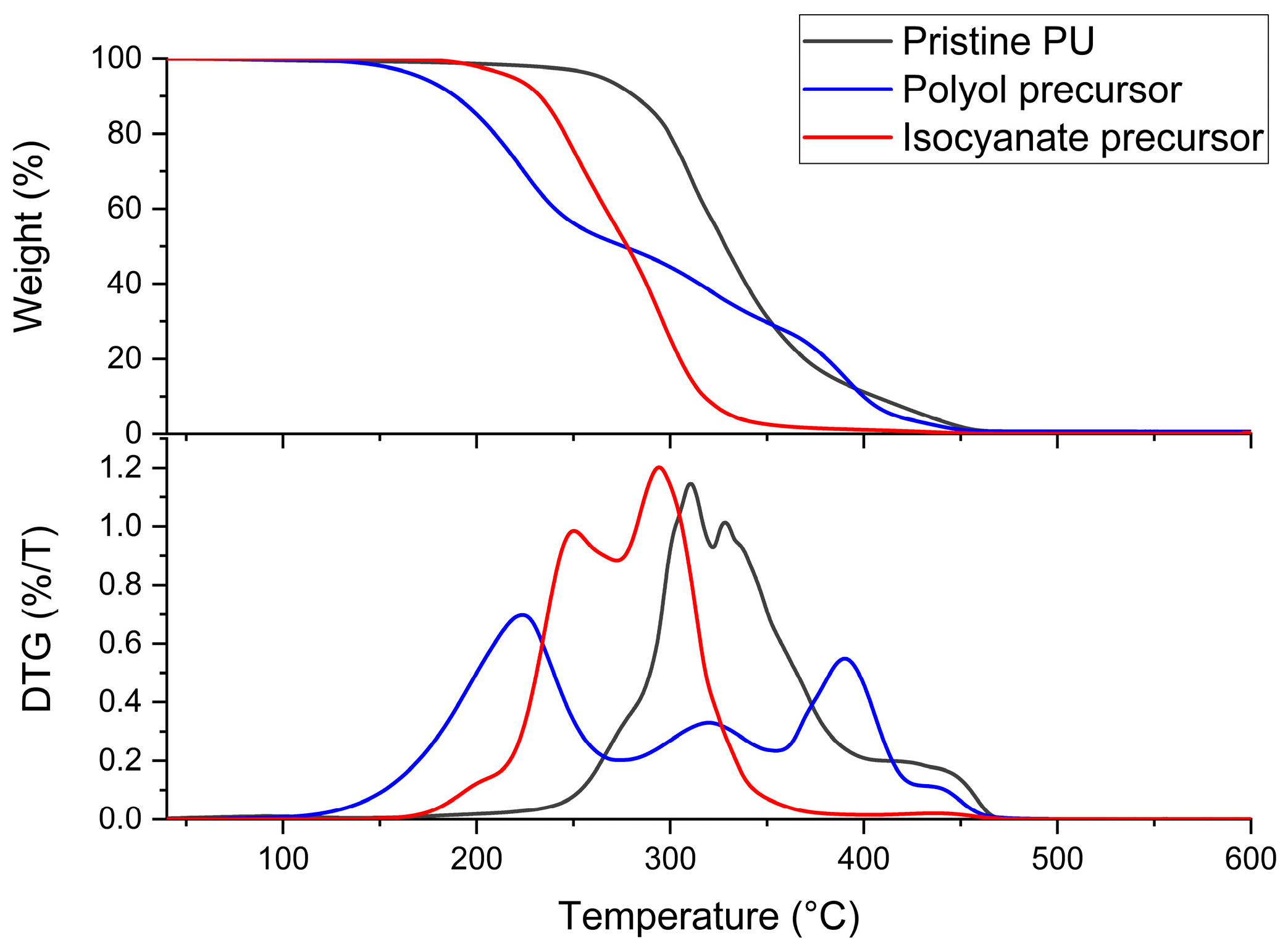
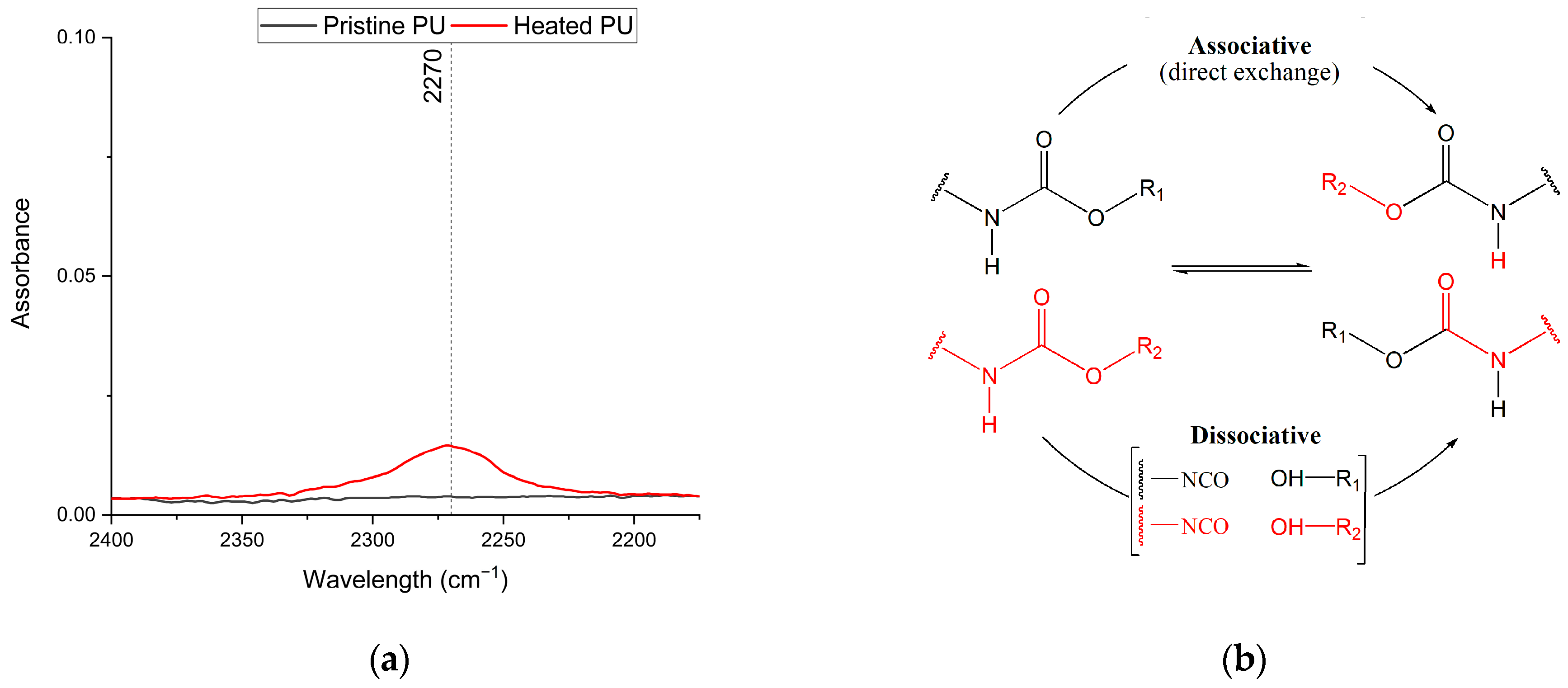
3.1. Characterization of Biobased PU Network
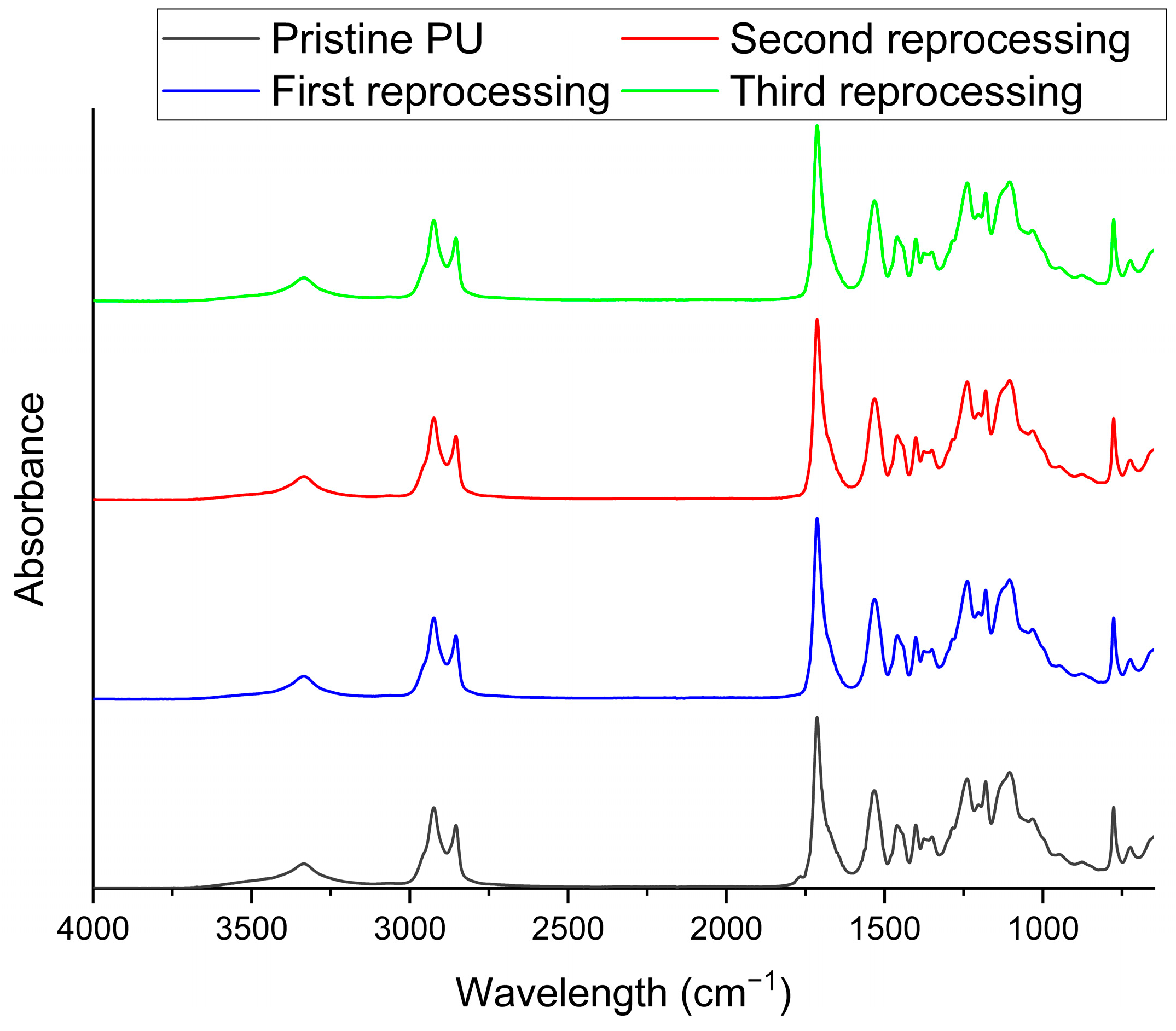
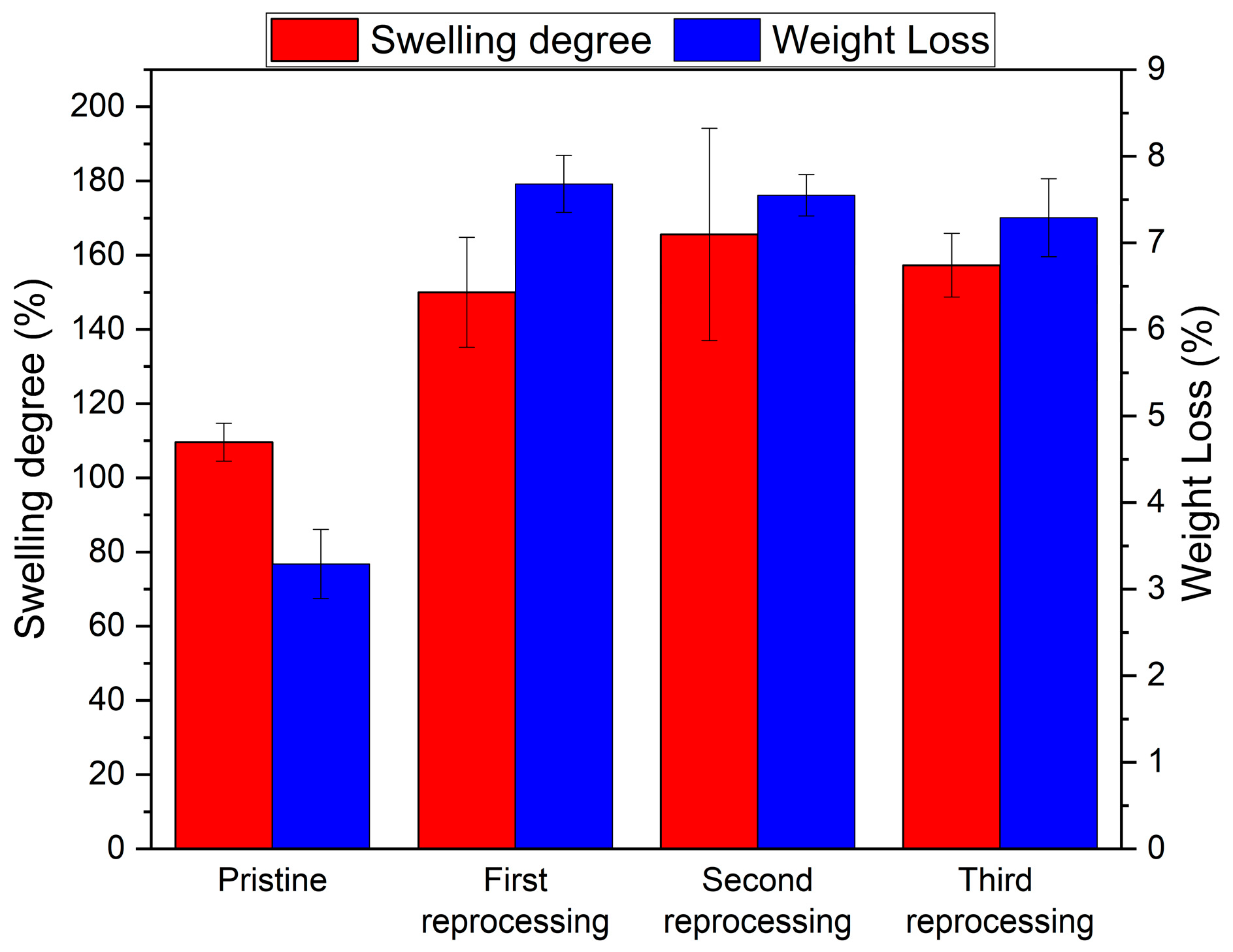
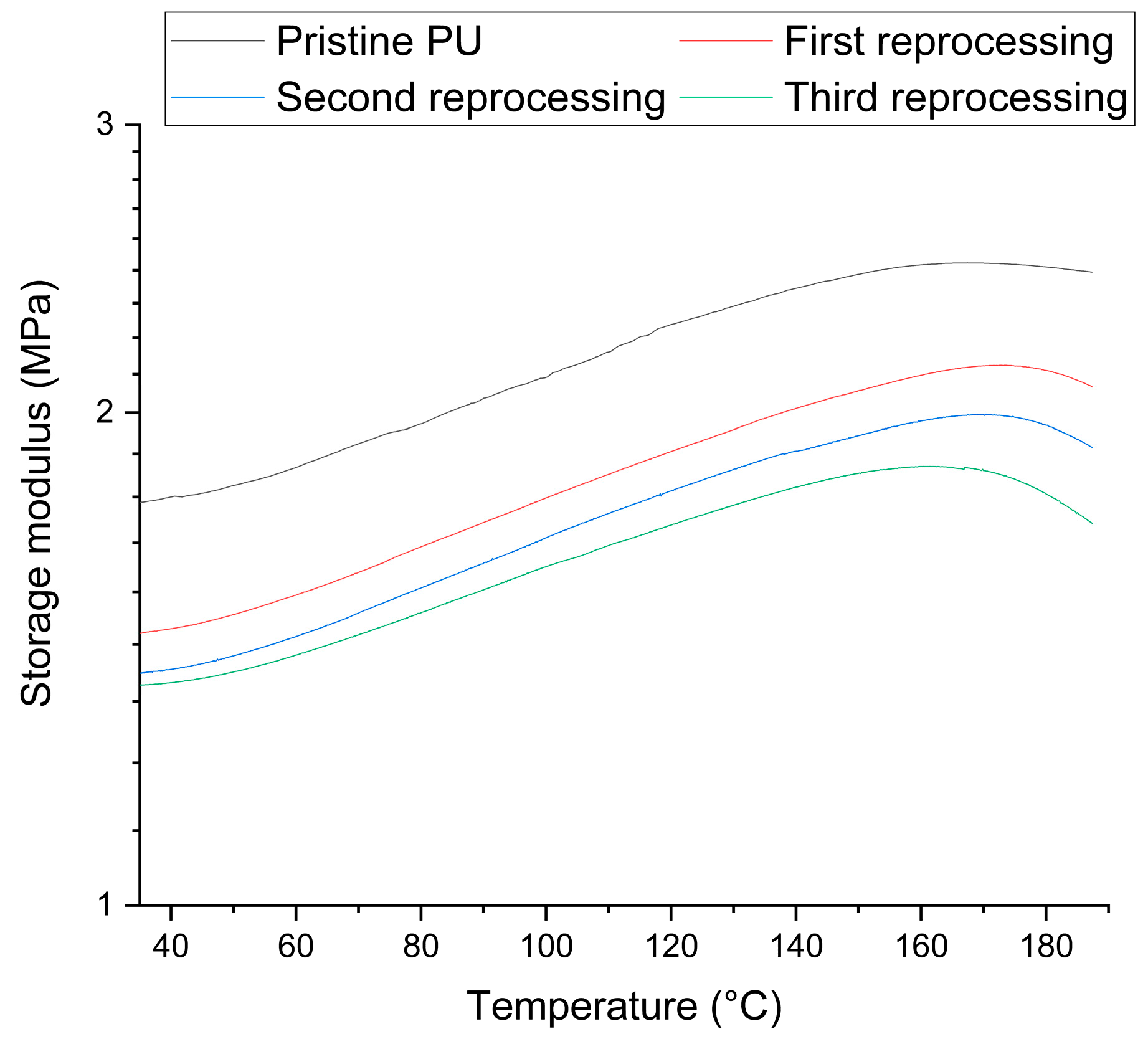
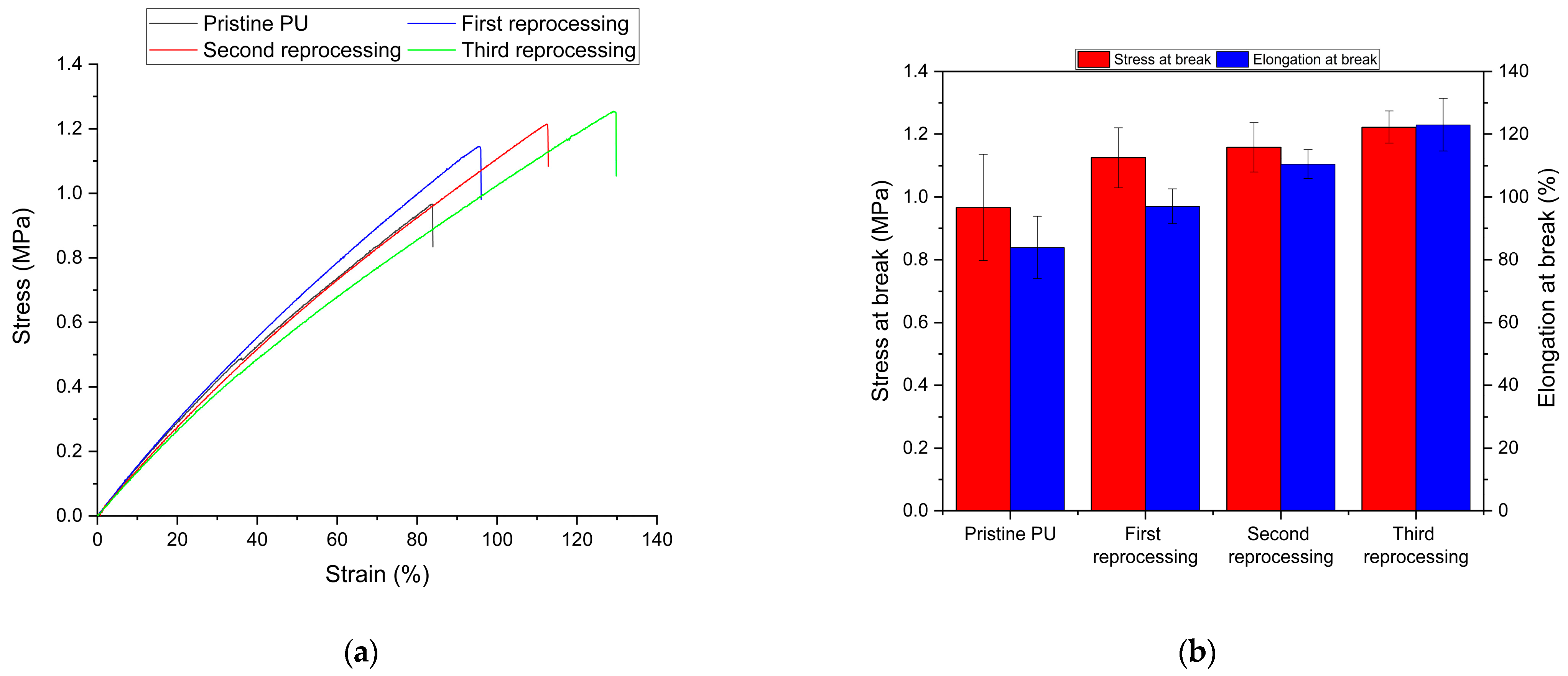
3.2. Netwok Reprocessing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Serrano, A.; Borreguero, A.M.; Garrido, I.; Rodríguez, J.F.; Carmona, M. Reducing Heat Loss through the Building Envelope by Using Polyurethane Foams Containing Thermoregulating Microcapsules. Appl. Therm. Eng. 2016, 103, 226–232. [Google Scholar] [CrossRef]
- Mekewi, M.A.; Ramadan, A.M.; ElDarse, F.M.; Abdel Rehim, M.H.; Mosa, N.A.; Ibrahim, M.A. Preparation and Characterization of Polyurethane Plasticizer for Flexible Packaging Applications: Natural Oils Affirmed Access. Egypt. J. Pet. 2017, 26, 9–15. [Google Scholar] [CrossRef]
- Daemi, H.; Rajabi-Zeleti, S.; Sardon, H.; Barikani, M.; Khademhosseini, A.; Baharvand, H. A Robust Super-Tough Biodegradable Elastomer Engineered by Supramolecular Ionic Interactions. Biomaterials 2016, 84, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Uscátegui, Y.L.; Díaz, L.E.; Valero, M.F. In Vitro and in Vivo Biocompatibility of Polyurethanes Synthesized with Castor Oil Polyols for Biomedical Devices. J. Mater. Res. 2019, 34, 519–531. [Google Scholar] [CrossRef]
- Plastic Europe Plastics—The Facts. 2022. Available online: https://plasticseurope.org/knowledge-hub/plastics-the-facts-2022/ (accessed on 15 June 2024).
- Mariotti, N.; Viada, G.; Galliano, S.; Menozzi, A.; Tammaro, F.; Gianelli, W.; Bonomo, M.; Barolo, C. Increasing Circular and Bio-Based Content of a Thermosetting Polyurethane for Encapsulation of Optoelectronic Devices: A Multivariate Investigation. J. Clean. Prod. 2023, 408, 137161. [Google Scholar] [CrossRef]
- Datta, J.; Kopczyńska, P.; Simón, D.; Rodríguez, J.F. Thermo-Chemical Decomposition Study of Polyurethane Elastomer Through Glycerolysis Route with Using Crude and Refined Glycerine as a Transesterification Agent. J. Polym. Env. Environ. 2018, 26, 166–174. [Google Scholar] [CrossRef]
- Madbouly, S.A. Novel Recycling Processes for Thermoset Polyurethane Foams. Curr. Opin. Green. Sustain. Chem. 2023, 42, 100835. [Google Scholar] [CrossRef]
- Simón, D.; Borreguero, A.M.; De Lucas, A.; Rodríguez, J.F. Glycolysis of Viscoelastic Flexible Polyurethane Foam Wastes. Polym. Degrad. Stab. 2015, 116, 23–35. [Google Scholar] [CrossRef]
- Heiran, R.; Ghaderian, A.; Reghunadhan, A.; Sedaghati, F.; Thomas, S.; Haghighi, A.h. Glycolysis: An Efficient Route for Recycling of End of Life Polyurethane Foams. J. Polym. Res. 2021, 28, 22. [Google Scholar] [CrossRef]
- Gama, N.; Godinho, B.; Marques, G.; Silva, R.; Barros-Timmons, A.; Ferreira, A. Recycling of Polyurethane Scraps via Acidolysis. Chem. Eng. J. 2020, 395, 125102. [Google Scholar] [CrossRef]
- Zhao, L.; Semetey, V. Recycling Polyurethanes through Transcarbamoylation. ACS Omega 2021, 6, 4175–4183. [Google Scholar] [CrossRef] [PubMed]
- Simón, D.; Borreguero, A.M.; de Lucas, A.; Rodríguez, J.F. Recycling of Polyurethanes from Laboratory to Industry, a Journey towards the Sustainability. Waste Manag. 2018, 76, 147–171. [Google Scholar] [CrossRef]
- Deng, Y.; Dewil, R.; Appels, L.; Ansart, R.; Baeyens, J.; Kang, Q. Reviewing the Thermo-Chemical Recycling of Waste Polyurethane Foam. J. Env. Environ. Manag. 2021, 278, 111527. [Google Scholar] [CrossRef] [PubMed]
- Zia, K.M.; Bhatti, H.N.; Bhatti, I.A. Methods for Polyurethane and Polyurethane Composites, Recycling and Recovery: A Review. React. Funct. Polym. 2007, 67, 675–692. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, Y.; Wang, D.; Yan, M.; Zhang, J.; Zhang, P.; Ding, T.; Chen, L.; Chen, C. Current Technologies for Plastic Waste Treatment: A Review. J. Clean. Prod. 2021, 282, 124523. [Google Scholar] [CrossRef]
- Kemona, A.; Piotrowska, M. Polyurethane Recycling and Disposal: Methods and Prospects. Polymers 2020, 12, 1752. [Google Scholar] [CrossRef] [PubMed]
- Bowman, C.; Du Prez, F.; Kalow, J. Introduction to Chemistry for Covalent Adaptable Networks. Polym. Chem. 2020, 11, 5295–5296. [Google Scholar] [CrossRef]
- Mcbride, M.K.; Worrell, B.T.; Brown, T.; Cox, L.M.; Sowan, N.; Wang, C.; Podgorski, M.; Martinez, A.M.; Bowman, C.N. Enabling Applications of Covalent Adaptable Networks. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 175–198. [Google Scholar] [CrossRef]
- Kloxin, C.J.; Bowman, C.N. Covalent Adaptable Networks: Smart, Reconfigurable and Responsive Network Systems. Chem. Soc. Rev. 2013, 42, 7161–7173. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-like Malleable Materials from Permanent Organic Networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef]
- Zheng, J.; Png, Z.M.; Ng, S.H.; Tham, G.X.; Ye, E.; Goh, S.S.; Loh, X.J.; Li, Z. Vitrimers: Current Research Trends and Their Emerging Applications. Mater. Today 2021, 51, 586–625. [Google Scholar] [CrossRef]
- Miravalle, E.; Bracco, P.; Brunella, V.; Barolo, C.; Zanetti, M. Improving Sustainability through Covalent Adaptable Networks in the Recycling of Polyurethane Plastics. Polymers 2023, 15, 3780. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Lu, D.; Zhao, X.; Li, Y.; Zeng, J. Sustainable and Malleable Polyurethane Networks from Castor Oil and Vanillin with Tunable Mechanical Properties. Ind. Crops Prod. 2021, 174, 114198. [Google Scholar] [CrossRef]
- Zhang, C.; Liang, H.; Liang, D.; Lin, Z.; Chen, Q.; Feng, P.; Wang, Q. Renewable Castor-Oil-Based Waterborne Polyurethane Networks: Simultaneously Showing High Strength, Self-Healing, Processability and Tunable Multishape Memory. Angew. Chem.-Int. Ed. 2021, 60, 4289–4299. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, W.; Ning, Z.; Yang, B.; Zeng, Y. Sustainable Polyurethane Networks Based on Rosin with Reprocessing Performance. Polymers 2021, 13, 3538. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, H.; Liu, W.; Huang, J.; Yang, D.; Qiu, X.; Zhao, L.; Hu, F.; Feng, Y. Solvent-Free Synthesis of High-Performance Polyurethane Elastomer Based on Low-Molecular-Weight Alkali Lignin. Int. J. Biol. Macromol. 2023, 225, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Yin, Q.; Han, F.; Cheng, J.; Zhao, J.; Zhang, J. A Bio-Based Healable/Renewable Polyurethane Elastomer Derived from L-Tyrosine/Vanillin/Dimer Acid. Chem. Eng. Sci. 2022, 258, 117736. [Google Scholar] [CrossRef]
- Kim, H.; Cha, I.; Yoon, Y.; Cha, B.J.; Yang, J.; Kim, Y.D.; Song, C. Facile Mechanochemical Synthesis of Malleable Biomass-Derived Network Polyurethanes and Their Shape-Memory Applications. ACS Sustain. Chem. Eng. 2021, 9, 6952–6961. [Google Scholar] [CrossRef]
- Yan, P.; Zhao, W.; Fu, X.; Liu, Z.; Kong, W.; Zhou, C.; Lei, J. Multifunctional Polyurethane-Vitrimers Completely on Transcarbamoylation of Carbamates: Thermally-Induced Dual-Shape Memory Effect and self-Welding†. RSC Adv. 2017, 7, 26858–26866. [Google Scholar] [CrossRef]
- Shi, J.; Zheng, T.; Zhang, Y.; Guo, B.; Xu, J. Reprocessable Cross-Linked Polyurethane with Dynamic and Tunable Phenol-Carbamate Network. ACS Sustain. Chem. Eng. 2020, 8, 1207–1218. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, C.; Song, F.; Shang, Q.; Hu, Y.; Jia, P.; Liu, C.; Hu, L.; Zhu, G.; Huang, J.; et al. Castor-Oil-Based, Robust, Self-Healing, Shape Memory, and Reprocessable Polymers Enabled by Dynamic Hindered Urea Bonds and Hydrogen Bonds. Chem. Eng. J. 2022, 429, 131848. [Google Scholar] [CrossRef]
- Sun, Y.; Sheng, D.; Wu, H.; Tian, X.; Xie, H.; Shi, B. Bio-Based Vitrimer-like Polyurethane Based on Dynamic Imine Bond with Antibacterial Ability. Polymers 2021, 233, 124208. [Google Scholar] [CrossRef]
- Debnath, S.; Tiwary, S.K.; Ojha, U. Dynamic Carboxylate Linkage Based Reprocessable and Self-Healable Segmented Polyurethane Vitrimers Displaying Creep Resistance Behavior and Triple Shape Memory Ability. ACS Appl. Polym. Mater. 2021, 3, 2166–2177. [Google Scholar] [CrossRef]
- Li, Q.; Ma, S.; Li, P.; Wang, B.; Feng, H.; Lu, N.; Wang, S.; Liu, Y.; Xu, X.; Zhu, J. Biosourced Acetal and Diels−Alder Adduct Concurrent Polyurethane Covalent Adaptable Network. Macromolecules 2021, 54, 1742–1753. [Google Scholar] [CrossRef]
- Wen, Z.; Raquez, J. Catalyst-Free Reprocessable Crosslinked Biobased Polybenzoxazine-Polyurethane Based on Dynamic Carbamate Chemistry. J. Appl. Polym. Sci. 2022, 139, 52120. [Google Scholar] [CrossRef]
- Levita, G.; De Petris, S.; Marchetti, A.; Lazzeri, A. Crosslink Density and Fracture Toughness of Epoxy Resins. J. Mater. Sci. 1991, 26, 2348–2352. [Google Scholar] [CrossRef]
- Schroeder, J.A.; Madsen, P.A.; Foister, R.T. Structure/Property Relationships for a Series of Crosslinked Aromatic/Aliphatic Epoxy Mixtures. Polymer 1987, 28, 929–940. [Google Scholar] [CrossRef]
- Chen, W.; Zhu, C.; Gu, X. Thermosetting Polyurethanes with Water-Swollen and Shape Memory Properties. J. Appl. Polym. Sci. 2002, 84, 1504–1512. [Google Scholar] [CrossRef]
- Trovati, G.; Sanches, E.A.; Neto, S.C.; Mascarenhas, Y.P.; Chierice, G.O. Characterization of Polyurethane Resins by FTIR, TGA, and XRD. J. Appl. Polym. Sci. 2010, 115, 263–268. [Google Scholar] [CrossRef]
- Hu, J.; Chen, Z.; He, Y.; Huang, H.; Zhang, X. Synthesis and Structure Investigation of Hexamethylene Diisocyanate (HDI)-Based Polyisocyanates. Res. Chem. Intermed. 2017, 43, 2799–2816. [Google Scholar] [CrossRef]
- Morales-Cerrada, R.; Tavernier, R.; Caillol, S. Fully Bio-Based Thermosetting Polyurethanes from Bio-Based Polyols and Isocyanates. Polymers 2021, 13, 1255. [Google Scholar] [CrossRef] [PubMed]
- Fridrihsone, A.; Stirna, U.; Lazdiņa, B.; Misane, M.; Vilsone, D. Characterization of Polyurethane Networks Structure and Properties Based on Rapeseed Oil Derived Polyol. Eur. Polym. J. 2013, 49, 1204–1214. [Google Scholar] [CrossRef]
- Głowińska, E.; Wolak, W.; Datta, J. Eco-Friendly Route for Thermoplastic Polyurethane Elastomers with Bio-Based Hard Segments Composed of Bio-Glycol and Mixtures of Aromatic–Aliphatic and Aliphatic–Aliphatic Diisocyanate. J. Polym. Env. Environ. 2021, 29, 2140–2149. [Google Scholar] [CrossRef] [PubMed]
- Jayavani, S.; Sunanda, S.; Varghese, T.O.; Nayak, S.K. Synthesis and Characterizations of Sustainable Polyester Polyols from Non-Edible Vegetable Oils: Thermal and Structural Evaluation. J. Clean. Prod. 2017, 162, 795–805. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, H.; Zhang, L.; Hu, L.; Zhou, Y. Study of the Mechanical, Thermal Properties and Flame Retardancy of Rigid Polyurethane Foams Prepared from Modified Castor-Oil-Based Polyols. Ind. Crops Prod. 2014, 59, 135–143. [Google Scholar] [CrossRef]
- Chattopadhyay, D.K.; Webster, D.C. Thermal Stability and Flame Retardancy of Polyurethanes. Prog. Polym. Sci. 2009, 34, 1068–1133. [Google Scholar] [CrossRef]
- Oenema, J.; Liu, H.; De Coensel, N.; Eschenbacher, A.; Van de Vijver, R.; Weng, J.; Li, L.; Wang, C.; Van Geem, K.M. Review on the Pyrolysis Products and Thermal Decomposition Mechanisms of Polyurethanes. J. Anal. Appl. Pyrolysis 2022, 168, 105723. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; Hillmyer, M.A.; Dichtel, W.R. Dichtel Structural Effects on the Reprocessability and Stress Relaxation of crosslinked Polyhydroxyurethanes. J. Appl. Polym. Sci. 2017, 134, 44984. [Google Scholar] [CrossRef]
- Dong, J.; Yan, H.; Lv, X.; Wang, Z.; Rao, Z.; Zhu, B.; Wu, J.; Zhou, Y.; Chen, H. Reprocessable Polyurethane Elastomers Based on Reversible Ketal Exchange: Dielectric Properties and Water Resistance. J. Mater. Chem. C Mater. 2022, 11, 1369–1380. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Yao, W.; Dong, Y.; Zhang, B.; Dong, X.; Fang, H.; Zhang, G.; Ding, Y. Dynamically Cross-Linked Waterborne Polyurethanes: Transalkylation Exchange of C-N Bonds Toward High Performance and Reprocessable Thermosets. ACS Appl. Polym. Mater. 2022, 4, 5920–5926. [Google Scholar] [CrossRef]
- Zheng, N.; Xu, Y.; Zhao, Q.; Xie, T. Dynamic Covalent Polymer Networks: A Molecular Platform for Designing Functions beyond Chemical Recycling and Self-Healing. Chem. Rev. 2021, 121, 1716–1745. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zheng, T.; Guo, B.; Xu, J. Solvent-Free Thermo-Reversible and Self-Healable Crosslinked Polyurethane with Dynamic Covalent Networks Based on Phenol-Carbamate Bonds. Polymer 2019, 181, 121788. [Google Scholar] [CrossRef]
- Shi, J.; Zheng, T.; Zhang, Y.; Guo, B.; Xu, J. Cross-Linked Polyurethane with Dynamic Phenol-Carbamate Bonds: Properties Affected by the Chemical Structure of Isocyanate. Polym. Chem. 2021, 12, 2421–2432. [Google Scholar] [CrossRef]
- Chiou, B.S.; Schoen, P.E. Effects of Crosslinking on Thermal and Mechanical Properties of Polyurethanes. J. Appl. Polym. Sci. 2002, 83, 212–223. [Google Scholar] [CrossRef]
- Huang, J.; Fu, P.; Li, W.; Xiao, L.; Chen, J.; Nie, X. Influence of Crosslinking Density on the Mechanical and Thermal Properties of Plant Oil-Based Epoxy Resin. RSC Adv. 2022, 12, 23048–23056. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Td5% (°C) | Tg (°C) | E′ (MPa) | Ve (mol/m3) | Tensile Strength (MPa) | Strain at Break (%) |
|---|---|---|---|---|---|---|
| Pristine | 266.4 | −28.0 | 1.77 ± 0.02 | 226.5 ± 3 | 0.99 ± 0.12 | 80.2 ± 11 |
| First Reprocessing | 259.4 | −27.6 | 1.54 ± 0.1 | 197.2 ± 12.2 | 1.09 ± 0.13 | 97.1 ± 5.5 |
| Second Reprocessing | 259.4 | −28.2 | 1.41 ± 0.02 | 181.1 ± 3.1 | 1.14 ± 0.08 | 110.5 ± 4.6 |
| Third Reprocessing | 258.8 | −28.3 | 1.36 ± 0.01 | 174.6 ± 1.5 | 1.17 ± 0.05 | 123 ± 8.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miravalle, E.; Viada, G.; Bonomo, M.; Barolo, C.; Bracco, P.; Zanetti, M. Recycling of Commercially Available Biobased Thermoset Polyurethane Using Covalent Adaptable Network Mechanisms. Polymers 2024, 16, 2217. https://doi.org/10.3390/polym16152217
Miravalle E, Viada G, Bonomo M, Barolo C, Bracco P, Zanetti M. Recycling of Commercially Available Biobased Thermoset Polyurethane Using Covalent Adaptable Network Mechanisms. Polymers. 2024; 16(15):2217. https://doi.org/10.3390/polym16152217
Chicago/Turabian StyleMiravalle, Edoardo, Gabriele Viada, Matteo Bonomo, Claudia Barolo, Pierangiola Bracco, and Marco Zanetti. 2024. "Recycling of Commercially Available Biobased Thermoset Polyurethane Using Covalent Adaptable Network Mechanisms" Polymers 16, no. 15: 2217. https://doi.org/10.3390/polym16152217
APA StyleMiravalle, E., Viada, G., Bonomo, M., Barolo, C., Bracco, P., & Zanetti, M. (2024). Recycling of Commercially Available Biobased Thermoset Polyurethane Using Covalent Adaptable Network Mechanisms. Polymers, 16(15), 2217. https://doi.org/10.3390/polym16152217