

Long-Chain Bio-Based Nylon 514 Salt: Crystal Structure, Phase Transformation, and Polymerization

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of the Crystal Forms
2.3. Single Crystal X-ray Diffraction (SCXRD)
2.4. Spectroscopy Analysis
2.5. Thermal Analysis
2.6. Hirshfeld Surface Analysis (HSs)
2.7. Computational Details
2.8. Stability Experiments
2.9. Polymerization Experiments
3. Results and Discussion
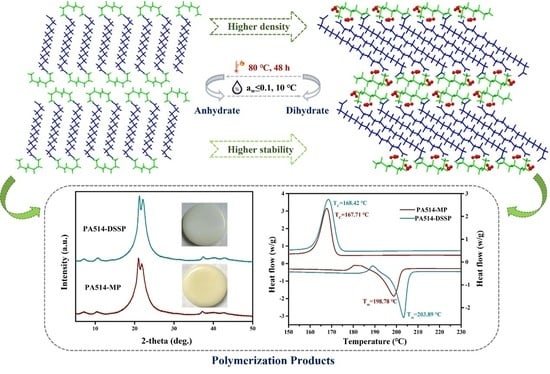
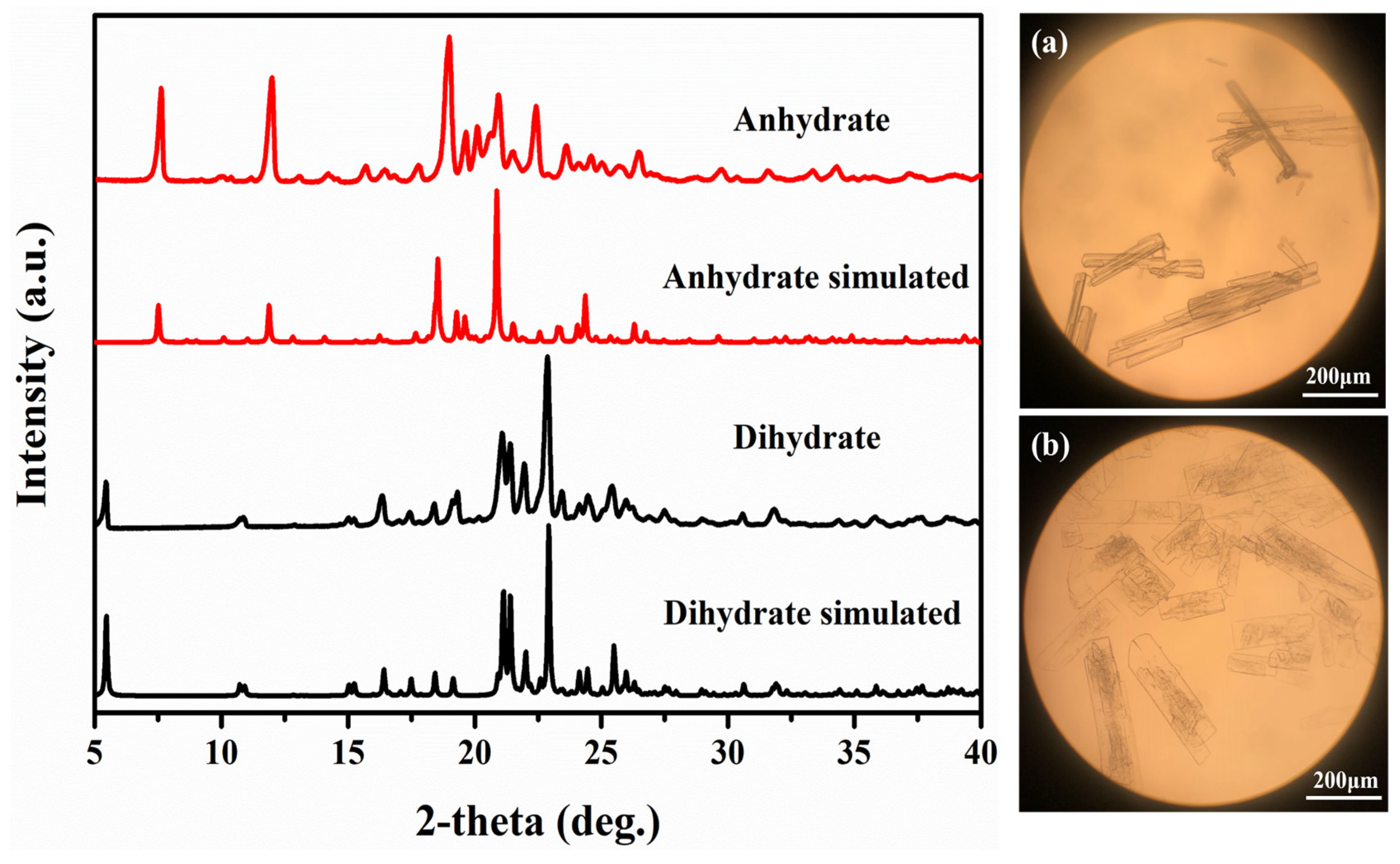
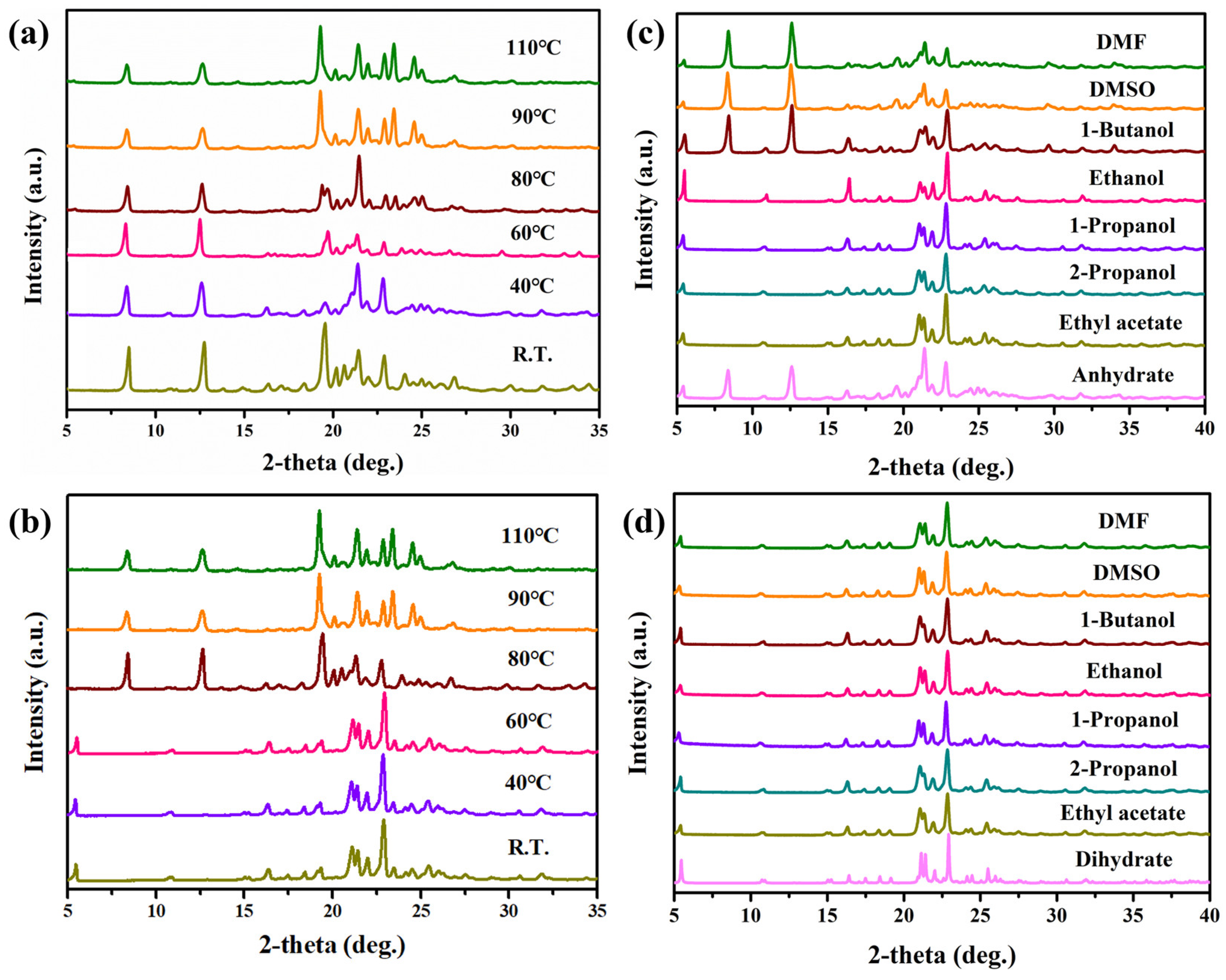
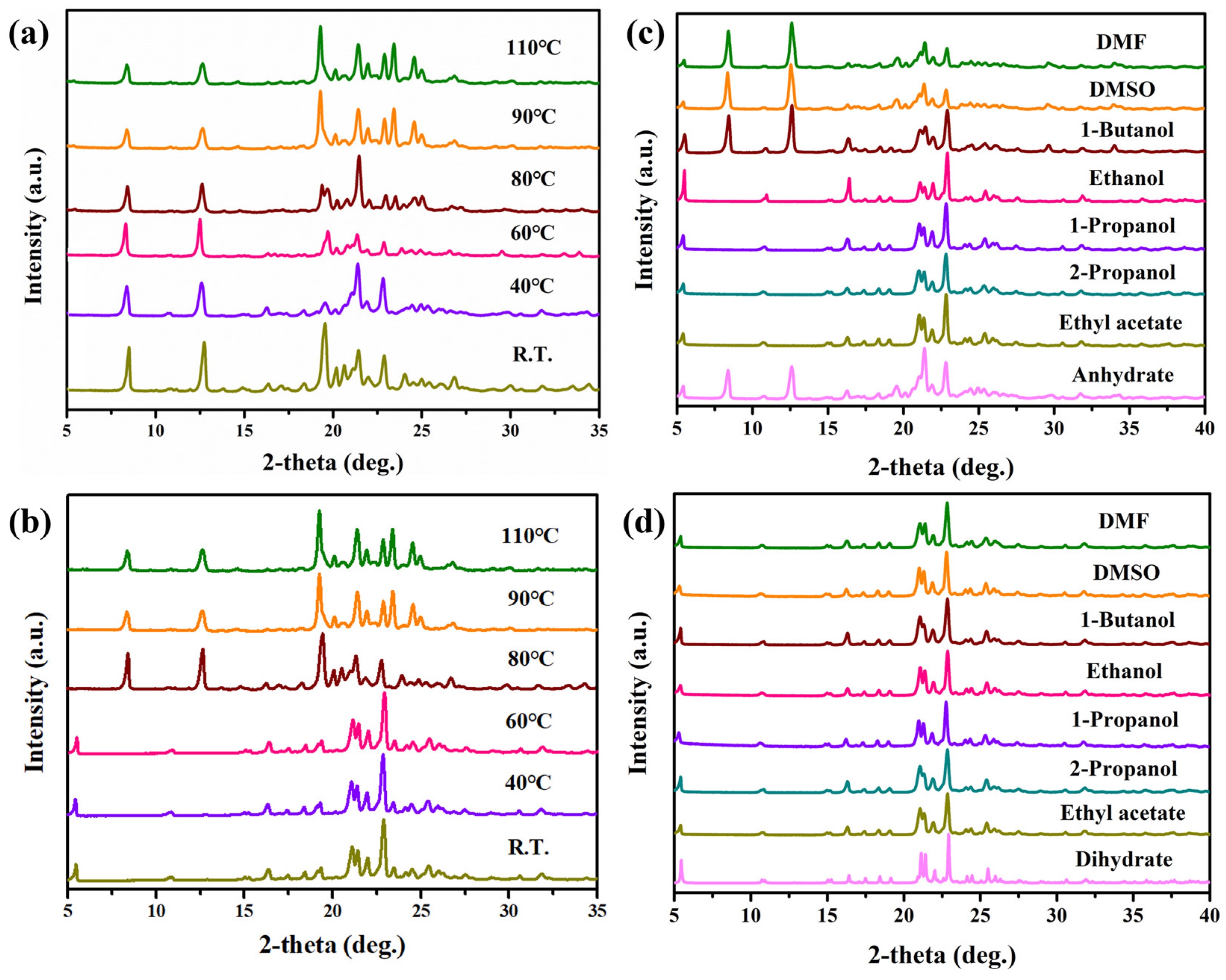
3.1. PXRD Analysis
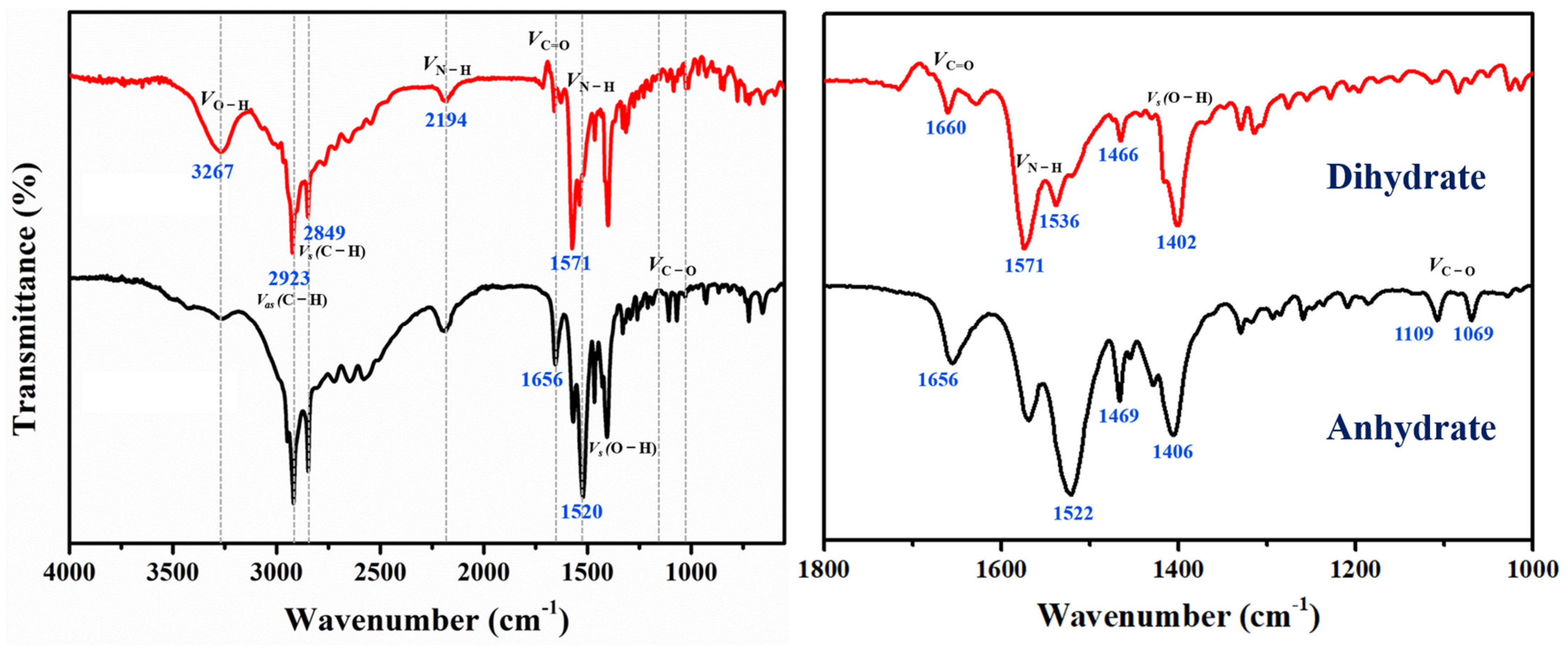
3.2. FTIR Spectroscopy Analysis
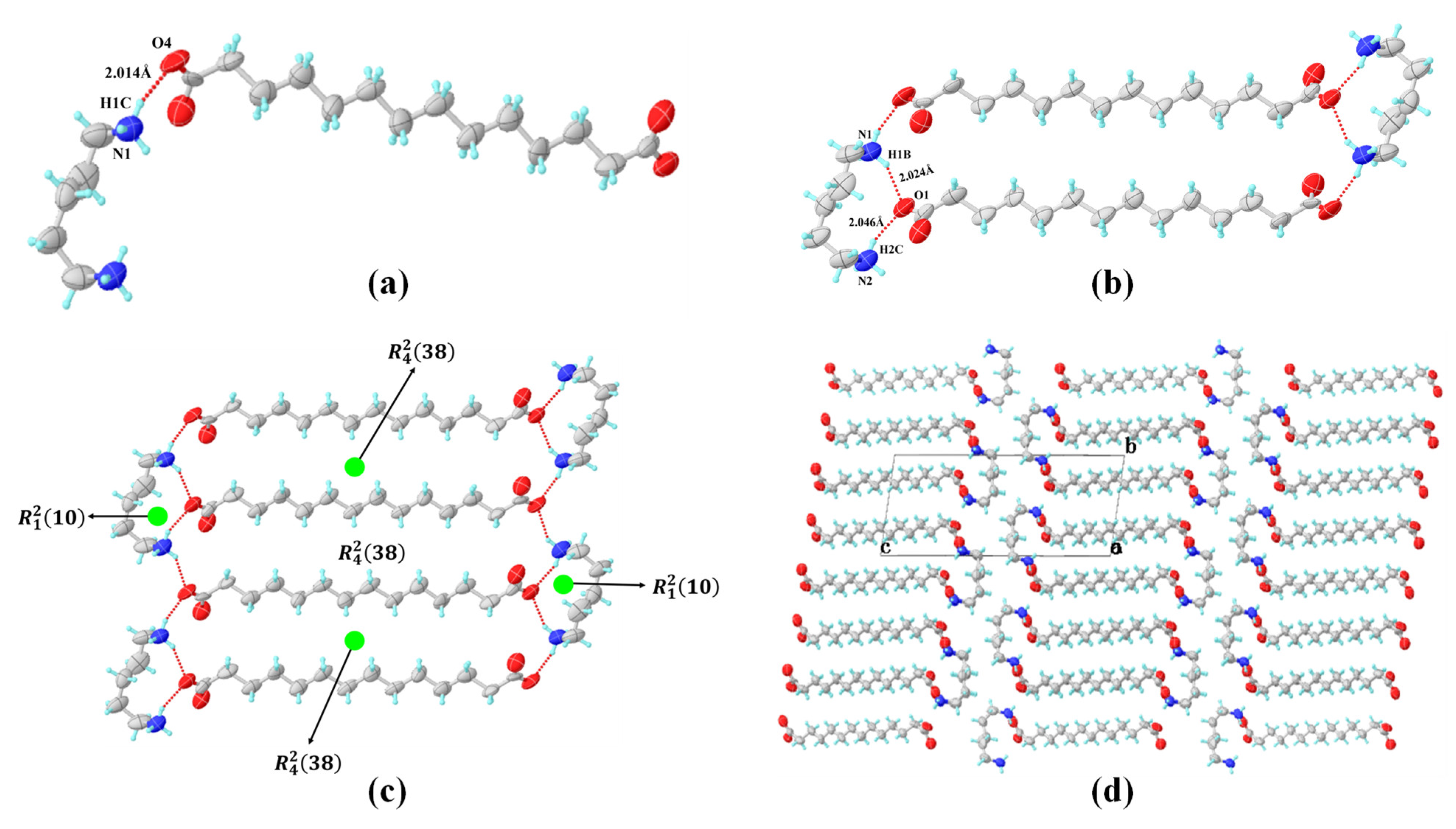
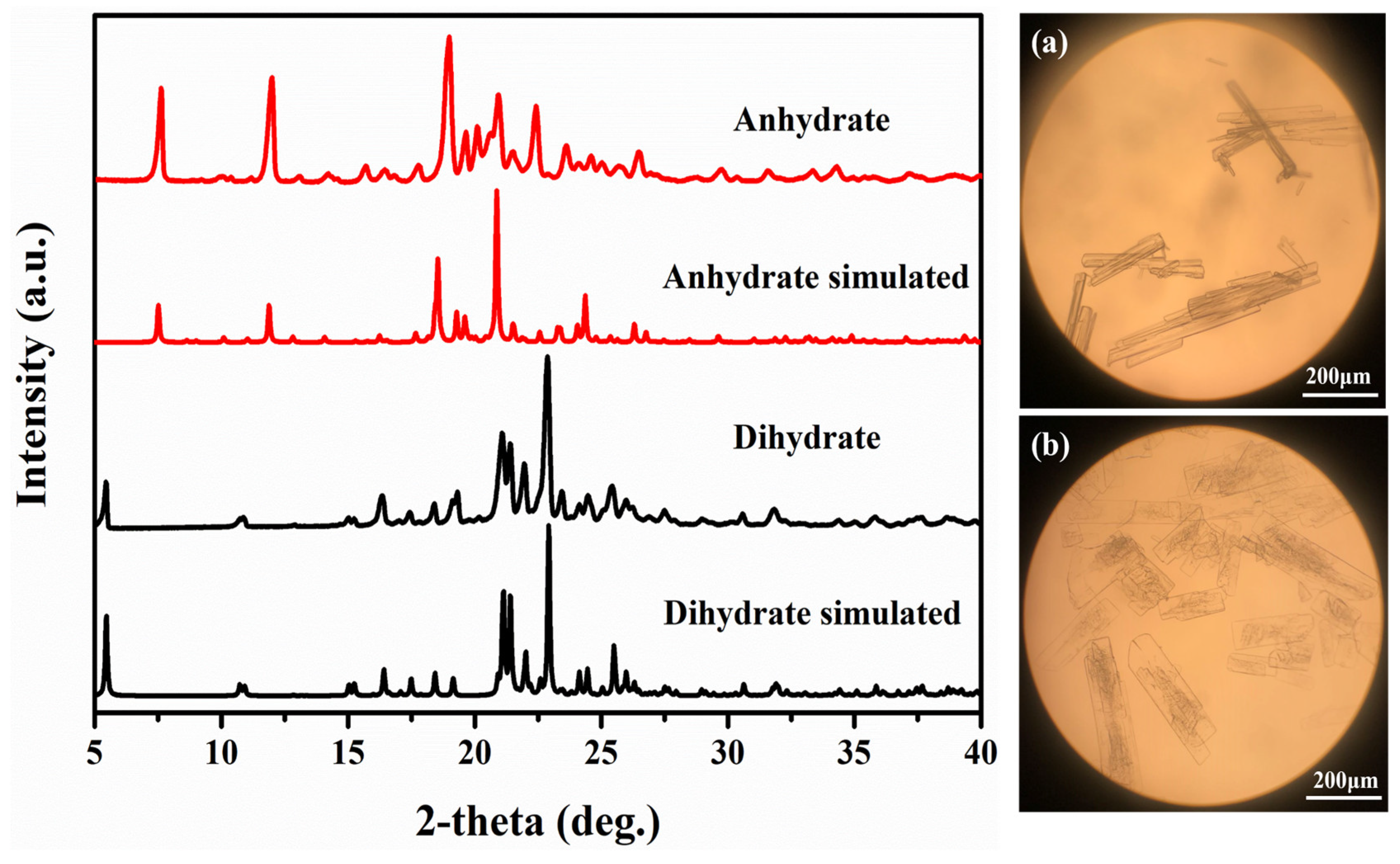
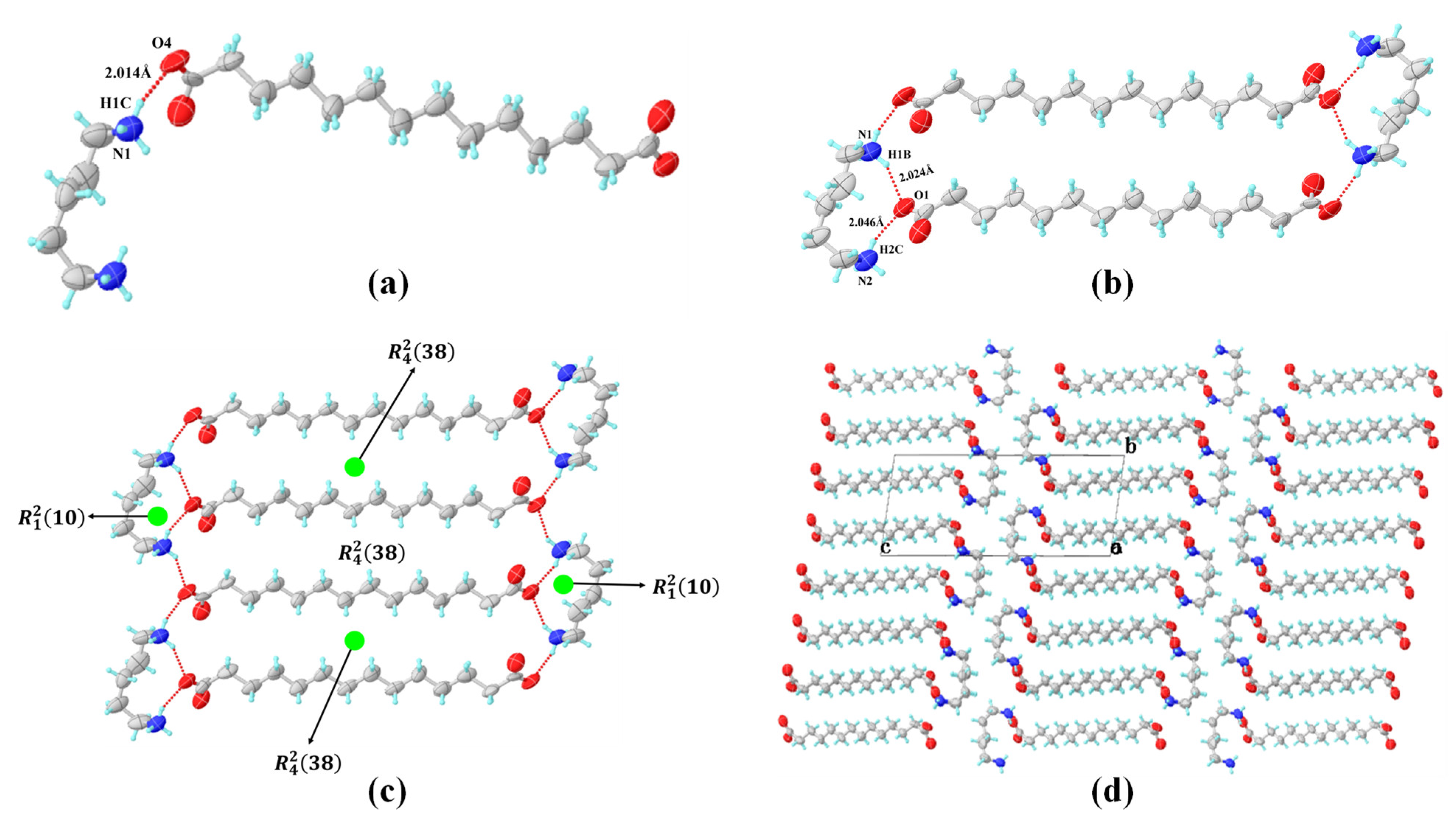
3.3. Crystal Structures Analysis of PDA-TDA Anhydrate and Dihydrate
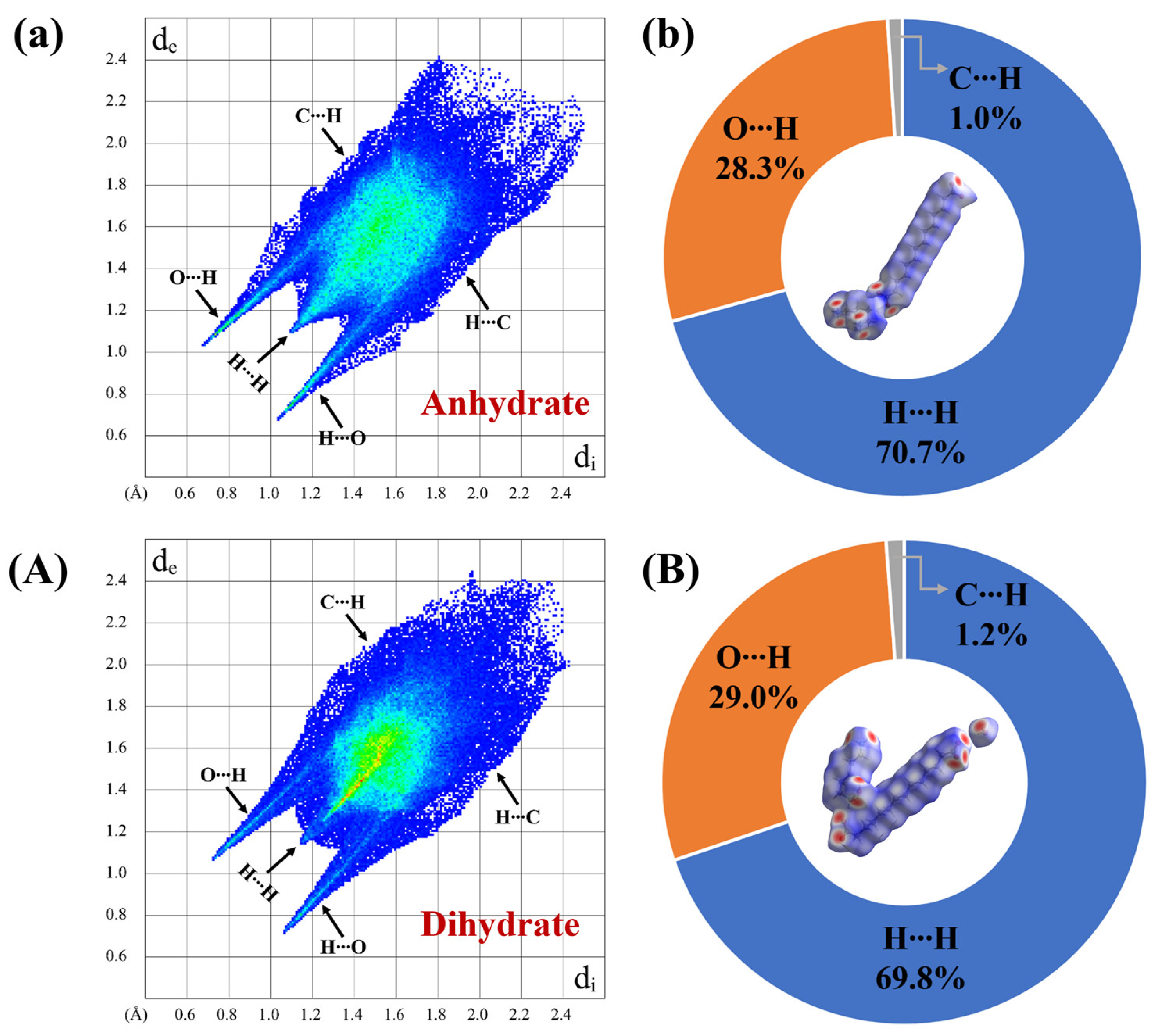
3.4. Hirshfeld Surface Analysis
3.5. Computational Analysis
3.6. Thermal Analysis
3.7. Stability Analysis
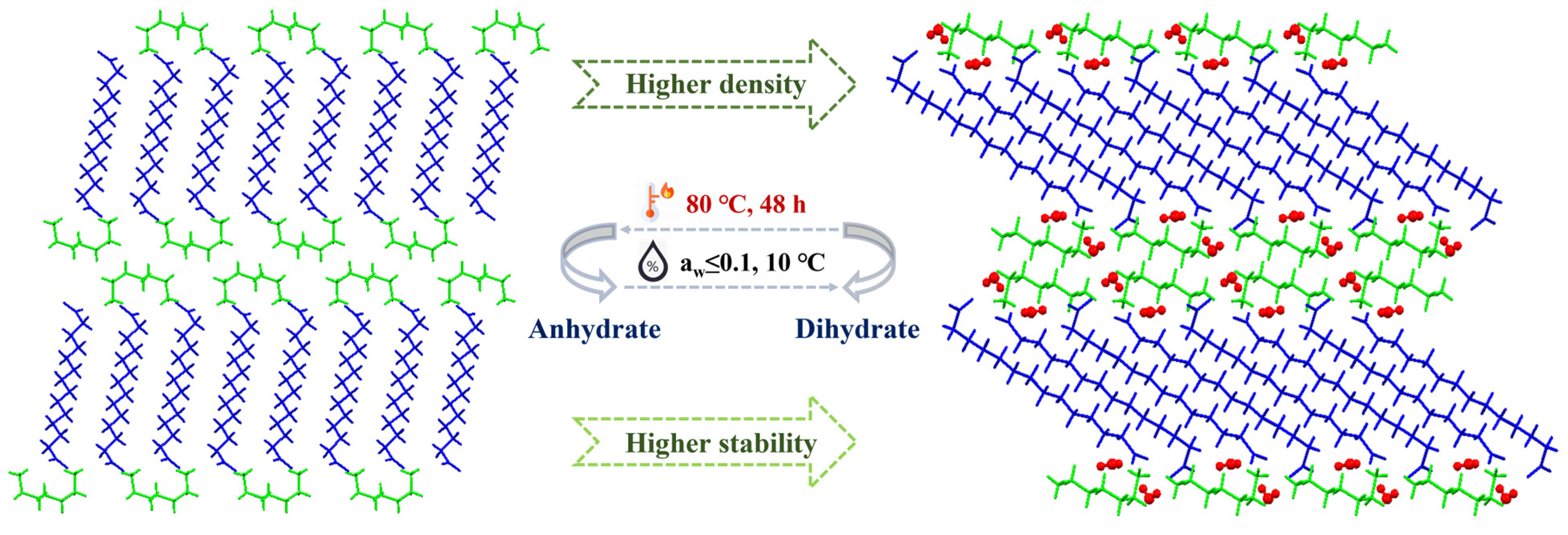
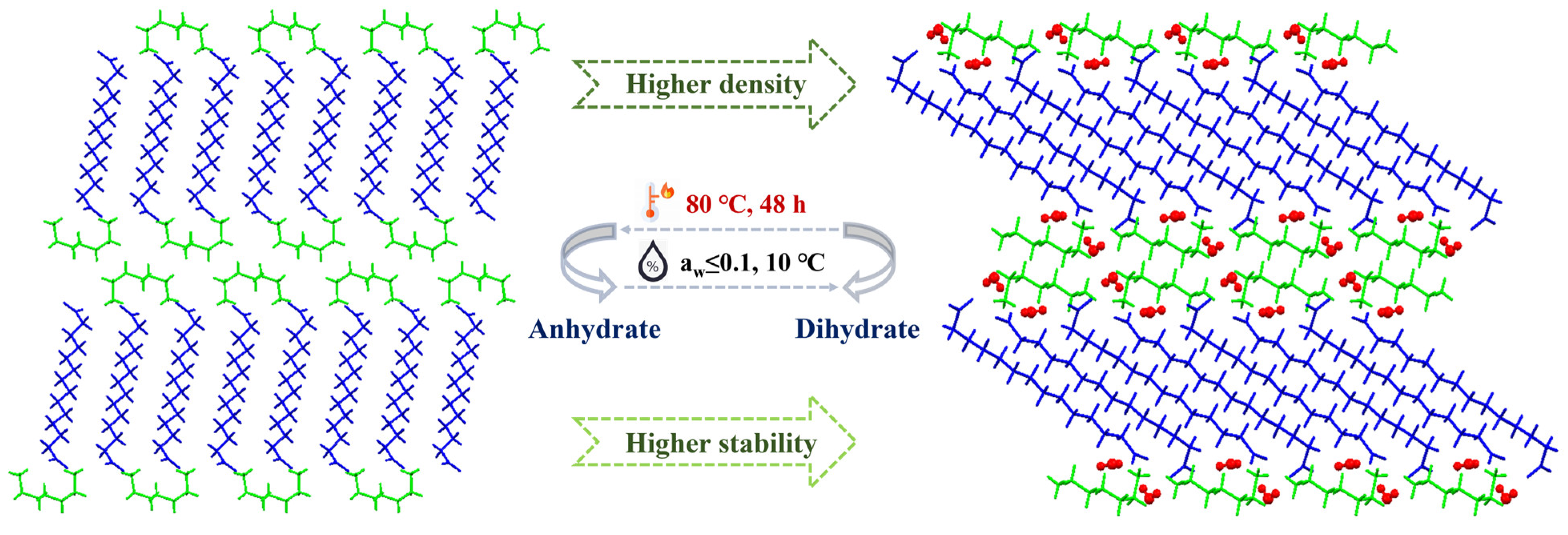
3.8. Transitions between the Dihydrate and Anhydrate
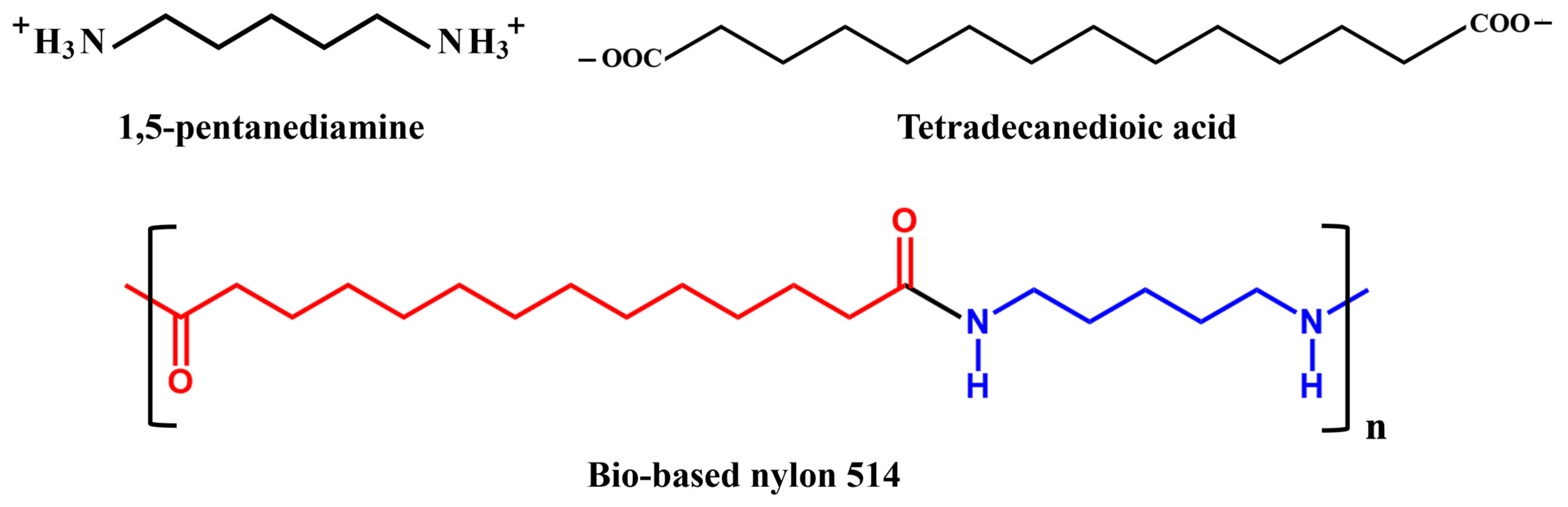
3.9. Polymerization Experiments Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Winnacker, M.; Rieger, B. Biobased Polyamides: Recent Advances in Basic and Applied Research. Macromol. Rapid Commun. 2016, 37, 1391–1413. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Ahn, J.H.; Kim, I.; Li, S.; Lee, S.Y. Synthesis, Characterization, and Application of Fully Biobased and Biodegradable Nylon-4,4 and -5,4. ACS Sustain. Chem. Eng. 2020, 8, 5604–5614. [Google Scholar] [CrossRef]
- Li, M. Study on melting and polymorphic behavior of poly(decamethylene terephthalamide). J. Polym. Sci. Part B Polym. Phys. 2019, 57, 465–472. [Google Scholar] [CrossRef]
- Deshmukh, Y.S.; Wilsens, C.H.R.M.; Verhoef, R.; Hansen, M.R.; Dudenko, D.; Graf, R.; Klop, E.A.; Rastogi, S. Conformational and Structural Changes with Increasing Methylene Segment Length in Aromatic–Aliphatic Polyamides. Macromolecules 2016, 49, 950–962. [Google Scholar] [CrossRef]
- Kim, Y.J.; Yohana, K.E.; Lee, H.-s.; Kim, J. Solid-State Polymerization of Semiaromatic Copolyamides of Nylon-4,T and Nylon-4,6: Composition Ratio Effect and Thermal Properties. Ind. Eng. Chem. Res. 2012, 51, 15801–15810. [Google Scholar] [CrossRef]
- Anwar, S.; Hassanpour Amiri, M.; Jiang, S.; Abolhasani, M.M.; Rocha, P.R.F.; Asadi, K. Piezoelectric Nylon-11 Fibers for Electronic Textiles, Energy Harvesting and Sensing. Adv. Funct. Mater. 2021, 31, 2004326. [Google Scholar] [CrossRef]
- Yanaka, A.; Sakai, W.; Kinashi, K.; Tsutsumi, N. Ferroelectric performance of nylons 6-12, 10-12, 11-12, and 12-12. RSC Adv. 2020, 10, 15740–15750. [Google Scholar] [CrossRef]
- Li, Y.-S.; Schwarz, G.; Kricheldorf, H.R. New polymers syntheses. 104. Synthesis of poly(ester-imide)s from nylon 6, nylon 11, or nylon 12. J. Polym. Sci. A Polym. Chem. 2000, 38, 1630–1638. [Google Scholar] [CrossRef]
- Bhushan, R.K.; Sharma, D. Green welding for various similar and dissimilar metals and alloys: Present status and future possibilities. Adv. Compos. Hybrid. Mater. 2019, 2, 389–406. [Google Scholar] [CrossRef]
- Liu, B.; Hu, G.; Zhang, J.; Yan, W. Non-isothermal crystallization, yellowing resistance and mechanical properties of heat-resistant nylon 10T/66/titania dioxide/glass fibre composites. RSC Adv. 2019, 9, 7057–7064. [Google Scholar] [CrossRef]
- Wang, T.; Li, H.; Diao, X.; Lu, X.; Ma, D.; Ji, N. Lignin to dispersants, adsorbents, flocculants and adhesives: A critical review on industrial applications of lignin. Ind. Crops Prod. 2023, 199, 116715. [Google Scholar] [CrossRef]
- Firdaus, M.; Meier, M.A.R. Renewable polyamides and polyurethanes derived from limonene. Green Chem. 2013, 15, 370–380. [Google Scholar] [CrossRef]
- Hashimoto, K.; Hashimoto, N.; Kamaya, T.; Yoshioka, J.; Okawa, H. Synthesis and properties of bio-based polyurethanes bearing hydroxy groups derived from alditols. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 976–985. [Google Scholar] [CrossRef]
- Miri, V.; Persyn, O.; Lefebvre, J.M.; Seguela, R. Effect of water absorption on the plastic deformation behavior of nylon 6. Eur. Polym. J. 2009, 45, 757–762. [Google Scholar] [CrossRef]
- Li, Z.; Li, S.; Yang, P.; Fang, X.; Wen, Q.; Li, M.; Zhuang, W.; Wu, J.; Ying, H. The effect of polymorphism on polymer properties: Crystal structure, stability and polymerization of the short-chain bio-based nylon 52 monomer 1,5-pentanediamine oxalate. IUCrJ 2023, 10, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Osswald, T.A.; García-Rodríguez, S. History of sustainable bio-based polymers. Green Chem. 2011, 1, 1–21. [Google Scholar]
- Yang, P.; Li, X.; Liu, H.; Li, Z.; Liu, J.; Zhuang, W.; Wu, J.; Ying, H. Thermodynamics, crystal structure, and characterization of a bio-based nylon 54 monomer. CrystEngComm 2019, 21, 7069–7077. [Google Scholar] [CrossRef]
- Ogunniyi, D.S. Castor oil: A vital industrial raw material. Bioresour. Technol. 2006, 97, 1086–1091. [Google Scholar] [CrossRef]
- Hong, S.H.; Kim, J.S.; Lee, S.Y.; In, Y.H.; Choi, S.S.; Rih, J.K.; Kim, C.H.; Jeong, H.; Hur, C.G.; Kim, J.J. The genome sequence of the capnophilic rumen bacterium Mannheimia succiniciproducens. Nat. Biotechnol. 2004, 22, 1275–1281. [Google Scholar] [CrossRef]
- Xue, Y.; Zhao, Y.; Ji, X.; Yao, J.; Busk, P.K.; Lange, L.; Huang, Y.; Zhang, S. Advances in bio-nylon 5X: Discovery of new lysine decarboxylases for the high-level production of cadaverine. Green Chem. 2020, 22, 8656–8668. [Google Scholar] [CrossRef]
- Mi, J.; Liu, S.; Qi, H.; Huang, J.; Lan, X.; Zhang, L. Cellular Engineering and Biocatalysis Strategies toward Sustainable Cadaverine Production: State of the Art and Perspectives. ACS Sustain. Chem. Eng. 2021, 9, 1061–1072. [Google Scholar] [CrossRef]
- Li, Y.; Song, X.; Xu, W.; Duan, X.; Shi, J.; Li, X. Preparation of biomass film from waste biomass energy corn stalk under carbon neutralization strategy. Mater. Today Commun. 2022, 32, 104001. [Google Scholar] [CrossRef]
- Ma, Z.; Qin, S.; Yao, Y.; Xin, Z.; Luan, L.; Zhang, Y.; Huang, Y. Directed synthesis of nylon 5X key monomer cadaverine with alkaline metal modified Ru@FAU catalysts. Appl. Catal. A—Gen. 2023, 658, 119172. [Google Scholar] [CrossRef]
- Li, Z.; Wang, K.; Xue, Y.; Lin, K.; Ji, X.; Huang, Y. Ionozymes for Efficient Synthesis of Cadaverine: Offering a Sustainable Way for Bio-nylon 5X Production. ACS Sustain. Chem. Eng. 2023, 11, 4197–4208. [Google Scholar] [CrossRef]
- Bosch, J. Modeling Techniques for Measuring Galaxy Properties in Multi-Epoch Surveys. J. Bus. Strategy 2011, 35, 49–57. [Google Scholar] [CrossRef]
- Burke, D.J.; Kawauchi, T.; Kade, M.J.; Leibfarth, F.A.; McDearmon, B.; Wolffs, M.; Kierstead, P.H.; Moon, B.; Hawker, C.J. Ketene-Based Route to rigid Cyclobutanediol Monomers for the Replacement of BPA in High Performance Polyesters. ACS Macro Lett. 2012, 1, 1228–1232. [Google Scholar] [CrossRef]
- Fleming, M.E.; Swift, J.A. Enhancement of Hydrate Stability through Substitutional Defects. Cryst. Growth Des. 2023, 23, 5860–5867. [Google Scholar] [CrossRef]
- Bui, M.; Nagapudi, K.; Chakravarty, P. Determination of BET Specific Surface Area of Hydrate-Anhydrate Systems Susceptible to Phase Transformation Using Inverse Gas Chromatography. AAPS PharmSciTech 2022, 23, 237. [Google Scholar] [CrossRef]
- Cui, P.; Yin, Q.; Guo, Y.; Gong, J. Polymorphic Crystallization and Transformation of Candesartan Cilexetil. Ind. Eng. Chem. Res. 2012, 51, 12910–12916. [Google Scholar] [CrossRef]
- Brittain, H.G. Polymorphism in Pharmaceutical Solids; Informa Healthcare: New York, NY, USA, 1999; Volume 192. [Google Scholar]
- Fujii, K.; Aoki, M.; Uekusa, H. Solid-State Hydration/Dehydration of ErythromycinA Investigated by ab Initio Powder X-ray Diffraction Analysis:Stoichiometric and Nonstoichiometric Dehydrated Hydrate. Cryst. Growth Des. 2013, 13, 2060–2066. [Google Scholar] [CrossRef]
- Liu, Z.; Yin, Q.; Zhang, X.; Gong, J.; Xie, C. Characterization and Structure Analysis of Cefodizime Sodium Solvates Crystallized from Water and Ethanol Binary Solvent Mixtures. Ind. Eng. Chem. Res. 2014, 53, 3373–3377. [Google Scholar] [CrossRef]
- SeethaLekshmi, S.; Kiran, M.S.R.N.; Ramamurty, U.; Varughese, S. Phase Transitions and Anisotropic Mechanical Response in a Water-rich Trisaccharide Crystal. Cryst. Growth Des. 2020, 20, 442–448. [Google Scholar] [CrossRef]
- Liu, F.; Hooks, D.E.; Li, N.; Mara, N.A.; Swift, J.A. Mechanical Properties of Anhydrous and Hydrated Uric Acid Crystals. Chem. Mater. 2018, 30, 3798–3805. [Google Scholar] [CrossRef]
- Noguerol, A.T.; Marta Igual, M.; Pagán, M.J. Developing psyllium fibre gel-based foods: Physicochemical, nutritional, optical and mechanical properties. Food Hydrocoll. 2022, 122, 107108. [Google Scholar] [CrossRef]
- Zhu, B.; Zhang, Q.; Ren, G.; Mei, X. Solid-State Characterization and Insight into Transformations and Stability of Apatinib Mesylate Solvates. Cryst. Growth Des. 2017, 17, 5994–6005. [Google Scholar] [CrossRef]
- Zhu, B.; Wang, J.-R.; Zhang, Q.; Li, M.; Guo, C.; Ren, G.; Mei, X. Stable Cocrystals and Salts of the Antineoplastic Drug Apatinib with Improved Solubility in Aqueous Solution. Cryst. Growth Des. 2018, 18, 4701–4714. [Google Scholar] [CrossRef]
- Kocian, Š.; Štejfa, V.; Rohlíček, J.; Červinka, C. Calorimetric and Crystallographic Phase-Behavior Study of Selected 1-Butylpyridinium Ionic Liquids. Cryst. Growth Des. 2023, 23, 5221–5235. [Google Scholar] [CrossRef]
- Du, W.; Yin, Q.; Hao, H.; Bao, Y.; Zhang, X.; Huang, J.; Li, X.; Xie, C.; Gong, J. Solution-Mediated Polymorphic Transformation of Prasugrel Hydrochloride from Form II to Form I. Ind. Eng. Chem. Res. 2014, 53, 5652–5659. [Google Scholar] [CrossRef]
- Steendam, R.R.E.; Khandavilli, U.B.R.; Keshavarz, L.; Frawley, P.J. Solution versus Crystal Hydration: The Case of γ-Amino Acid Pregabalin. Cryst. Growth Des. 2019, 19, 4483–4488. [Google Scholar] [CrossRef]
- Boeckmann, J.; Näther, C. Solid-state transformation of [Co(NCS)2(pyridine)4] into [Co(NCS)2(pyridine)2]n: From Curie-Weiss paramagnetism to single chain magnetic behaviour. Daltont 2010, 39, 11019–11026. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Sommer, F.; Bos, C.; Mittemeijer, E.J. Analysis of solid state phase transformation kinetics: Models and recipes. Metall. Rev. 2007, 52, 193–212. [Google Scholar] [CrossRef]
- Zou, F.; Chen, Q.; Yang, P.; Zhou, J.; Wu, J.; Zhuang, W.; Ying, H. Solution-Mediated Polymorphic Transformation: From Amorphous to Crystals of Disodium Guanosine 5′-Monophosphate in Ethanol. Ind. Eng. Chem. Res. 2017, 56, 8274–8282. [Google Scholar] [CrossRef]
- Maher, A.; Seaton, C.C.; Hudson, S.; Croker, D.M.; Rasmuson, Å.C.; Hodnett, B.K. Investigation of the Solid-State Polymorphic Transformations of Piracetam. Cryst. Growth Des. 2012, 12, 6223–6233. [Google Scholar] [CrossRef]
- Chee-wei Jennifer, L.; Ronald, W.R. Solubilities of and Transformations between the Anhydrous and Hydrated Forms of L-Serine in Water−Methanol Solutions. Cryst. Growth Des. 2006, 6, 1808–1812. [Google Scholar]
- Maruyama, S.; Ooshima, H.; Kato, J. Crystal structures and solvent-mediated transformation of Taltireline polymorphs. Chem. Eng. J. 1999, 75, 193–200. [Google Scholar] [CrossRef]
- Bērziņš, A.; Trimdale-Deksne, A.; Belyakov, S.; ter Horst, J.H. Switching Nitrofurantoin Polymorphic Outcome in Solvent-Mediated Phase Transformation and Crystallization Using Solvent and Additives. Cryst. Growth Des. 2023, 23, 5469–5476. [Google Scholar] [CrossRef]
- Bruker. SAINT and SMART-1000; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Usón, I.; Sheldrick, G.M. An introduction to experimental phasing of macromolecules illustrated by SHELX: New autotracing features. Acta Cryst. 2018, 74, 106. [Google Scholar]
- Atwood, J.L.; Barbour, L.J. Molecular Graphics: From Science to Art. Cryst. Growth Des. 2003, 3, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Gavezzotti, A.; Filippini, G. Geometry of the Intermolecular X-H.cntdot..cntdot..cntdot.Y (X, Y = N, O) Hydrogen Bond and the Calibration of Empirical Hydrogen-Bond Potentials. J. Phys.Chem. 1994, 98, 4831–4837. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2010, 42, 339–341. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A. Molecules in crystals. Phys. Scr. 2013, 87, 048103. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; The University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S. Density functional theory with London dispersion corrections. WIREs Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Interaction Region Indicator: A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions. ChemRxiv 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Kalra, A.; Tishmack, P.; Lubach, J.W.; Munson, E.J.; Taylor, L.S.; Byrn, S.R.; Li, T. Impact of Supramolecular Aggregation on the Crystallization Kinetics of Organic Compounds from the Supercooled Liquid State. Mol. Pharm. 2017, 14, 2126–2137. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Li, M.; Wang, M.; Liu, Y.; Ouyang, R.; Liu, M.; Han, D.; Gong, J. Co-amorphization Story of Furosemide-Amino Acid Systems: Protonation and Aromatic Stacking Insights for Promoting Compatibility and Stability. Cryst. Growth Des. 2021, 21, 3280–3289. [Google Scholar] [CrossRef]
- Singh, M.K.; Banerjee, A. Role of Solvent and External Growth Environments to Determine Growth Morphology of Molecular Crystals. Cryst. Growth Des. 2013, 13, 2413–2425. [Google Scholar] [CrossRef]
- Yang, P.; Lin, C.; Zhuang, W.; Wen, Q.; Zou, F.; Zhou, J.; Wu, J.; Ying, H. Insight into a direct solid–solid transformation: A potential approach for the removal of residual solvents. CrystEngComm 2016, 18, 1699–1704. [Google Scholar] [CrossRef]
- Wei, X.-W.; Huang, G.; Wang, J.; Meng, X.; Zhou, Q.; Ye, H.-M. Tailoring Crystallization of Random Terpolyester: Combination of Isodimorphism and Isomorphism. Macromolecules 2020, 53, 8918–8927. [Google Scholar] [CrossRef]
- Jamieson, C.S.; Mebel, A.M.; Kaiser, R.I. Understanding the Kinetics and Dynamics of Radiation-induced Reaction Pathways in Carbon Monoxide Ice at 10 K. Astrophys. J. Suppl. Ser. 2006, 163, 184–206. [Google Scholar] [CrossRef]
- Boussia, A.C.; Vouyiouka, S.N.; Porfiris, A.D.; Papaspyrides, C.D. Long-Aliphatic-Segment Polyamides: Salt Preparation and Subsequent Anhydrous Polymerization. Macromol. Mater. Eng. 2010, 295, 812–821. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Zhang, X.; Zhou, L.; Wang, C.; Li, Y.; Wu, Y.; Zhang, M.; Yin, Q. Insight into the role of hydrogen bonding playing in the molecular self-assembly process of sulfamethazine solvates. Cryst. Growth Des. 2017, 17, 6151–6157. [Google Scholar] [CrossRef]
- Papaspyrides, C.D.; Porfyris, A.D.; Vouyiouka, S.; Rulkens, R.; Grolman, E.; Poel, G.V. Solid state polymerization in a micro-reactor: The case of poly(tetramethylene terephthalamide). J. Appl. Polym. Sci. 2016, 133, 43271. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, S.; Yang, W.; Bao, Y.; Hou, B.; Zhou, L.; Yin, Q. Insights into Intermolecular Interactions of Spironolactone Solvates. Cryst. Growth Des. 2021, 21, 3677–3688. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anhydrate | Dihydrate | |
|---|---|---|
| Empirical formula | C14H24O4, C5H16N2 | C14H24O4, C5H16N2, 2H2O |
| Formula weight | 360.53 | 396.53 |
| Crystal system | triclinic | triclinic |
| Space group | P-1 | P-1 |
| a (Å) | 5.7530(5) | 8.4524(7) |
| b (Å) | 9.5901(9) | 8.5034(8) |
| c (Å) | 21.1402(19) | 16.9887(14) |
| α (deg) | 96.590(2) | 75.787(1) |
| β (deg) | 94.309(2) | 78.920(1) |
| γ (deg) | 103.272(3) | 86.523(2) |
| Volume (Å3) | 1121.46(18) | 1161.49(17) |
| Z | 2 | 2 |
| Dcalc.(g/cm3) | 1.068 | 1.134 |
| μ (mm−1) | 0.074 | 0.083 |
| F (000) | 400 | 440 |
| Observed data [I >2σ (I)] | 1869 | 2554 |
| R(int) | 0.000 | 0.028 |
| R1 [I >2σ (I)] | 0.2660 | 0.0549 |
| wR2 | 0.5903 | 0.1604 |
| GOF on F2 | 1.83 | 1.02 |
| Diff density (e/Å3) | −0.56, 0.76 | −0.18, 0.24 |
| CCDC | 2112957 | 2112969 |
| Torsion Angles (°) | Anhydrate | Dihydrate |
|---|---|---|
| O1-C1-C2-C3 | −133.8(10) | 29.7(3) |
| O2-C1-C2-C3 | 54.9(13) | −153.1(2) |
| C1-C2-C3-C4 | 178.7(9) | 68.4(3) |
| C11-C12-C13-C14 | −177.9(11) | −173.2(2) |
| C12-C13-C14-O3 | −57.2(14) | 167.0(2) |
| C12-C13-C14-O4 | 131.5(11) | −12.2(3) |
| N1-C15-C16-C17 | −52.9(15) | −56.6(3) |
| C15-C16-C17-C18 | −66.9(13) | −177.19(19) |
| C16-C17-C18-C19 | −179.5(10) | −177.42(19) |
| C17-C18-C19-N2 | −58.5(16) | −177.15(18) |
| Loss of Water Content | Theoretical Water Content | Tonset/°C | ∆Heva/J/g | a Elatt (kJ/mol) | |
|---|---|---|---|---|---|
| Anhydrate | / | / | 106.7 | 272.0 | −169.3 |
| Dihydrate | 9.31% | 9.08% | 117.9 | 295.8 | −164.8 |
| Mn × 104/Da | Mw × 104/Da | PDI | [η]/dL/g | Water Absorption/% | Tm/°C | Tc/°C | |
|---|---|---|---|---|---|---|---|
| PA514-MP | 1.43 | 3.30 | 2.31 | 0.20 | 1.61 | 198.78 | 167.71 |
| PA514-DSSP | 1.85 | 3.53 | 1.91 | 0.37 | 0.06 | 203.89 | 168.42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Zhang, L.; Zhang, X.; Chen, T.; Yang, P.; Chen, Y.; Lin, H.; Zhuang, W.; Wu, J.; Ying, H. Long-Chain Bio-Based Nylon 514 Salt: Crystal Structure, Phase Transformation, and Polymerization. Polymers 2024, 16, 480. https://doi.org/10.3390/polym16040480
Li Z, Zhang L, Zhang X, Chen T, Yang P, Chen Y, Lin H, Zhuang W, Wu J, Ying H. Long-Chain Bio-Based Nylon 514 Salt: Crystal Structure, Phase Transformation, and Polymerization. Polymers. 2024; 16(4):480. https://doi.org/10.3390/polym16040480
Chicago/Turabian StyleLi, Zihan, Lei Zhang, Xiaohan Zhang, Tianpeng Chen, Pengpeng Yang, Yong Chen, Huajie Lin, Wei Zhuang, Jinglan Wu, and Hanjie Ying. 2024. "Long-Chain Bio-Based Nylon 514 Salt: Crystal Structure, Phase Transformation, and Polymerization" Polymers 16, no. 4: 480. https://doi.org/10.3390/polym16040480
APA StyleLi, Z., Zhang, L., Zhang, X., Chen, T., Yang, P., Chen, Y., Lin, H., Zhuang, W., Wu, J., & Ying, H. (2024). Long-Chain Bio-Based Nylon 514 Salt: Crystal Structure, Phase Transformation, and Polymerization. Polymers, 16(4), 480. https://doi.org/10.3390/polym16040480