3.1.1. Thermal Behavior and Miscibility
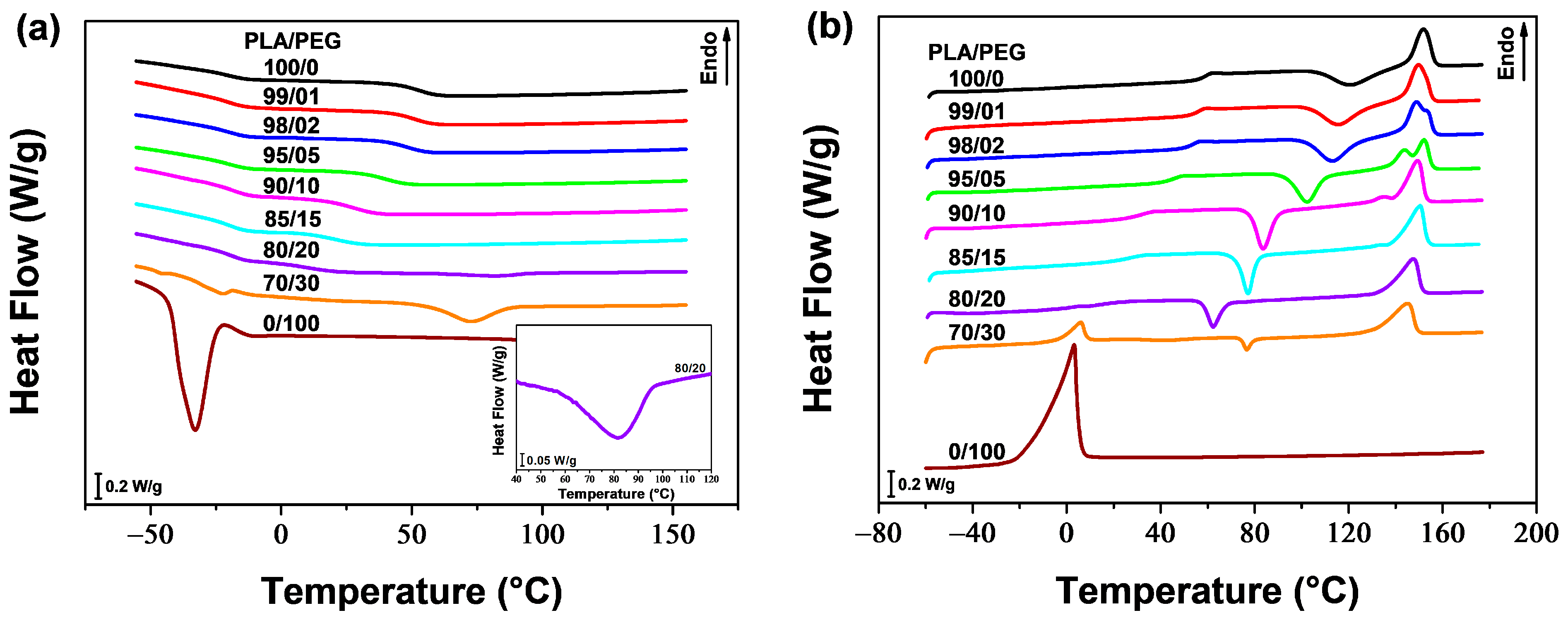
The thermal properties of neat PLA, neat PEG, and their blends were analyzed using DSC.
Figure 1a,b show the DSC thermograms recorded during cooling and second heating, respectively. The values of the glass transition temperature (
Tg), the cold crystallization temperature (
Tcc), the melting temperatures (
Tm1 and
Tm2) as well as the cold crystallization enthalpy (Δ
Hcc) and the melting enthalpy (Δ
Hm), were determined from the second heating scan.
Table 1 displays the corresponding results, including the calculated crystallinity degree (
Xc) using Equation (7).
where
is the weight fraction of PLA in the blend and
represents the enthalpy of fusion of fully crystalline PLA, which was reported by Fischer et al. [
60] to be 93.1 J/g.
Figure 1a shows that neat PLA and PLA plasticized with 1, 2, 5, 10, and 15 wt% of PEG did not exhibit any crystallization peak during cooling from the melt (at a cooling rate of 20 °C/min). However, a weak and broad crystallization peak of the PLA phase was observed for PLA/PEG-80/20 near 82 °C (see insert in
Figure 1a), and a clear and well-defined peak for PLA/PEG-70/30 at around 73 °C. Upon cooling, neat PEG demonstrated a strong ability to crystallize, with a distinct peak at approximately −33 °C. In addition to the PLA phase’s crystallization peak, a small peak was detected at around −22 °C for the blend containing 30 wt% of PEG, indicating phase separation at this concentration. Li et al. [
28] found similar behavior when plasticizing PLA with PEG-10,000. They observed crystallization peaks of the PEG phase in the DSC cooling thermograms when the plasticizer content exceeded 10 wt%. This suggests a phase separation, which was further confirmed by means of transmission electron microscopy.
The thermograms of the second heating scan for neat PLA and PLA plasticized with up to 20 wt% of PEG (
Figure 1b) show the cold crystallization of the PLA phase above the glass transition, followed by the melting of PLA crystals at higher temperatures (130–160 °C). PLA plasticized with 1, 2, 5, 10, and 15 wt% of PEG exhibit a double melting peak, with the lower temperature peak gradually fading away as the plasticizer content increases and the higher temperature peak becomes dominant. The first peak corresponds to the melting of the original α-form crystals that developed during non-isothermal cold crystallization. The second peak is associated with the melting of the disordered α′-form, which transformed to the ordered α-form through a melt-recrystallization process [
46,
53,
61]. It is noteworthy that increasing the plasticizer content from 1 to 15 wt% leads to a decrease in the α to α′ ratio. However, when the PEG content reached 20 wt% and beyond, only one melting peak was observed, indicating the formation of α-form crystals. This double melting behavior of PLA when a plasticizer is added has been reported by several authors [
6,
14,
26,
62].
Figure 1b and
Table 1 demonstrate that PLA samples containing up to 20 wt% of PEG exhibit a single
Tg. This
Tg decreases with increasing plasticizer content, from 59.3 °C for neat PLA to 16.5 °C for PLA/PEG-80/20 (i.e., a decrease of 43 °C), indicating the miscibility of the blends. Similarly,
Tcc decreases with increasing PEG content up to 20 wt%. The cold crystallization peak of neat PLA is centered at 121.5 °C, while those of plasticized samples become sharper and shift to lower temperatures. The gradual decrease in
Tcc as PEG concentration increases, reaching 62.3 °C for PLA/PEG-80/20 (i.e., a decrease of 58 °C), indicates miscibility of the PLA/PEG systems up to 20 wt% of PEG content. The melting temperatures, however, did not significantly change with plasticizer content, but all blends exhibited a lower
Tm1 value compared to neat PLA.
A small crystallization peak was observed during the second heating scan for the blend containing 30 wt% of PEG. This is because the majority of crystallization occurred during the cooling process from the melt. The Tcc of this blend did not follow the same trend as the ones of PLA/PEG blends with lower PEG content, and increased to 76.7 °C. The DSC heating thermogram showed two distinct endothermic peaks for this sample. These peaks correspond to the melting peaks of the PEG phase and the PLA phase, respectively, confirming the phase separation of the blend. The Tg, however, was not clearly defined due to the overlap of the PLA glass transition with the PEG melting peak.
The Fox formula [
63] was used to confirm the miscibility of mixtures containing up to 20 wt% of PEG.
where
Tg is the glass transition temperature of the blends,
Tg1 and
Tg2 are those of the blends’ components, and
w1 and
w2 are their weight fractions (with
w1 +
w2 = 1). Subscripts 1 and 2 refer to PLA and PEG, respectively. The
Tg2 of PEG could not be determined within the range of our DSC experiments; however, a value of −78 °C was reported for PEG-400 in the literature [
64].
Figure 2a shows the application of the Fox equation to the
Tgs measured by DSC for PLA/PEG blends. The Fox equation was constructed using the measured
Tg of PLA (
Tg1 = 59.3 °C) and the
Tg reported in the literature for PEG 400 (
Tg2 = −78 °C). The measured
Tgs conform well to the Fox equation, indicating that the PLA/PEG blends in this study are miscible up to at least 20 wt% of PEG content.
Additionally, miscibility can be examined without the need for the
Tg of PEG. In fact, Equation (8) can be rewritten as:
Equation (9) shows that 1/
Tg has a linear relationship with
w2, indicating miscibility when the reciprocals of the measured
Tgs vary linearly with the plasticizer content.
Figure 2b demonstrates good linearity between the reciprocals of
Tgs and the change in PEG content (
R2 = 0.99), confirming miscibility within the plasticizer concentration range of 0–20 wt%. Furthermore, the
Tg of PEG can be estimated from the slope of the linear regression curve, and was found to be −83.4 °C, slightly lower than the experimental value reported in the literature (−78 °C).
The degree of crystallinity (
Xc), calculated using Equation (7), increased with higher PEG content, as shown in
Figure 3. The plasticizer enhanced the crystallization ability of PLA by increasing polymer chain mobility. In the concentration range of 1–15 wt% of plasticizer, there was a significant increase in
Xc, from 14.8 wt% for neat PLA to 22.2 wt% for PLA/PEG-90/15. An inflection point was noticed at 15 wt% of PEG content, and for high PEG content,
Xc continued to increase. The increase in crystallinity in the plasticizer content range of 1–15 wt% was accompanied by a decrease in the α to α′ ratio, but outside this range, only the α crystals developed. PLA/PEG-70/30 reached the highest value of
Xc, which was of 26.1%, indicating evidence of phase separation.
The decrease of
Tg and
Tcc, along with the increase in
Xc as the plasticizer level increased, were attributed to the improved molecular chain mobility of PLA resulting from the lubricating effect of PEG. The molecules of PEG diffused into the PLA matrix, penetrating between the polymer chains. This increased the free volume and decreased the intermolecular polymer chain interactions, resulting in improved chain mobility at lower temperatures [
20,
21].
3.1.2. Non-Isothermal Cold Crystallization Kinetics
Crystallization is a first order phase transition in which a material undergoes a transition from an amorphous state to a crystalline phase. The crystallization enthalpy can be determined by measuring the area under the exothermic peak during the crystallization process. The equation for expressing the relative crystallinity (
XT) as a function of temperature is as follows [
65]:
where
T0 is the temperature at the crystallization onset,
T represents a temperature during the crystallization process, d
Hcc corresponds to the enthalpy of crystallization released during an infinitesimal temperature range d
t, and Δ
Hcc stands for the overall heat released during the crystallization process.
Assuming minimal thermal lag between the sample and the DSC furnace, the relationship between crystallization time
and sample temperature
can be formulated as follows [
66]:
where
ϕ represents the heating rate.
Figure 4 shows the change in relative crystallinity over time for PLA/PEG blends that were non-isothermally cold crystallized at a heating rate of 10 °C/min, with plasticizer content ranging from 0–15 wt%. All curves have a sigmoidal shape. The plateau of the curves in the early stage of crystallization reflects the induction period during which nuclei were formed. Subsequently, the crystals grew, as evidenced by the ascending part of the curves. During the final stage of crystallization, the upward curvature of the plots is attributed to secondary crystallization resulting from the impingement of spherulites. The crystallization rate decreased in this stage, but the crystallinity continued to increase slowly until the completion of the crystallization process [
67,
68].
Several models have been proposed in the literature for the quantitative description of crystallization kinetics, the most common approach being that of Avrami [
69]:
where
Xt stands for the relative crystallinity depending on time
t,
k is the crystallization rate constant depending on the nucleation and growth rate, and
n represents the Avrami exponent depending on the nucleation type and the growth geometry of the crystals. The Avrami equation was proposed for isothermal crystallization. For non-isothermal processes, the parameter
k should be appropriately corrected because the temperature constantly changes during the measurements, affecting the rates of both nuclei formation and spherulite growth, which are temperature-dependent [
45]. Assuming a constant heating rate (
ϕ), Jeziorny [
70] proposed a correction for the crystallization rate constant as follows:
where
kc is the corrected crystallization rate constant. The study utilized the Avrami equation to conduct an analysis by fitting the experimental
Xt data to Equation (12), as illustrated in
Figure 4.
Table 2 lists the values of the kinetic parameters (
n,
k and
kc) along with the
R2 parameter. The Avrami method offers a suitable description of the non-isothermal crystallization for the samples under study. However, minor deviations were observed at high
Xt (>90%) due to the neglect of the significant role of the secondary crystallization process [
45].
Table 2 shows that
n values increase with higher plasticizer content, indicating that the addition of PEG affects the mechanisms of nucleation and growth of PLA crystals. The
n parameter ranges from 3.34 to 4.73, suggesting three-dimensional spherulitic growth with sporadic or simultaneous nucleation types [
71]. The
kc parameter increases with increasing plasticizer content, indicating that the crystallization process of PLA was accelerated by the addition of PEG. This phenomenon was also observed in PLA plasticized with jojoba oil [
45] and PLA plasticized with thermoplastic starch [
72]. The plasticizer enhances the chain mobility of PLA, which in turn enhances the crystallization rate by reducing the energy required for the chain folding process during crystallization.
An important kinetic parameter, the crystallization half-time (
t1/2), defined as the time required to reach half crystallinity (
Xt = 0.5), can be calculated from the Avrami parameters as follows:
The values of
t1/2 obtained, as shown in
Table 2, decrease with increasing plasticizer level. This demonstrates that the crystallization rate increased as the plasticizer content increased.
3.1.3. Dynamic Mechanical Behavior
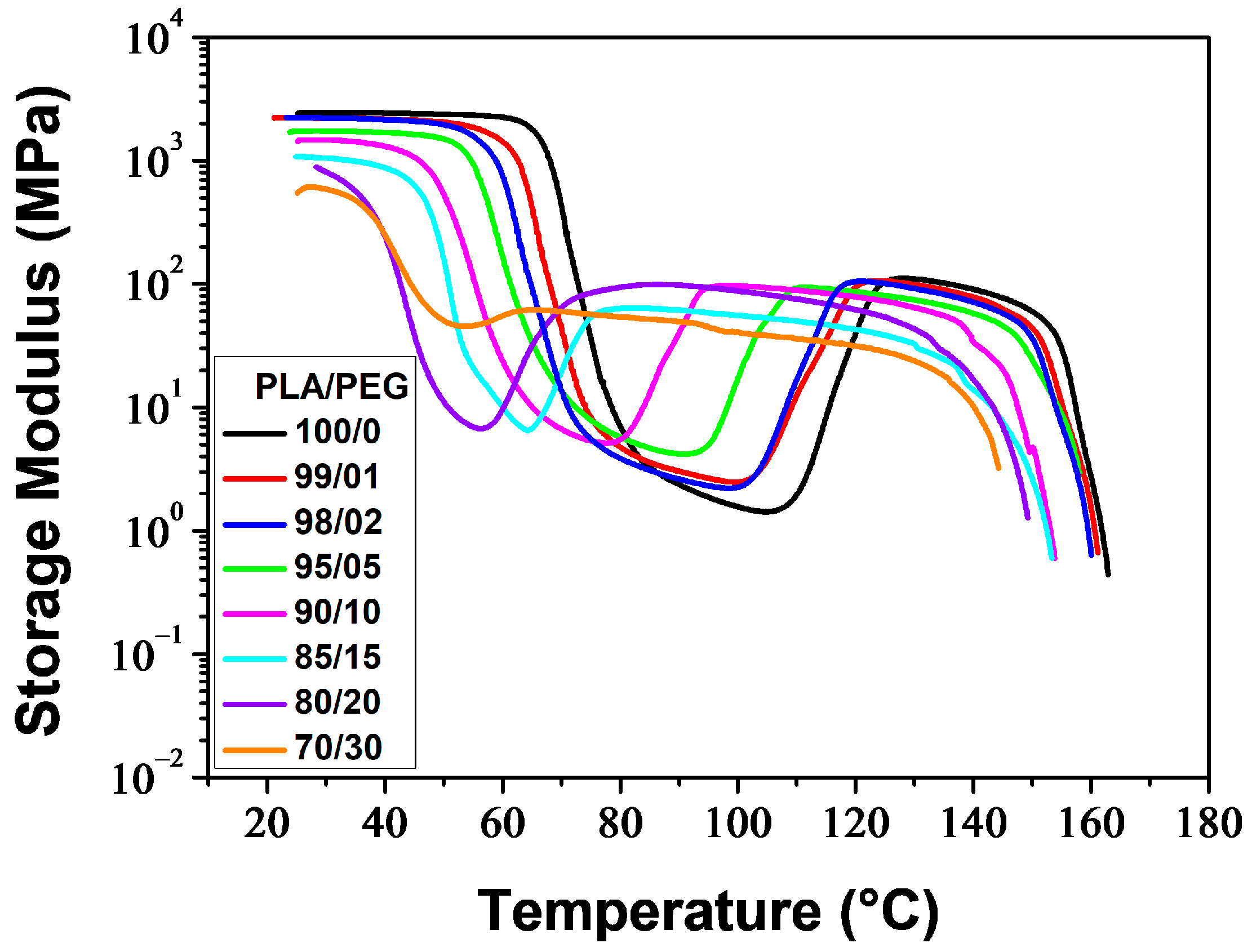
Figure 5 and
Figure 6 illustrate the temperature dependence of the storage modulus (
E′) and loss factor (tan
δ) for the melt-quenched PLA/PEG blends. At low temperatures,
E′ displayed a glassy plateau (
Eg), characterizing the glassy rigid state. As the temperature increased,
E′ decreased as the samples entered the glass transition region. The decrease of
E′ was observed at lower temperatures as the PEG content increased. Both neat PLA and PLA plasticized with up to 20 wt% of PEG showed a significant decrease in
E′ (two-three orders of magnitude) to a minimum value of
, followed by a sharp increase due to cold crystallization. After the glass transition, PLA chains became mobile enough to form crystalline regions, which was facilitated by a slow heating rate (3 °C/min) and sinusoidal solicitation. The temperature at which
E′ starts to increase is considered the temperature of the onset of crystallization (
Tcc′). The significant decrease of
E′ in the glass transition region, followed by the sharp increase resulting from the cold crystallization for the compositions containing up to 20 wt% of PEG, indicates the amorphous state of these materials before analysis. The blend containing 30 wt% of PEG exhibited a reduced decline of
E′ in the glass transition region, followed by a slight increase. This suggests that most of the crystallization had already occurred before analysis and the sample was not amorphous. The crystallization caused a stiffening of the macromolecular chains, resulting in an increase of
E′ up to a rubbery plateau (
EN), which characterizes the rubbery elastic state. The value and length of the rubbery plateau depend on the PEG content. At higher temperatures,
E′ decreased as the materials began to flow due to the melting of crystals.
One notable observation from
E′ versus temperature curves is that the rubbery plateau expands as the PEG level increases. This means that the rubbery elastic state is reached more quickly with higher plasticizer content. Another significant effect is the reduction in the size of the hollow formed in the glass transition and cold crystallization regions (due to the decrease and increase of
E′), as the PEG content increases. Additionally, the hollow shifts towards lower temperatures with a decrease in
Tcc′, which is attributed to the improvement of PLA chain mobility. Kang et al. [
41] reported similar behavior for PLA plasticized with cardanol (CD). They observed that as the CD content increased up to 15 wt%,
E′ gradually decreased at the glassy state and the temperature at which
E′ began to rise shifted to a lower temperature. This indicates that the introduction of CD enhanced the cold crystallization ability of PLA.
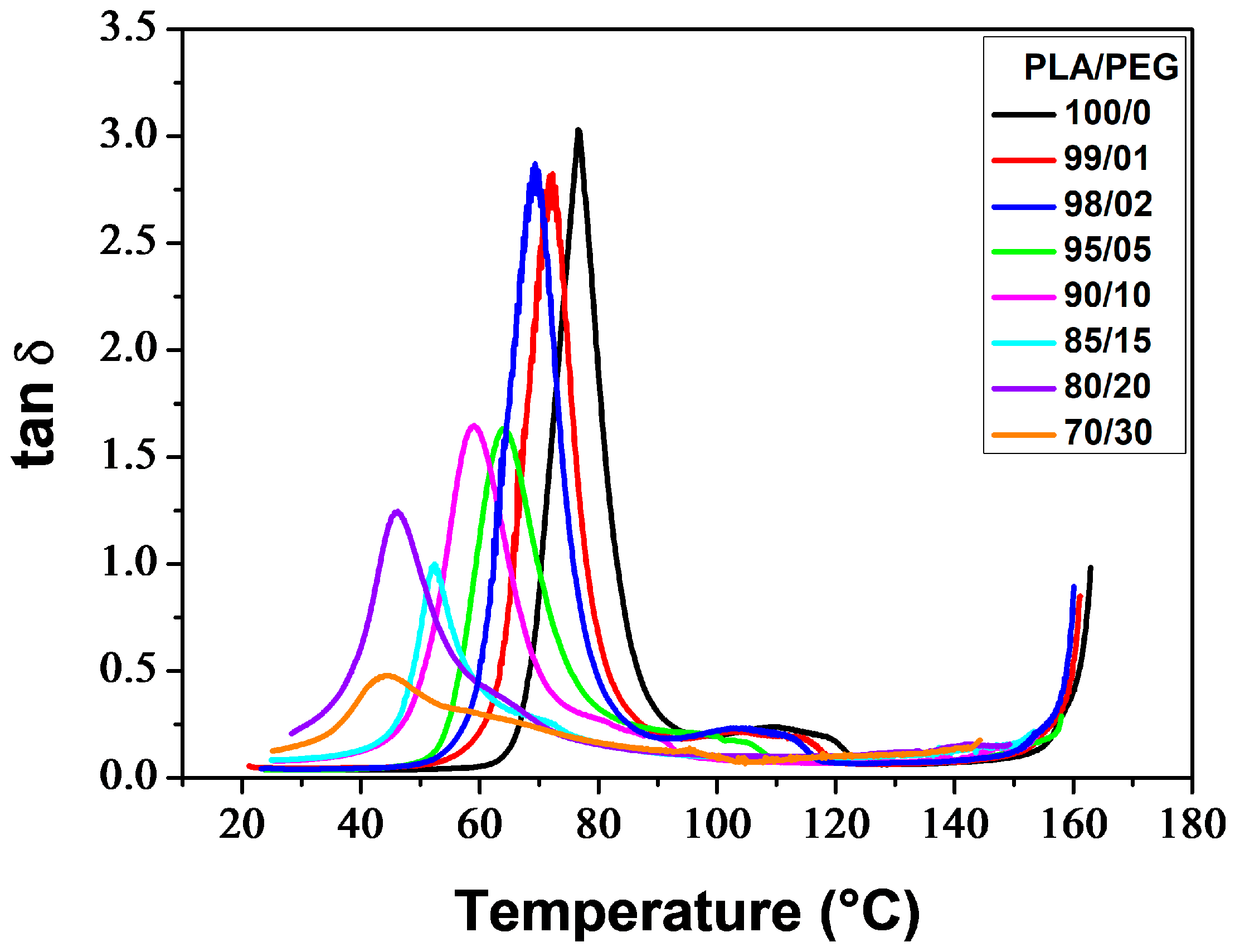
Figure 6 thermograms illustrate the α-relaxation process, which is associated with the glass transition, for neat and plasticized PLA. The glass transition temperature (
Tα), determined at the maximum of the tan
δ peak, decreased with increasing plasticizer level. Additionally, the incorporation of PEG resulted in lower peaks. PLA/PEG-70/30 exhibited a broad and low-intensity peak, which was observed due to crystallization before analysis. The tan
δ curves also displayed bumps after the glass transition peaks (for low PEG content) or shoulders on the downward side of the peaks (for high PEG content), which were associated with the cold-crystallization process [
73,
74].
Table 3 reports the main thermo-mechanical properties resulting from the DMA thermograms shown in
Figure 5 and
Figure 6. These properties include the glassy plateau modulus (
Eg), the rubbery plateau modulus (
EN), the glass transition temperature (
Tα), the temperature of the onset of crystallization (
Tcc′), and the extent in temperature of the rubbery plateau (Δ
TN).
Figure 7 illustrates the impact of PEG content on
Eg,
EN and Δ
TN. As the PEG content increased, the
Eg of PLA/PEG blends decreased from 2428 MPa for neat PLA to 611 MPa for PLA/PEG-70/30, indicating a reduction in rigidity. The
EN values were relatively low and decreased slightly from 107 MPa for neat PLA to 58 MPa for PLA/PEG-70/30 with the addition of PEG. The rigidity of the materials in the rubbery plateau region came from the crystallites, which behaved like crosslinking nodes. Meanwhile, the Δ
TN significantly increased from 22.2 °C for neat PLA to 68.2 °C for PLA/PEG-70/30 due to the shift of
Tcc′ towards lower temperatures with increasing PEG content.
Figure 8 shows that increasing the plasticizer concentration decreased
Tα and
Tcc′ in a monotonous manner until reaching a plateau value at around 20 wt% PEG content. This indicates phase separation in the blend containing 30 wt% PEG, as demonstrated by the presence of two melting peaks in the respective DSC thermogram.
Tα and
Tcc′ exhibited similar trends to
Tg and
Tcc as determined by DSC technique (
Table 1), but with
Tα values approximately 15 °C higher than
Tg values. Averous et al. [
75] attribute the difference between the temperatures corresponding to the transitions observed by DMA and DSC to the frequency of the analysis method.
3.1.4. Rheological Properties
The melt viscosity of polymers is highly sensitive to changes in the structure of macromolecular chains and the addition of plasticizers. Plasticizers, by definition, increase the free volume of the polymer and the mobility of the chains. Therefore, rheological measurements are both of practical and fundamental interest in this study. The study measured the storage modulus (G′), loss modulus (G″), and complex viscosity (η*) of PLA/PEG blends at a temperature of 180 °C and a strain of 5% over an angular frequency range of ω = 600–0.1 rad/s.
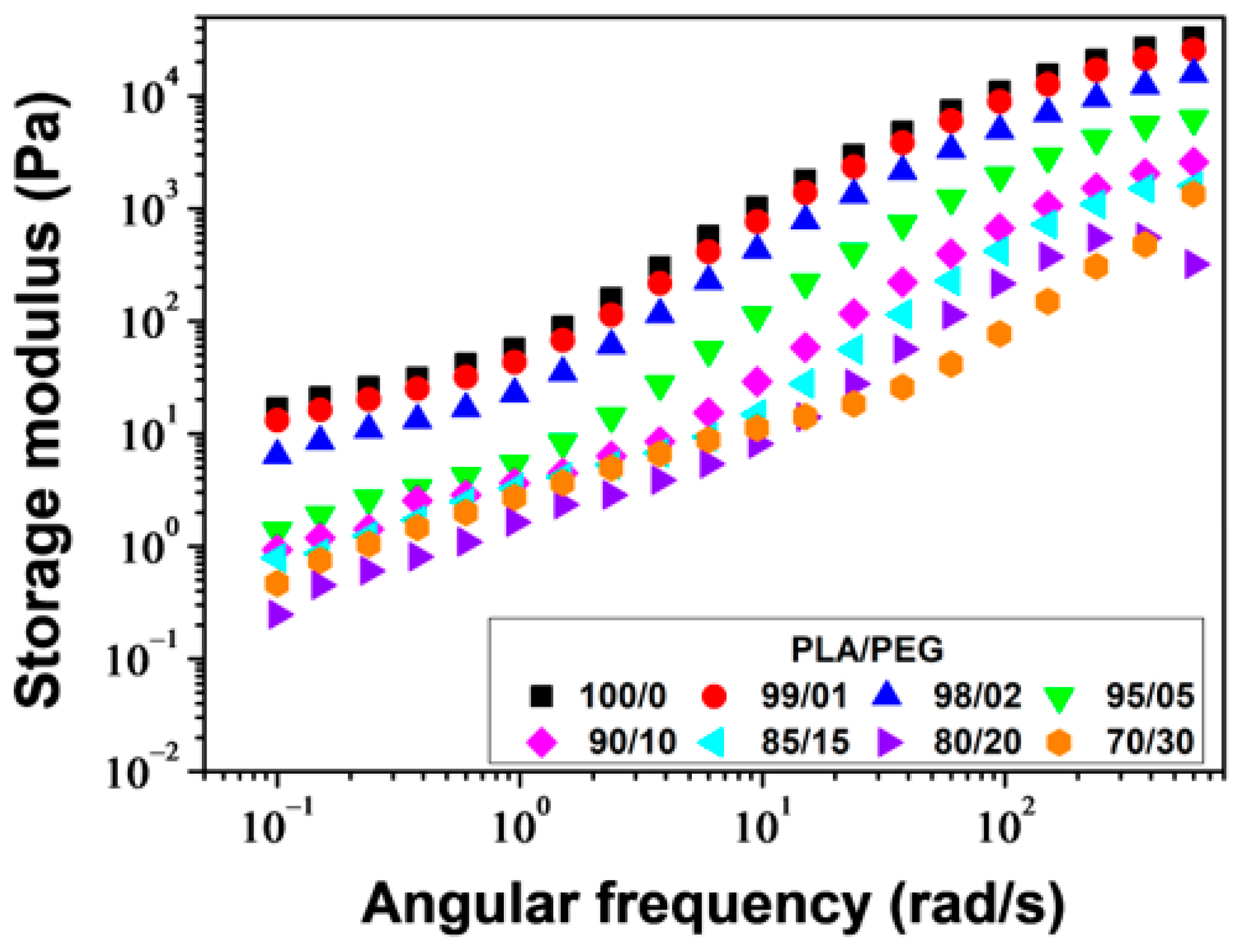
Figure 9 shows the plots of
G′ versus angular frequency for PLA/PEG blends. For all compositions,
G′ increased as the angular frequency increased, but decreased with increasing PEG content. The decrease in
G′ with increasing PEG content was due to a decrease in molecular entanglements. PLA samples plasticized with up to 20 wt% of PEG showed parallel log
G′ versus log
ω slopes beyond
ω ≈ 2 rad/s. Below
ω ≈ 2 rad/s, a decrease in the slopes was observed due to the re-entanglement of the molecular chains, resulting in excess elasticity. For PLA/PEG-70/30, a significant change in the slope occurred around
ω ≈ 30 rad/s, due to a high rate of re-entanglement, which can be attributed to the phase separation of this mixture, as described in previous sections.
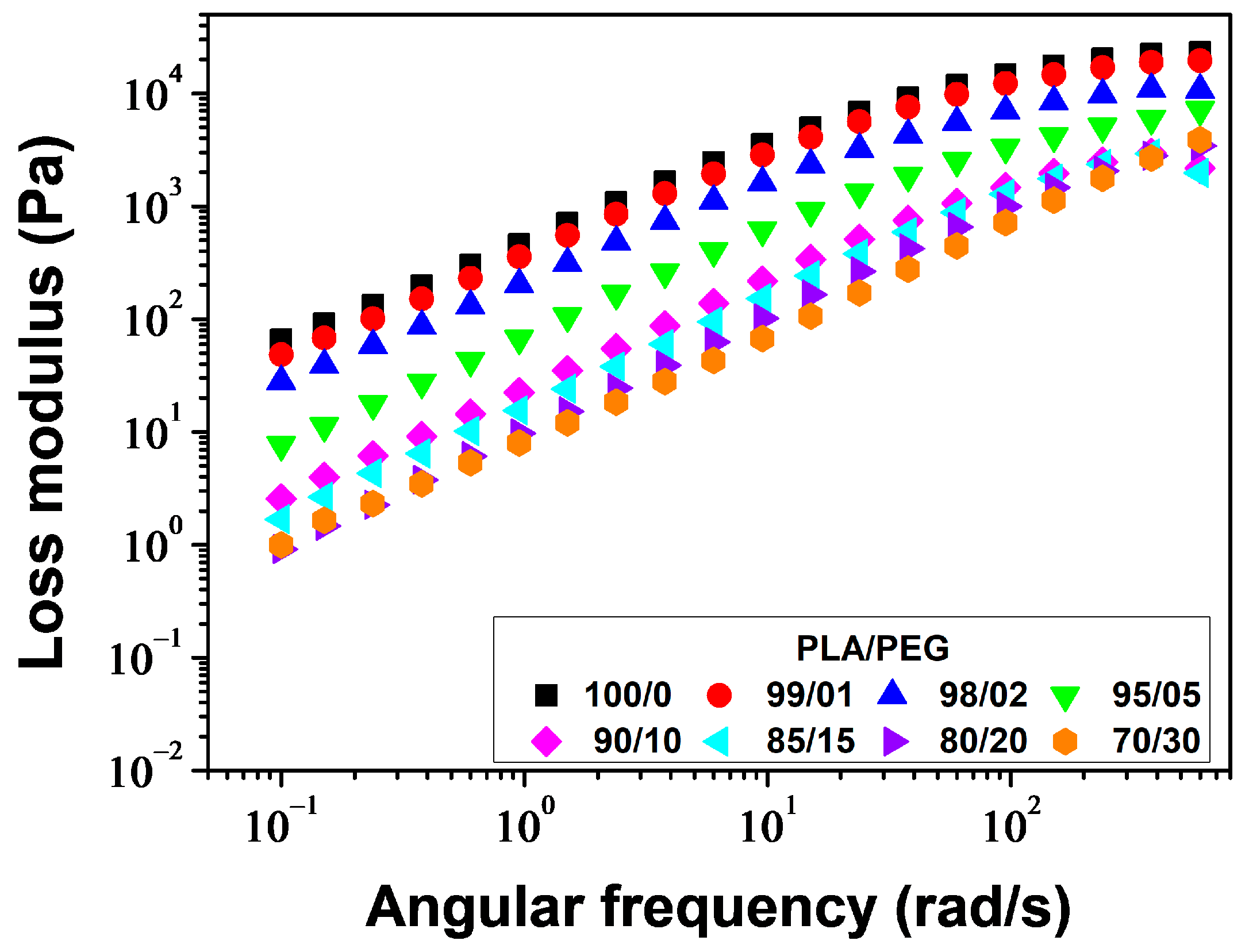
Figure 10 displays the plots of
G″ versus angular frequency.
G″ increased linearly with increasing angular frequency for all samples but decreased with increasing PEG content. The decrease of
G″ with increasing PEG concentration indicates a positive plasticization effect.
Figure 11 shows the angular frequency dependence of complex viscosity (
η*) for various samples. The samples containing 0–20 wt% of PEG exhibit a Newtonian plateau, followed by a shear thinning behavior above
ω ≈ 30 rad/s. Neat PLA and PLA plasticized with 1 and 2 wt% of PEG exhibit an increase in viscosity with decreasing angular frequency below
ω ≈ 1 rad/s. This behavior is described using the Carreau–Yasuda model with yield stress [
58,
59]:
where
σe is the yield stress,
represents the zero-shear viscosity,
λ stands for the relaxation time which corresponds to the frequency of the onset of the shear thinning behavior,
a corresponds to the Yasuda parameter which indicates the width of the transition region between the Newtonian and shear-thinning behavior and
n is the power law index. The values of these parameters, along with the
R2 parameter, are presented in
Table 4. The model accurately fit the complex viscosities of PLA/PEG blends containing up to 20 wt% of PEG. The plasticizing effect of PEG was demonstrated by a significant decrease in
from 490 to 10 Pa·s,
σe from 19.5 to 0 Pa, and
λ from 0.0251 to 0.0045 s. The curve of
η* versus
ω confirms the phase separation of PLA/PEG-70/30, as already demonstrated by DSC. It shows two relaxation times: the first between
ω ≈ 0.1 and
ω ≈ 9.5 rad/s with a relaxation time
λ1 ≈ 2.6420 s, and the second between
ω ≈ 9.5 and
ω ≈ 230 rad/s with a relaxation time
λ2 ≈ 0.0042 s. This observation indicates the presence of two distinct relaxation processes in the material, confirming the phase separation effect of PLA/PEG-70/30 already demonstrated by DSC.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}