
Impact of Copolymer Architecture on Demicellization and Cargo Release via Head-to-Tail Depolymerization of Hydrophobic Blocks or Branches

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Model
2.1. Coarse-Grained Molecular Dynamics Simulation Details
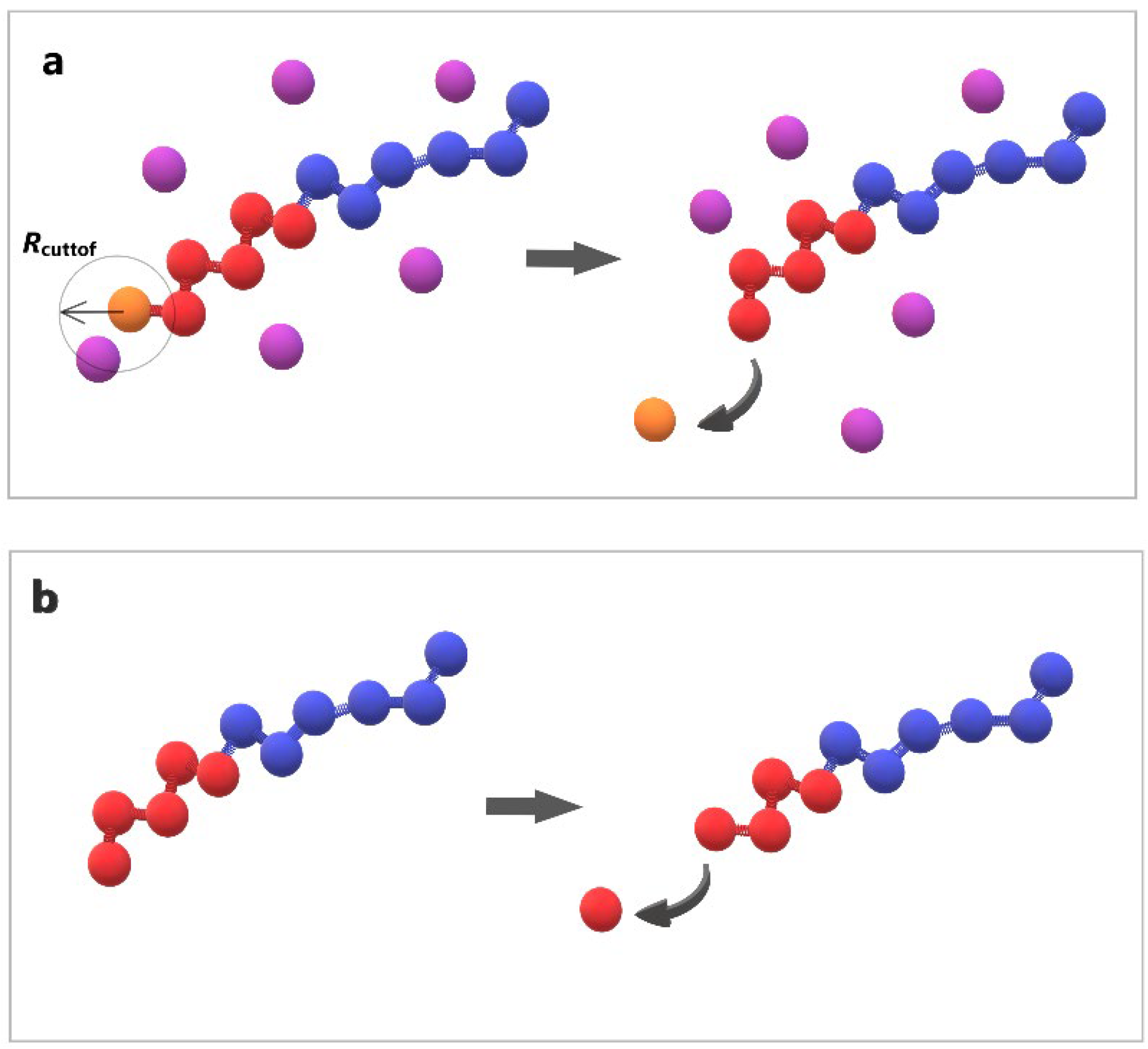
2.2. Depolymerization and Cluster Analysis
3. Results and Discussion
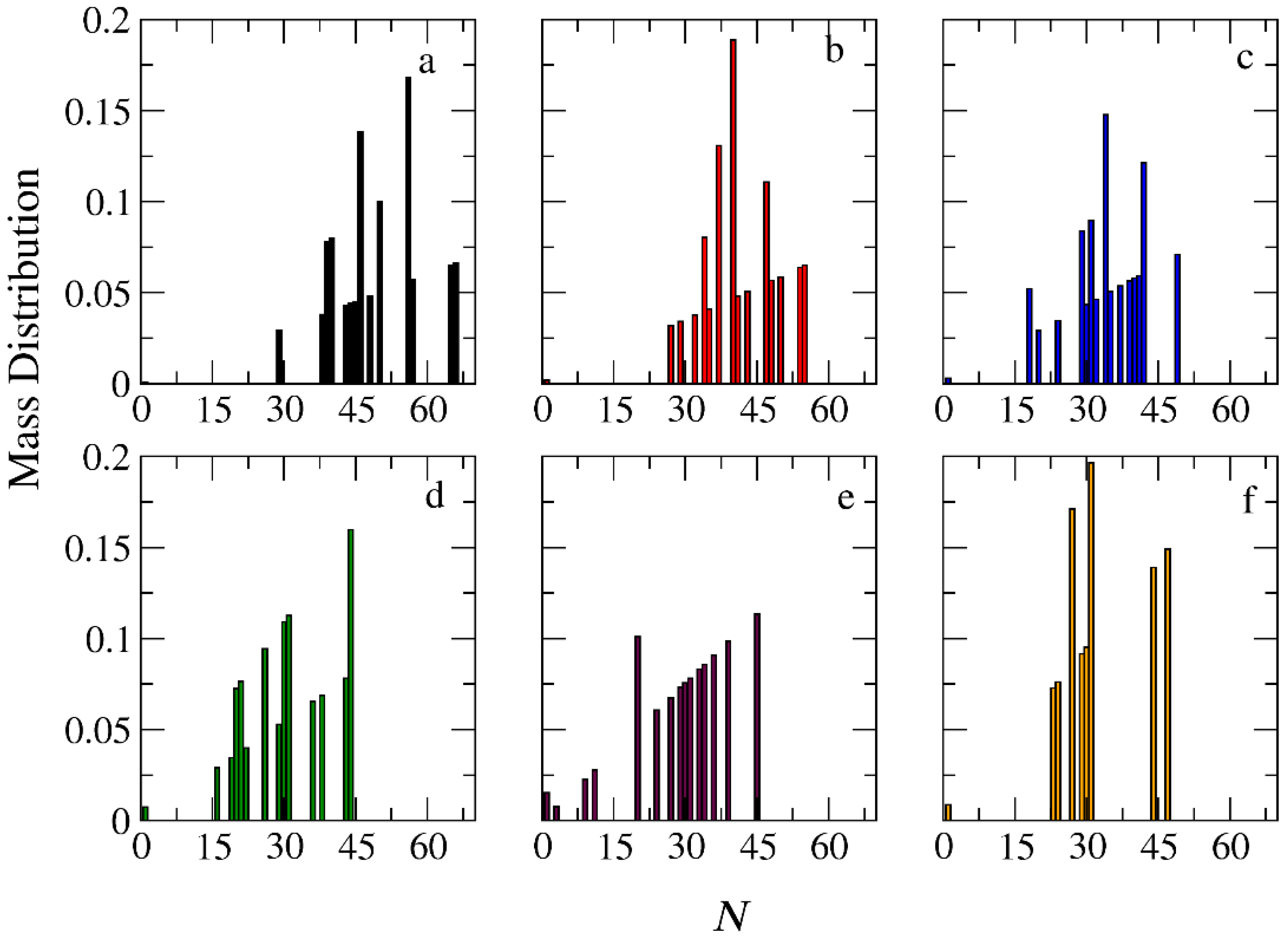
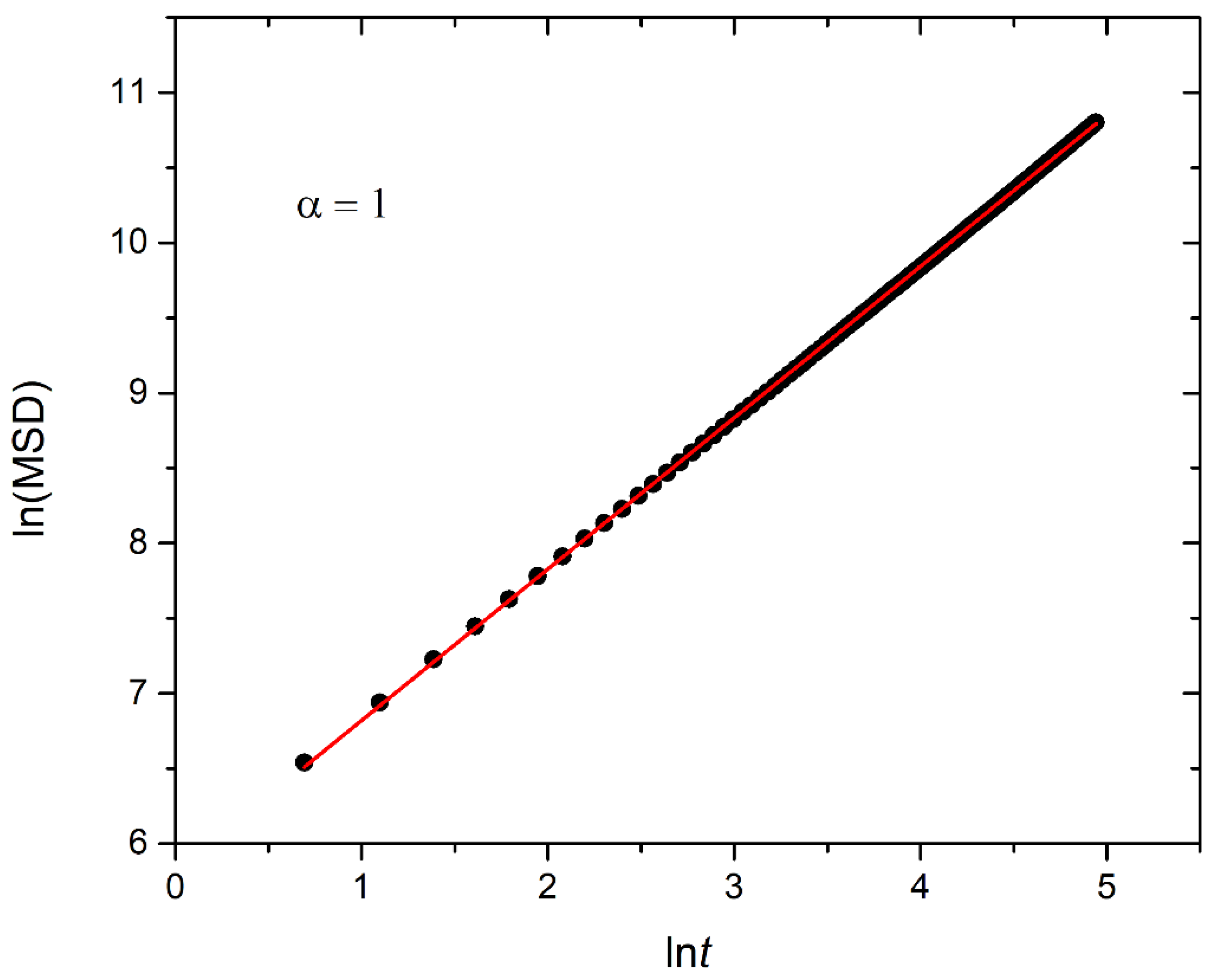
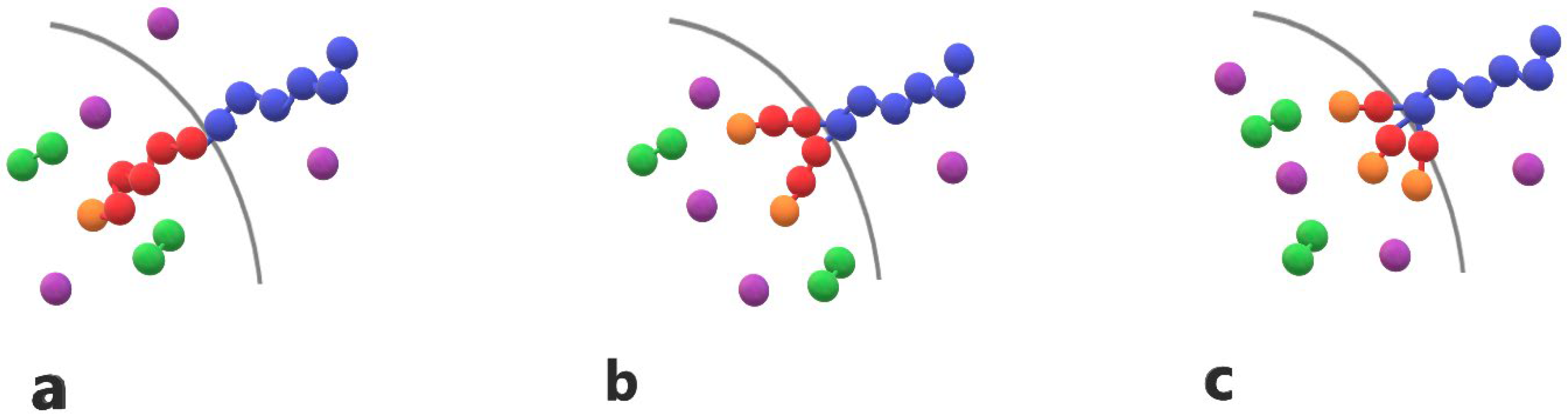
3.1. Micellization and Cargo Encapsulation
3.2. Kinetics of Depolymerization
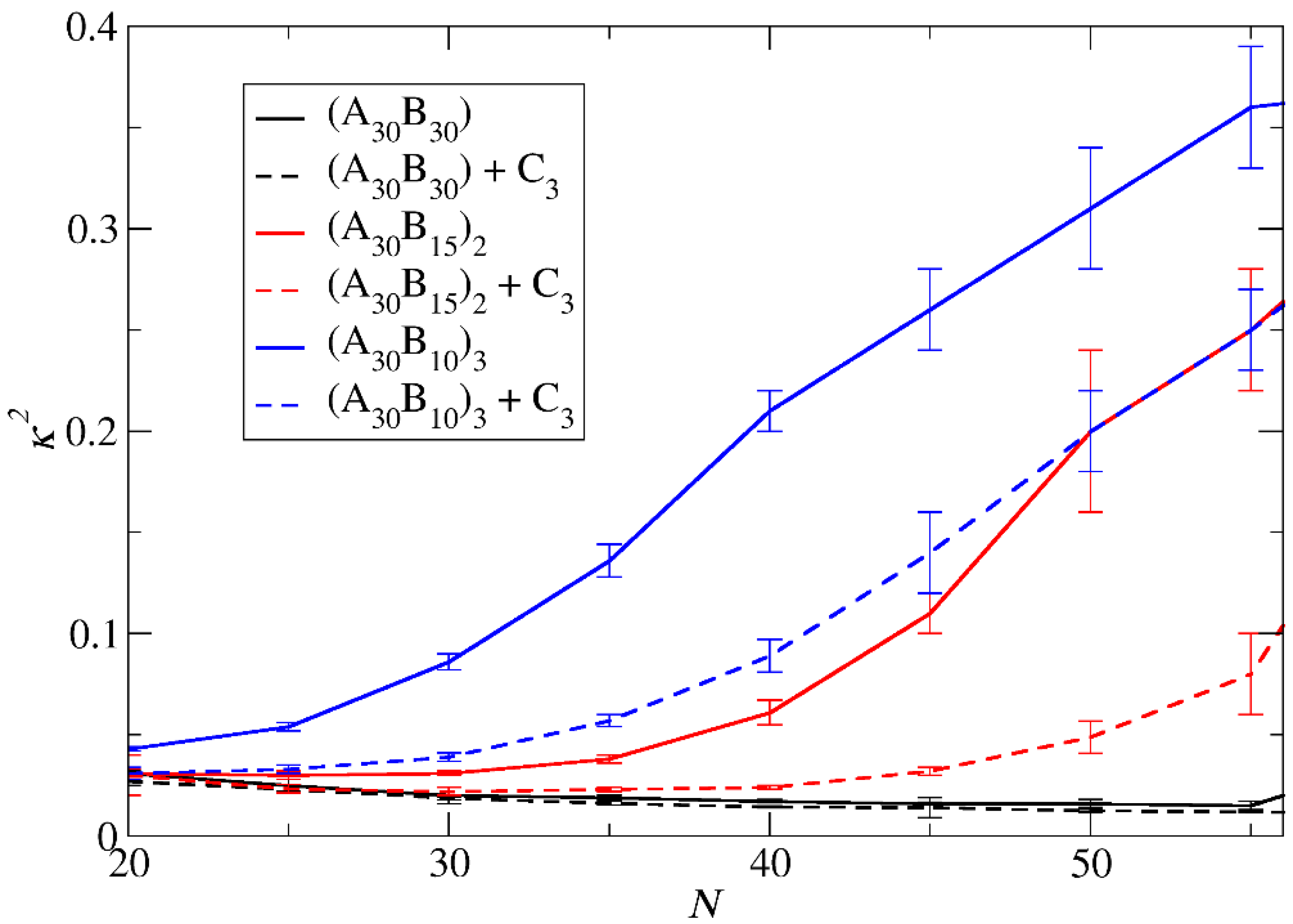
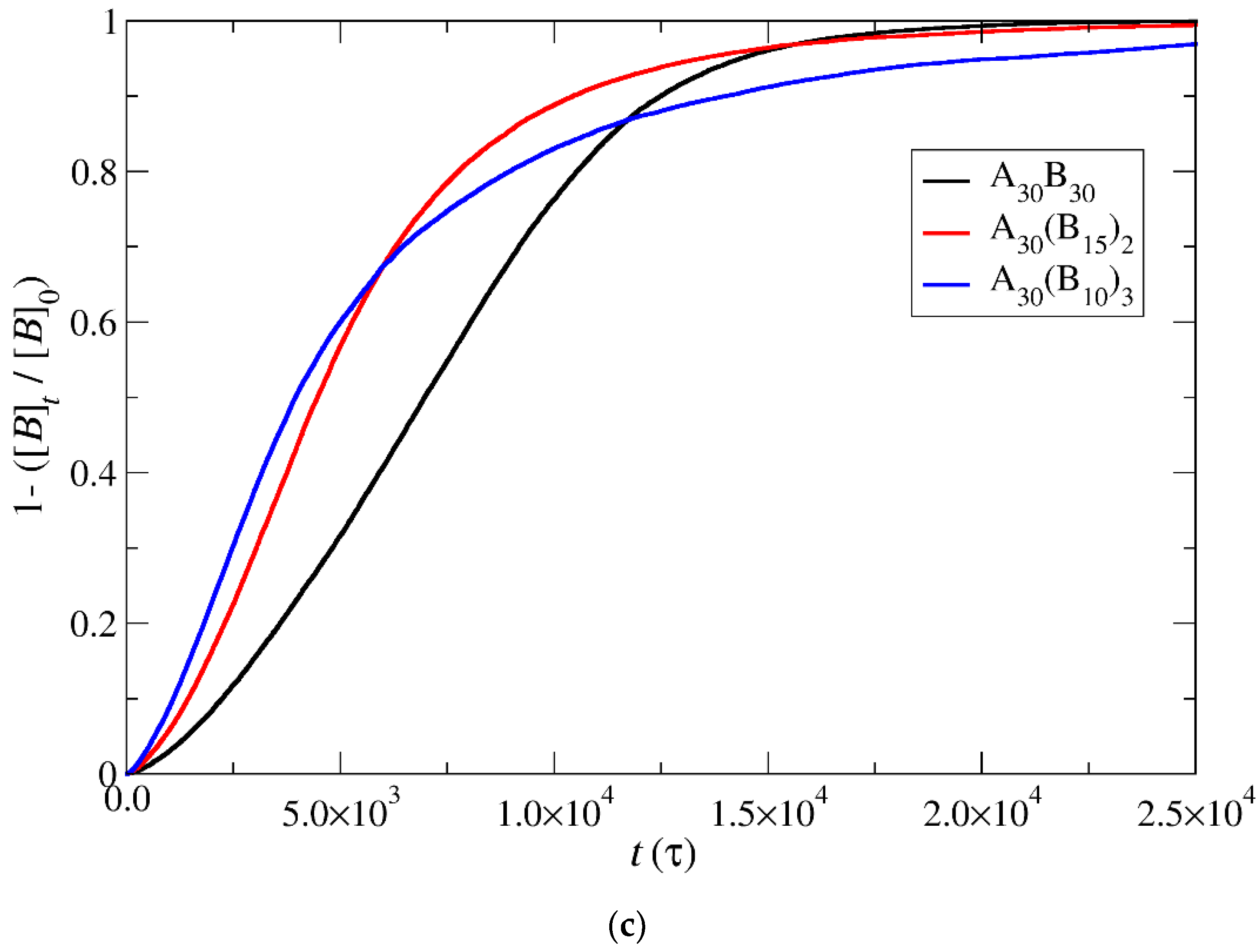
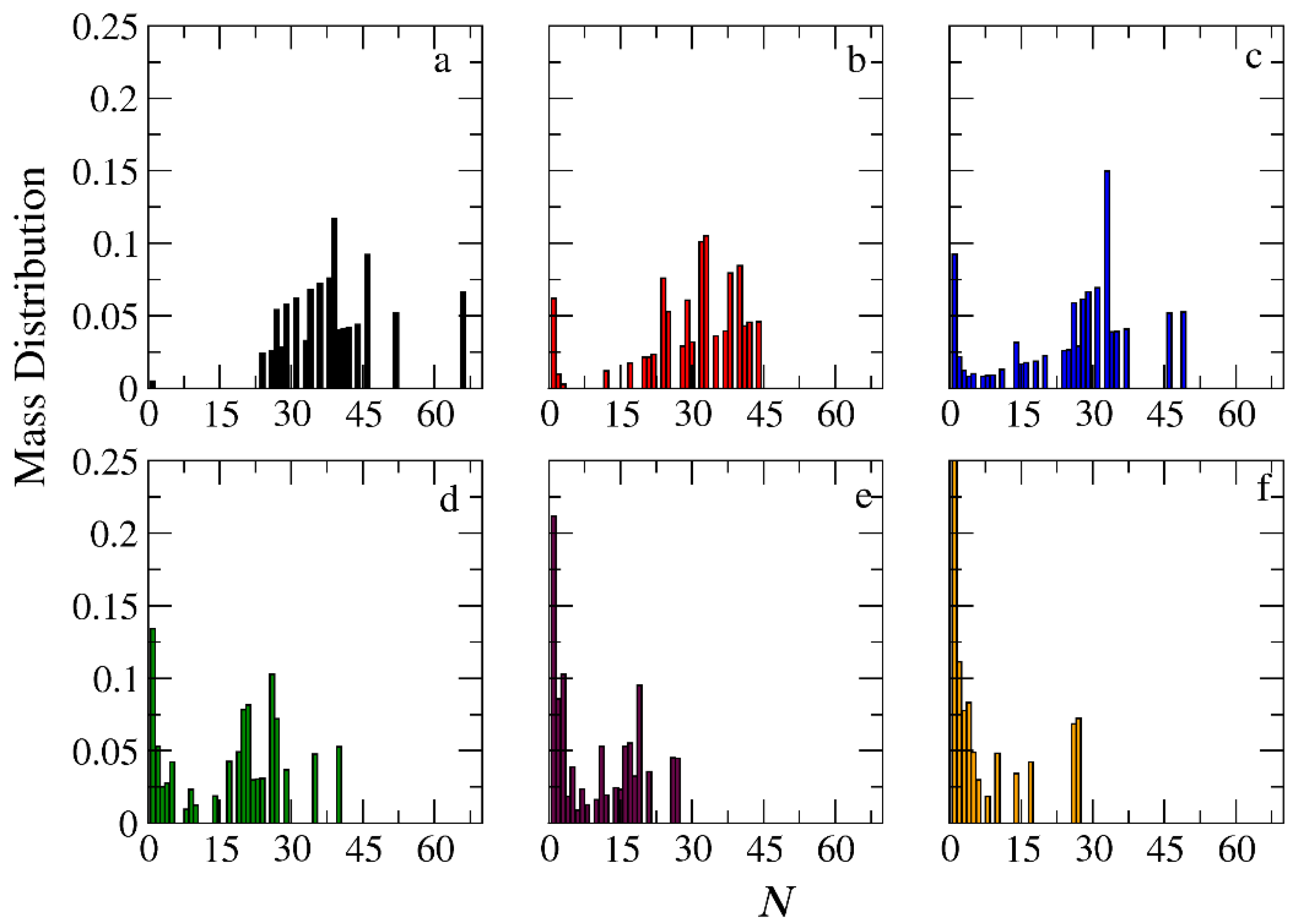
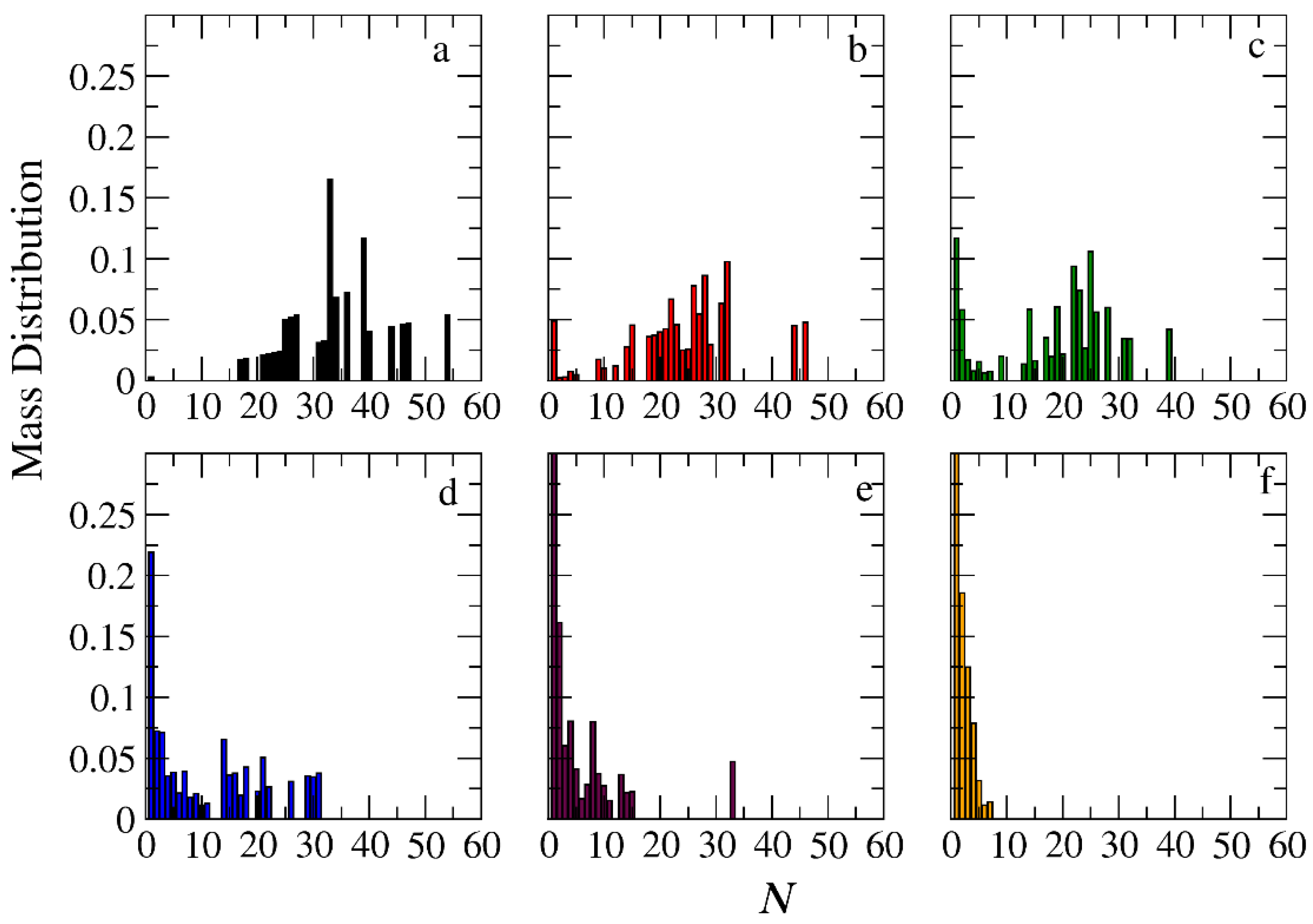
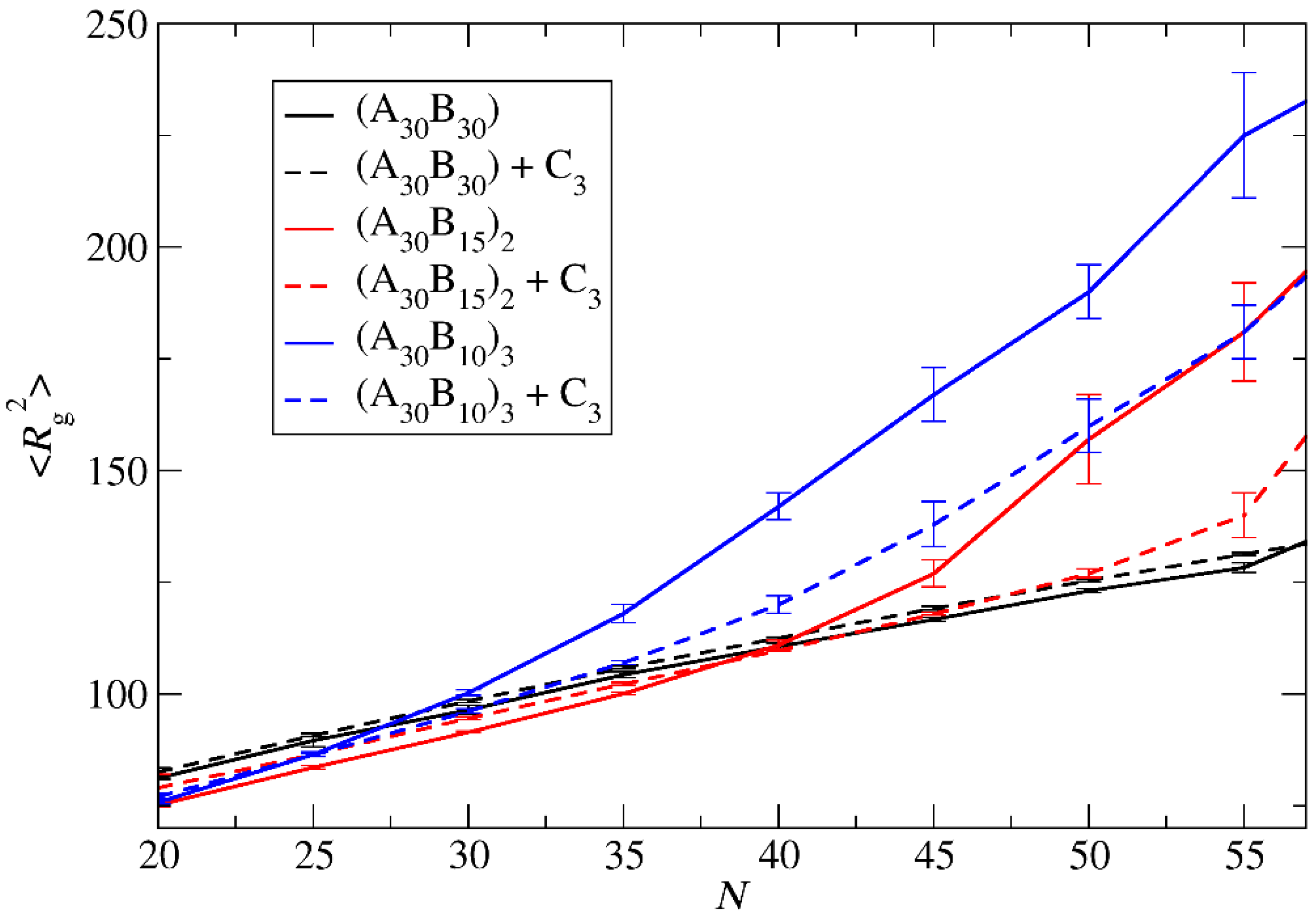
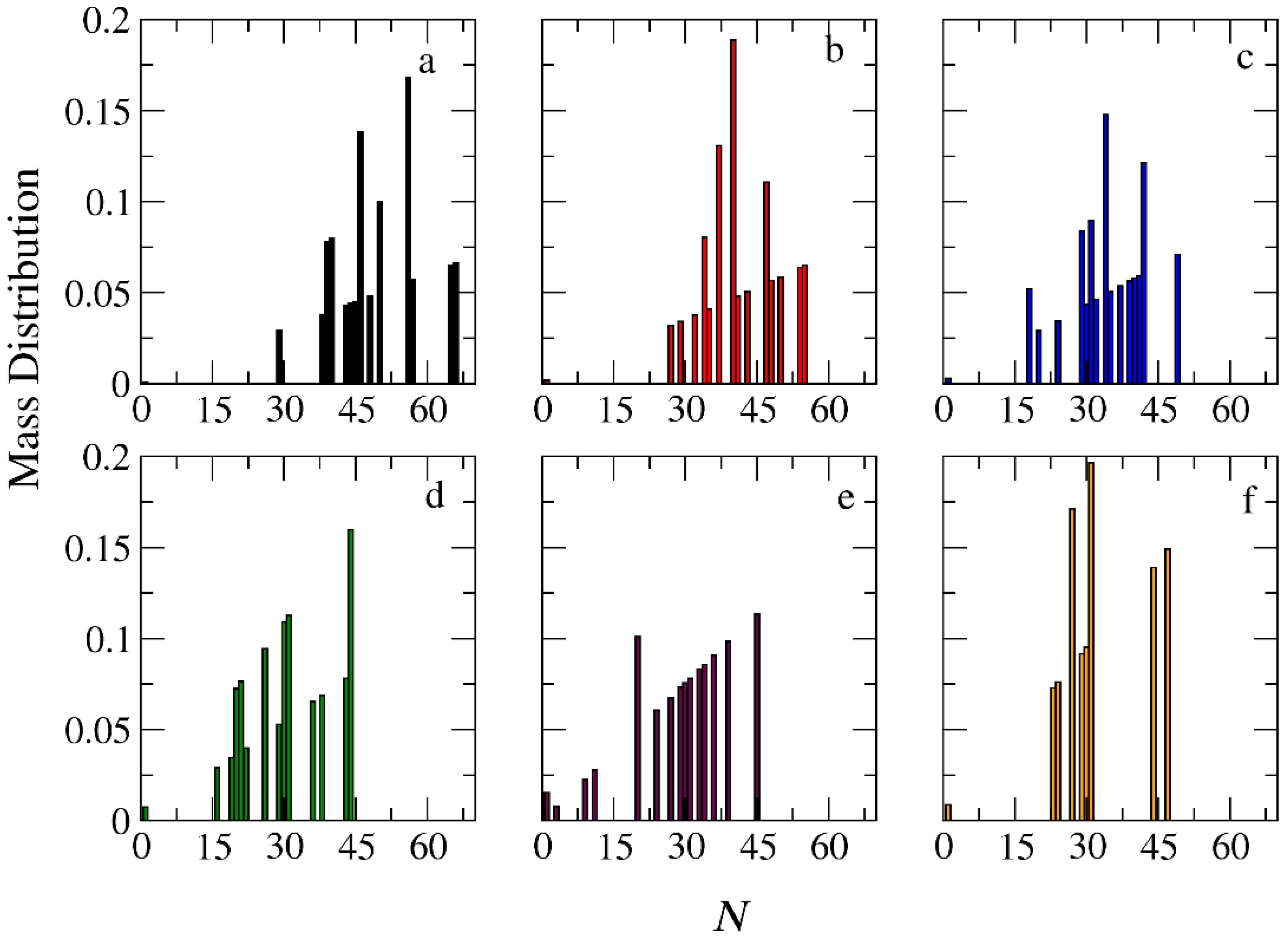
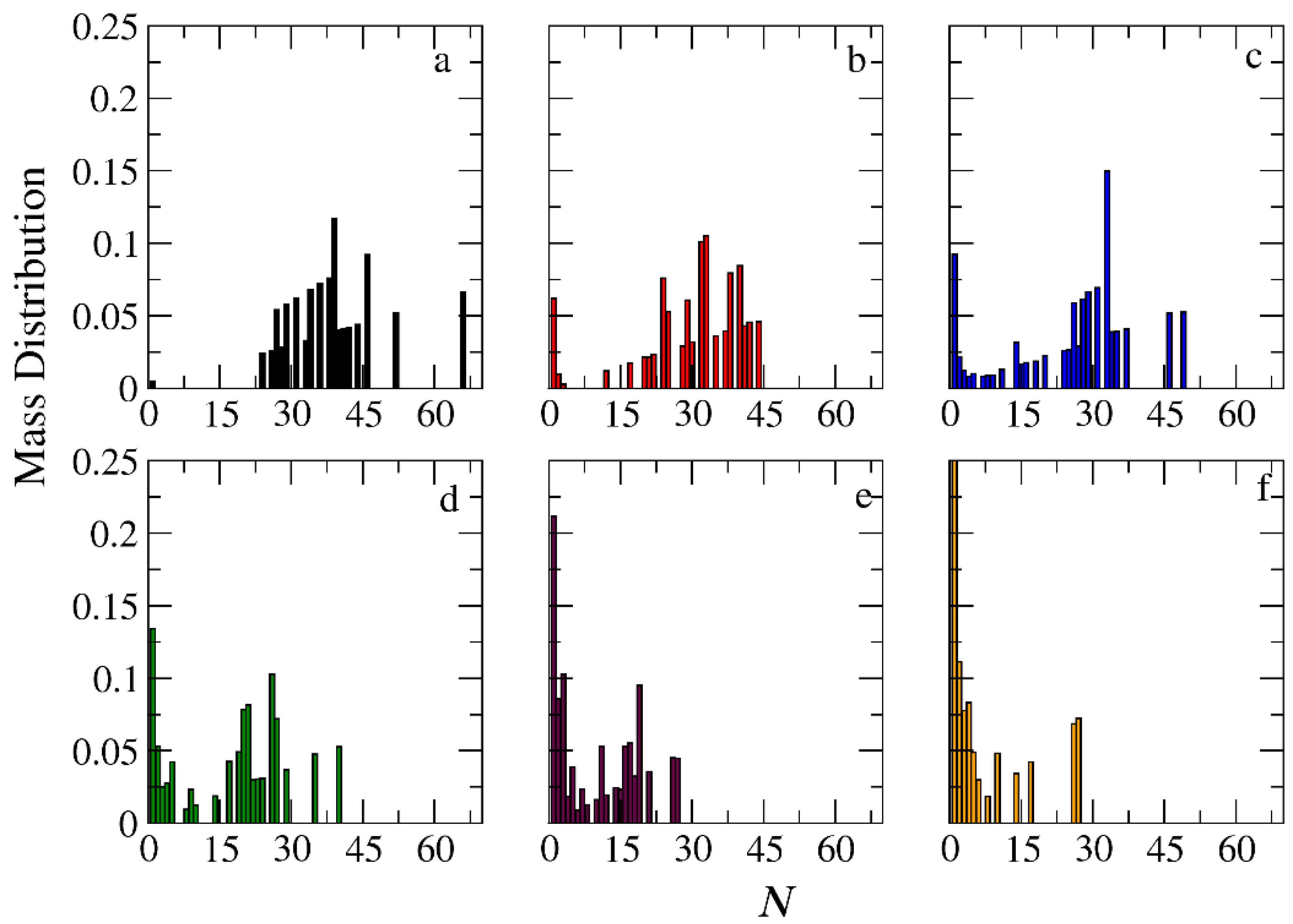
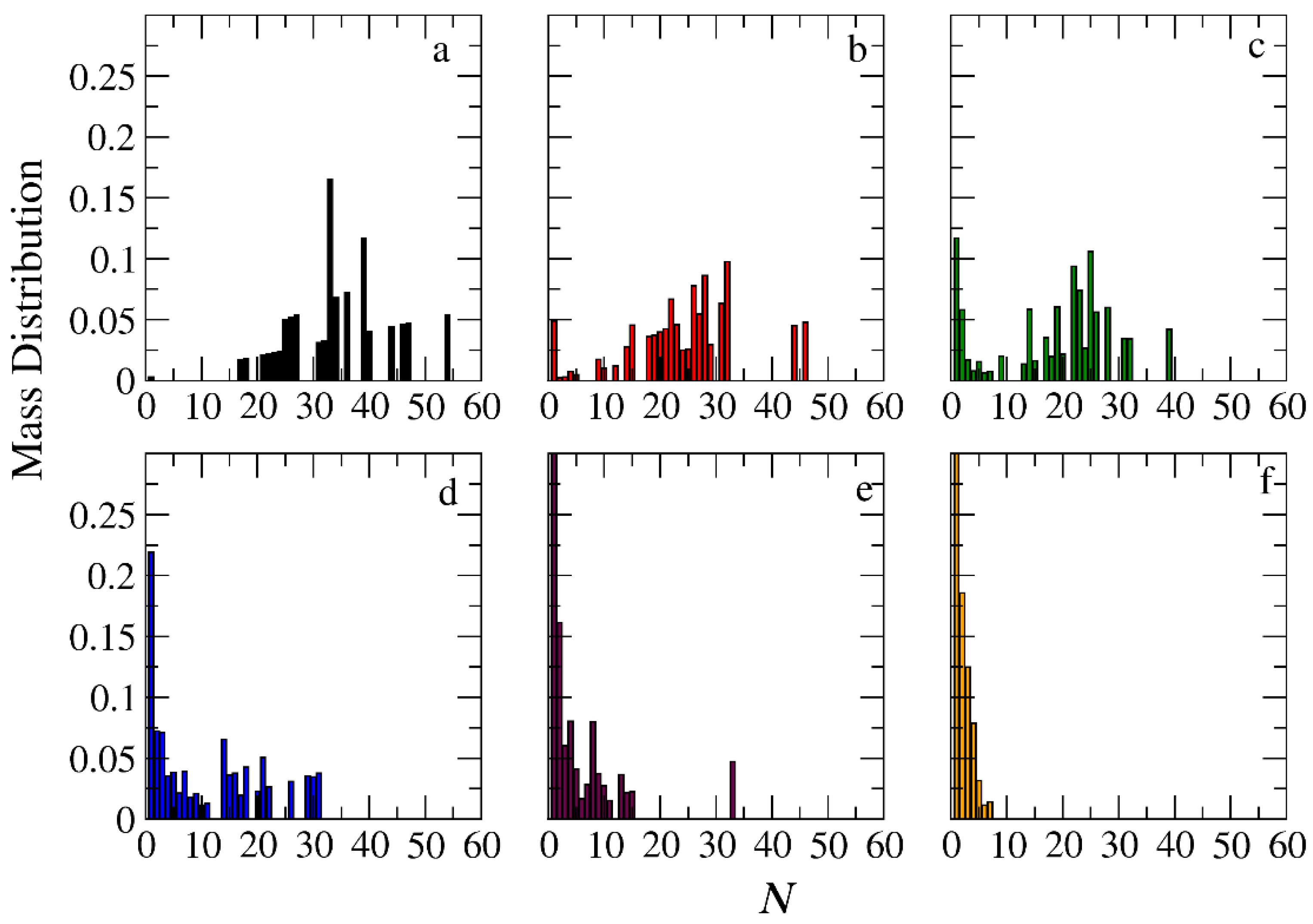
3.3. Demicellization
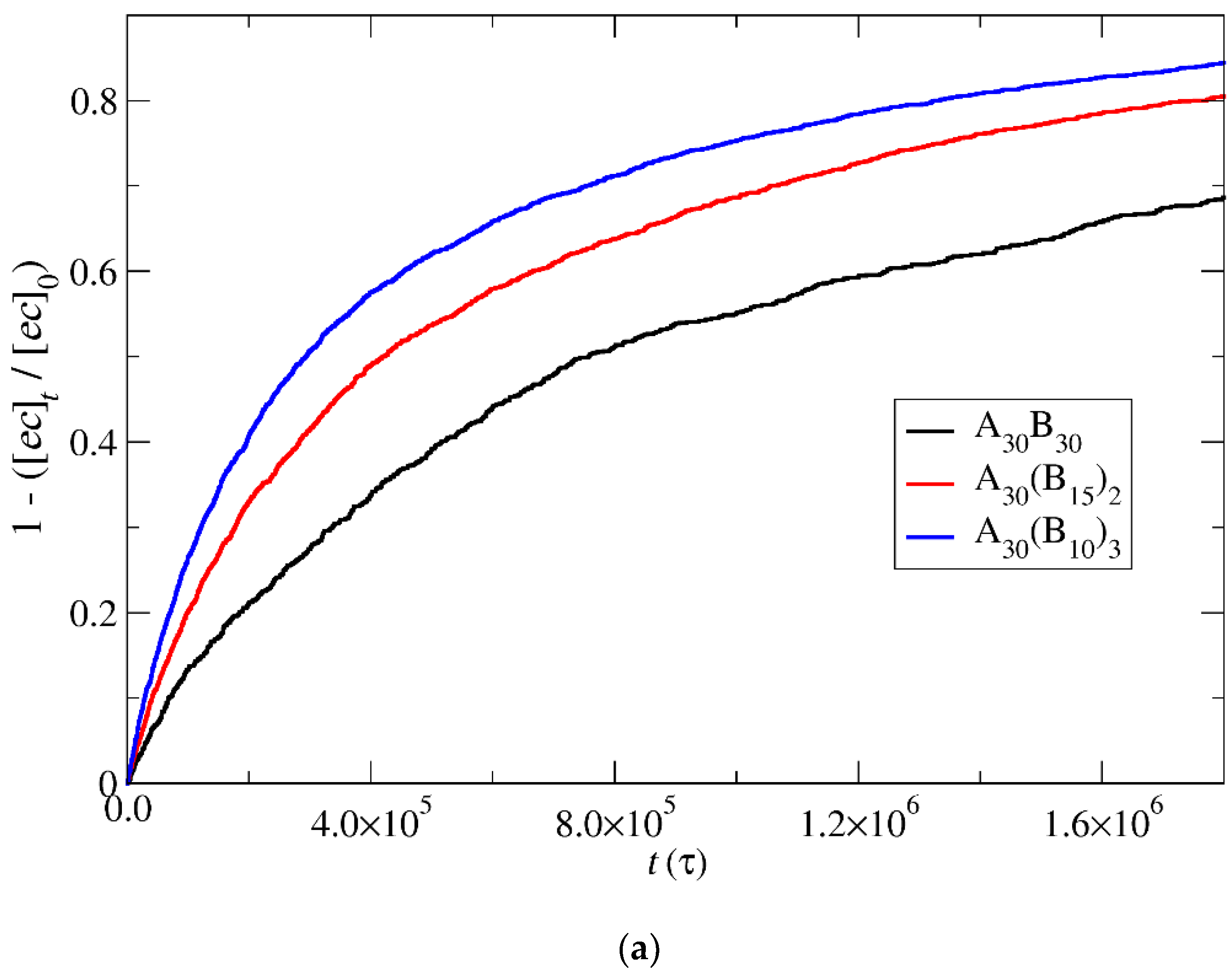
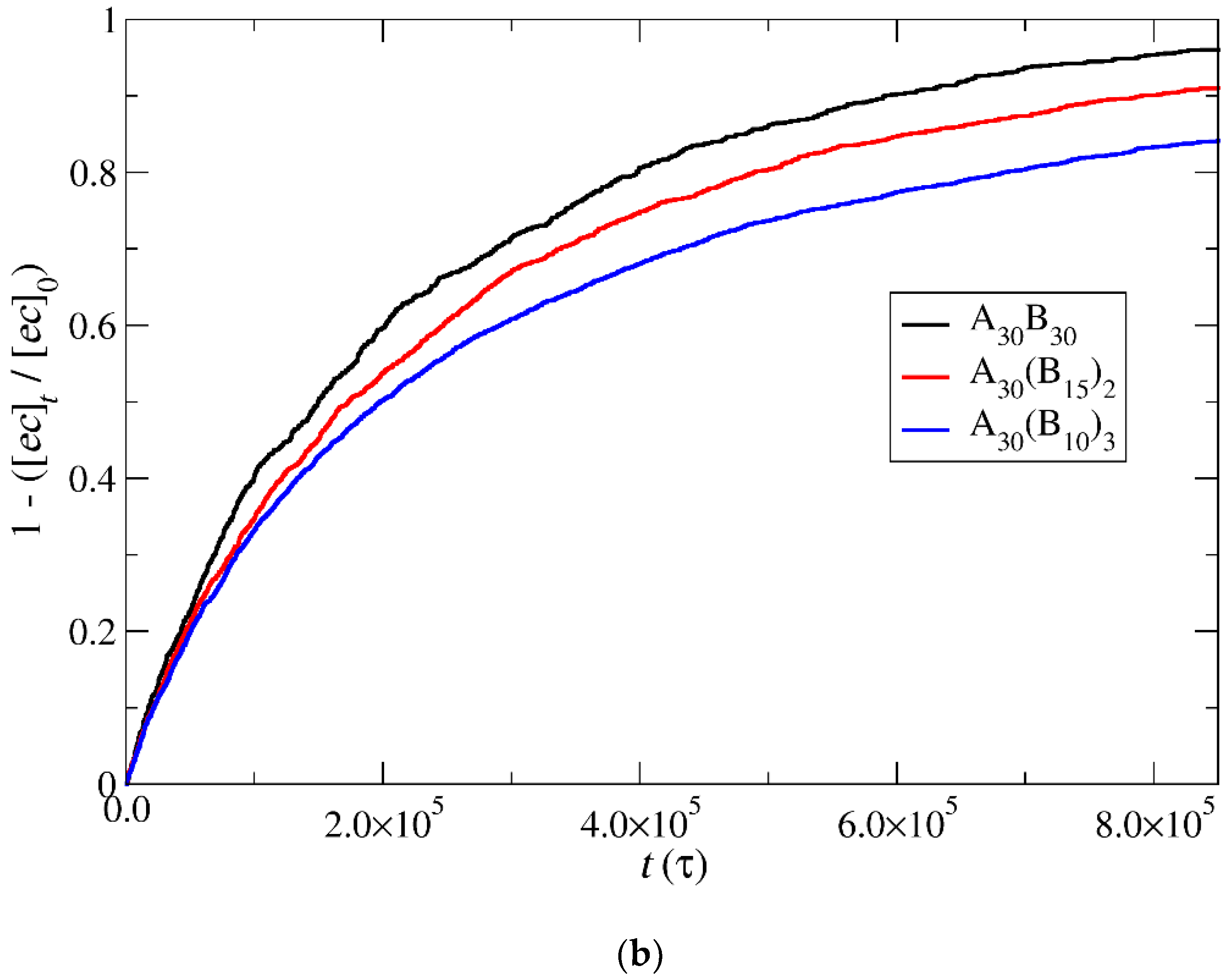
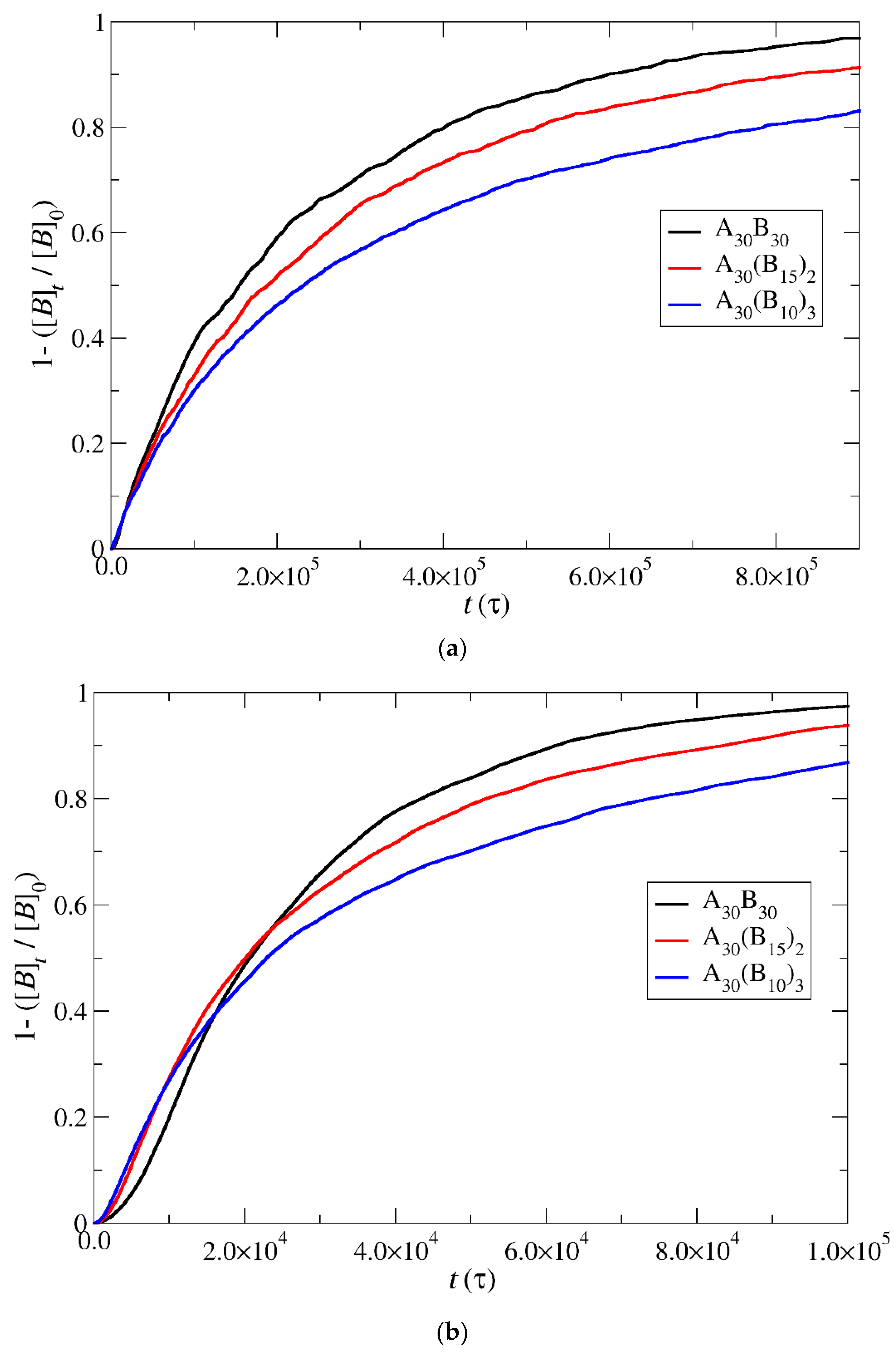
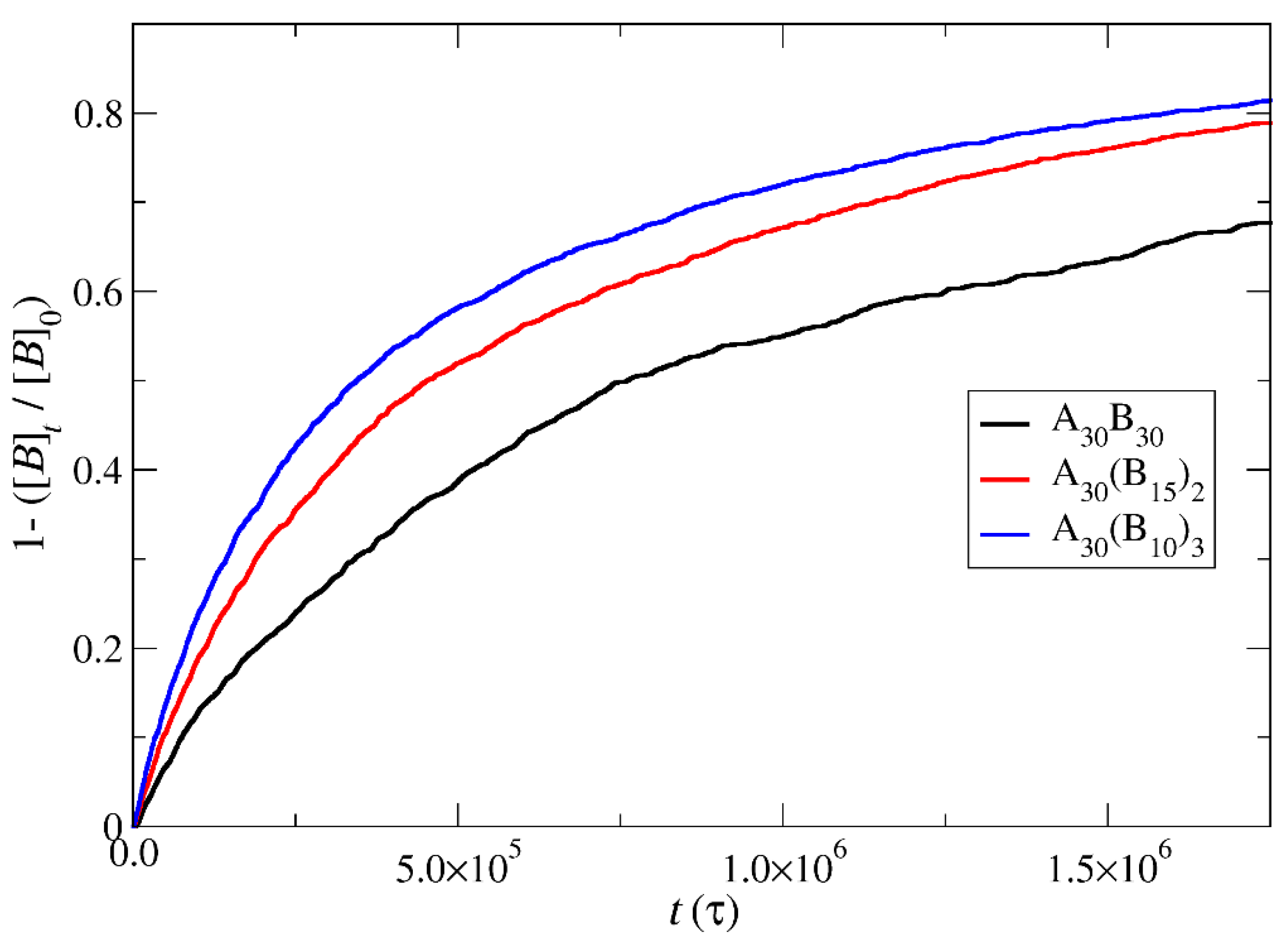
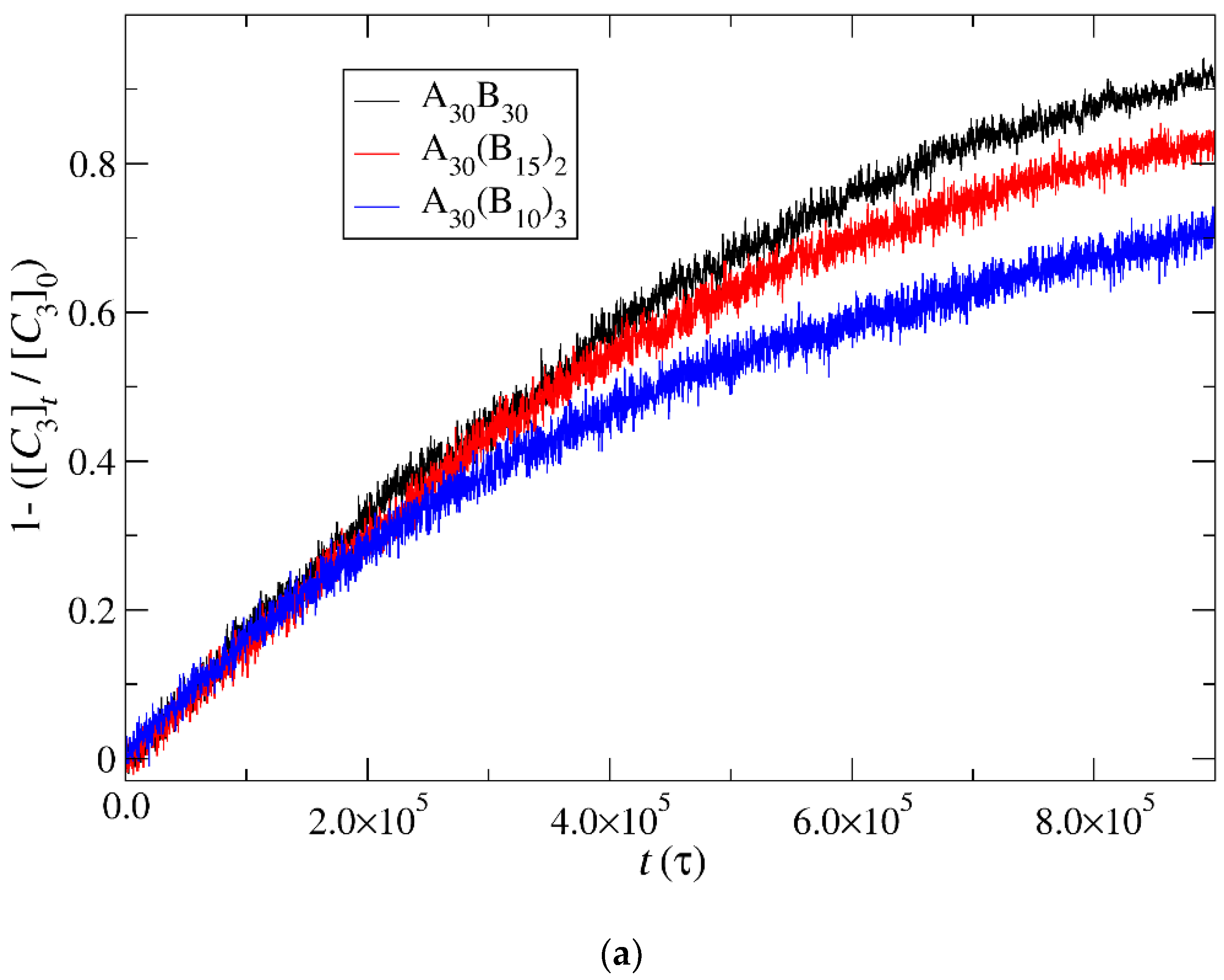
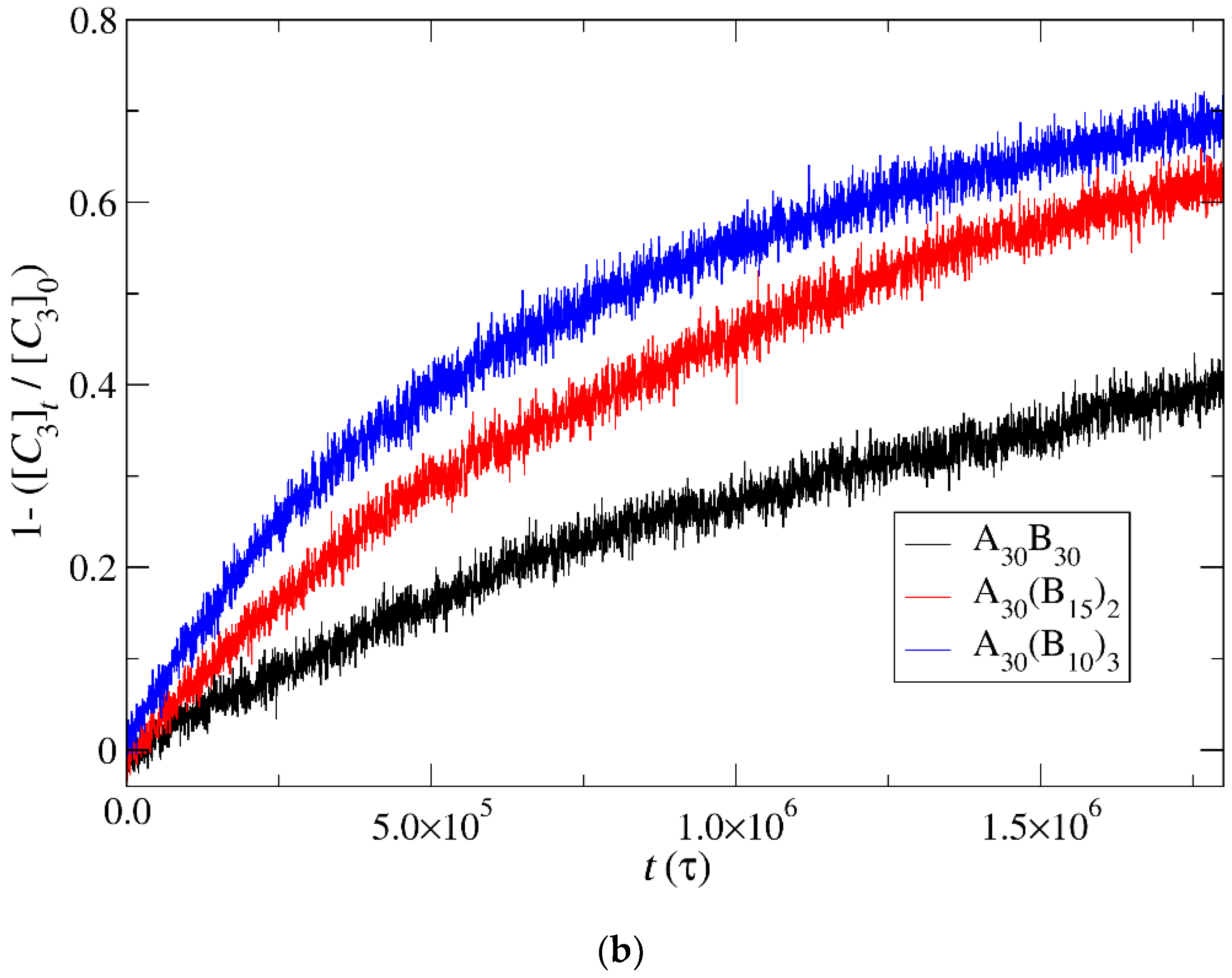
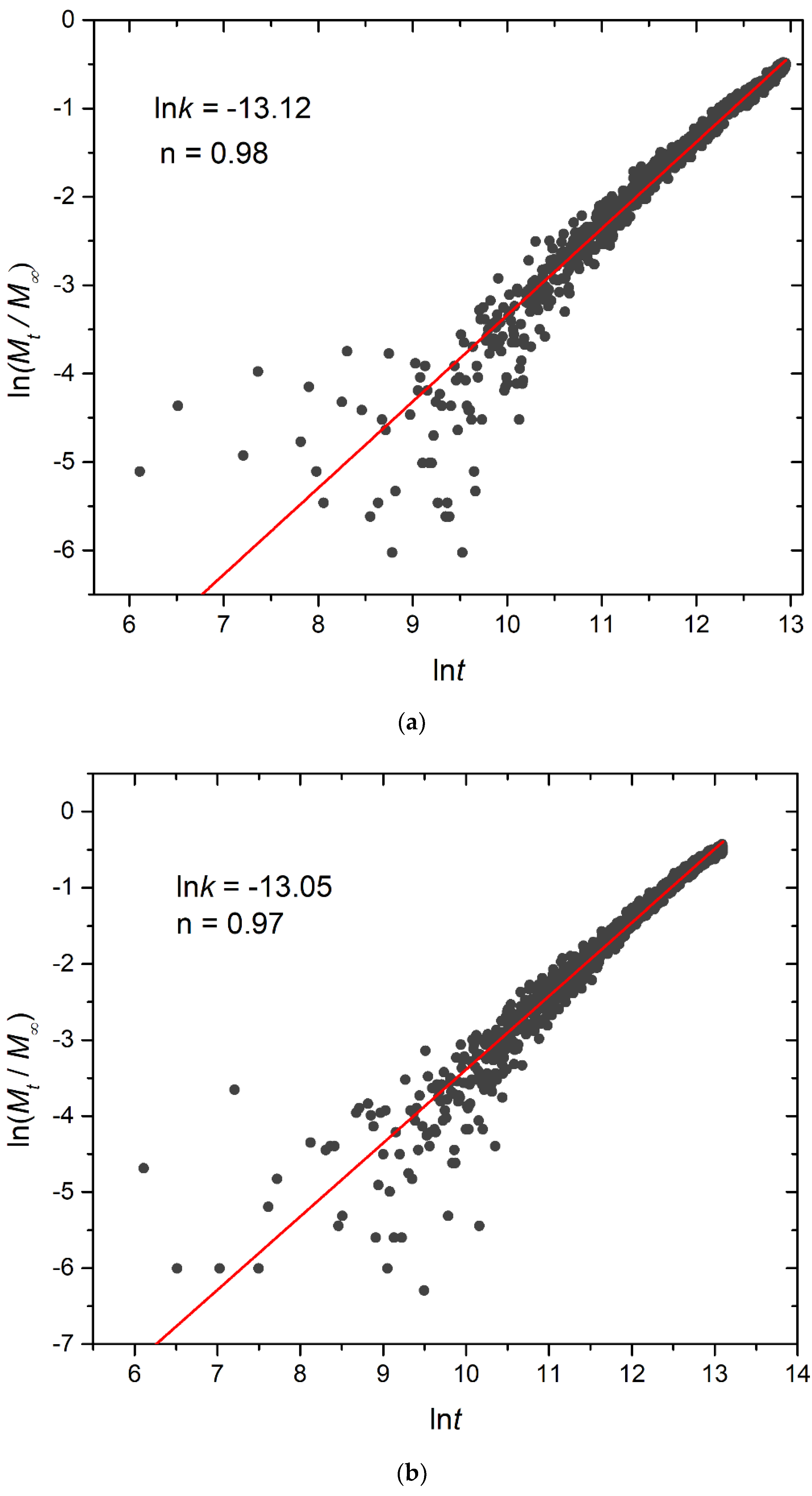
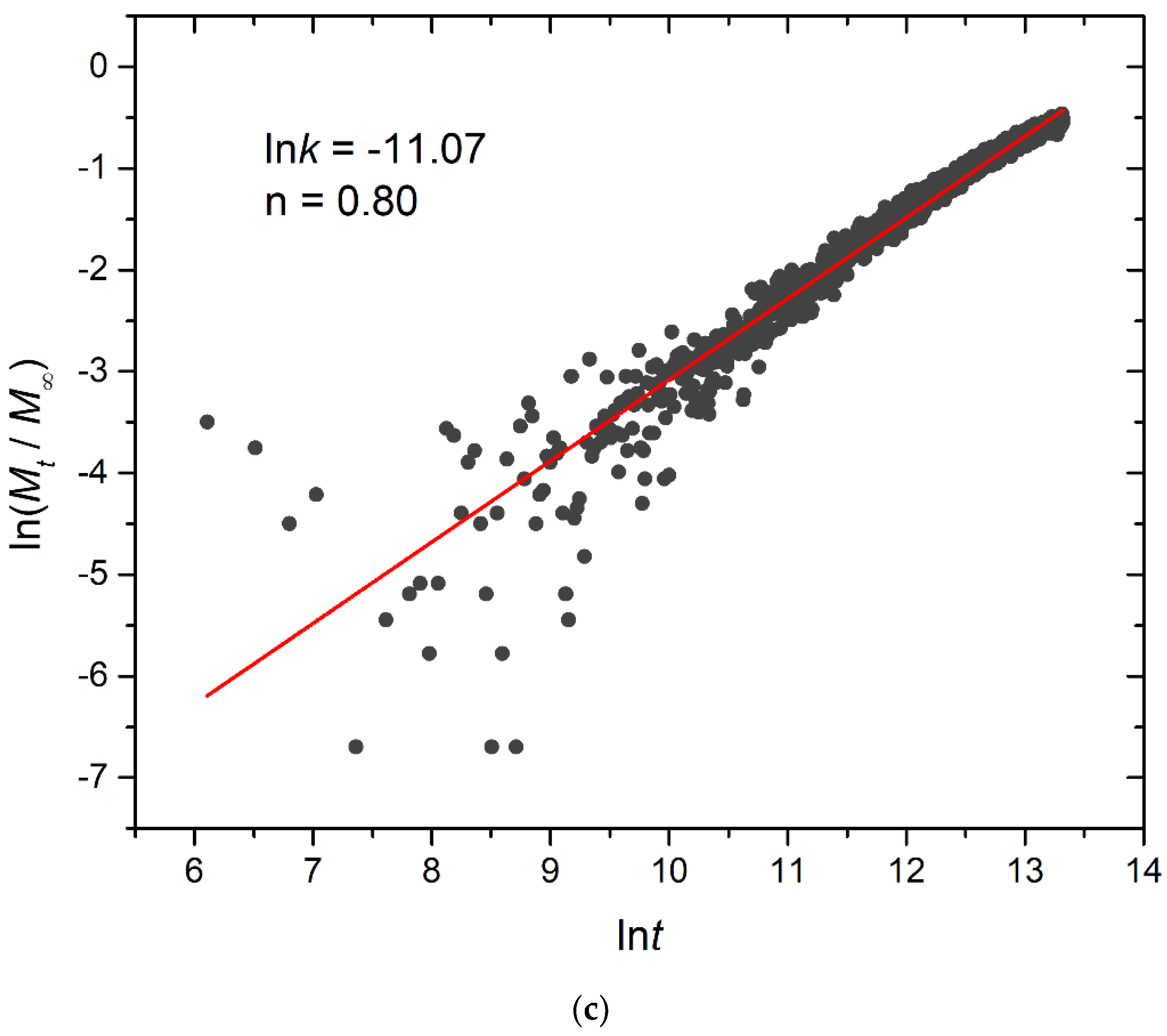
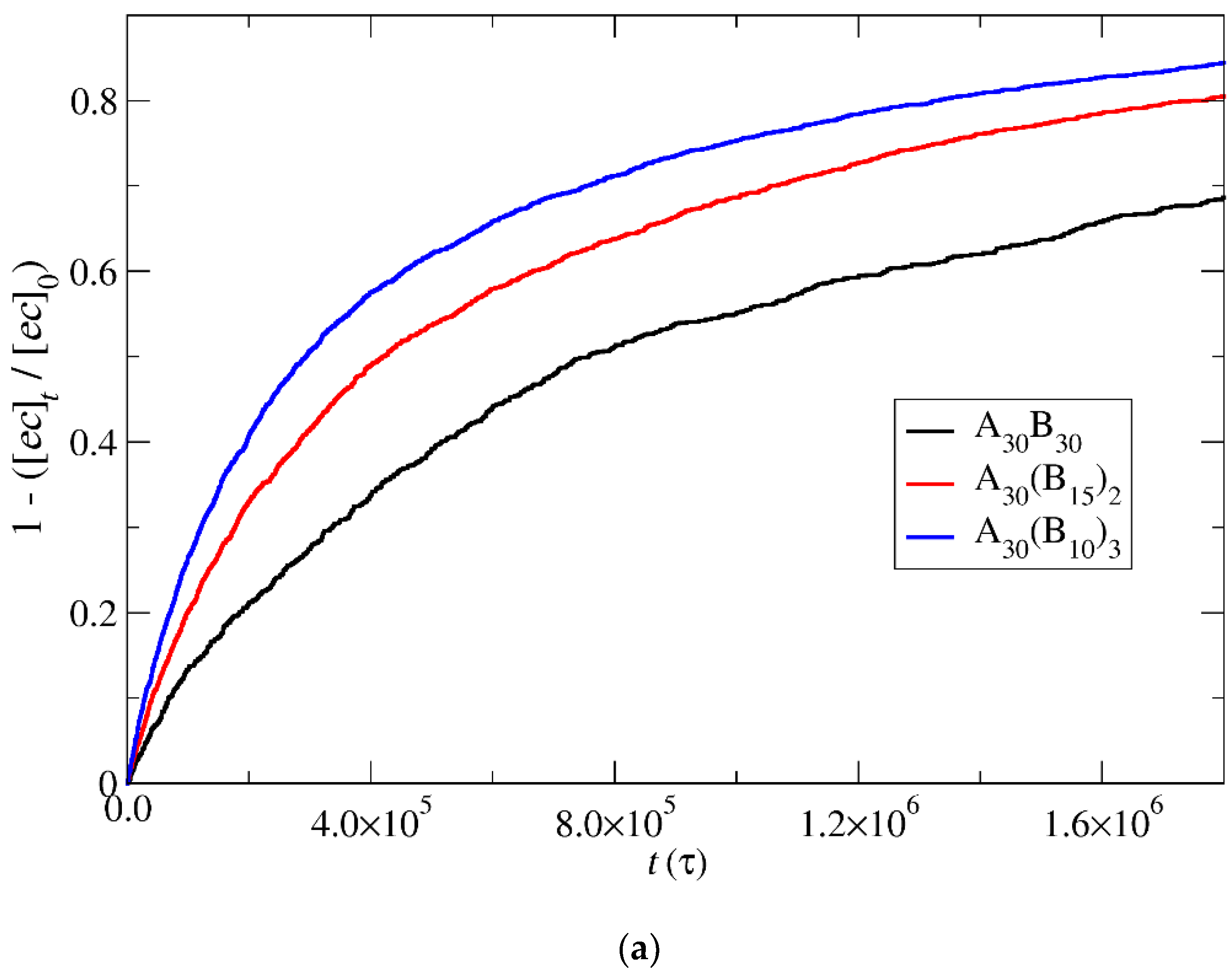
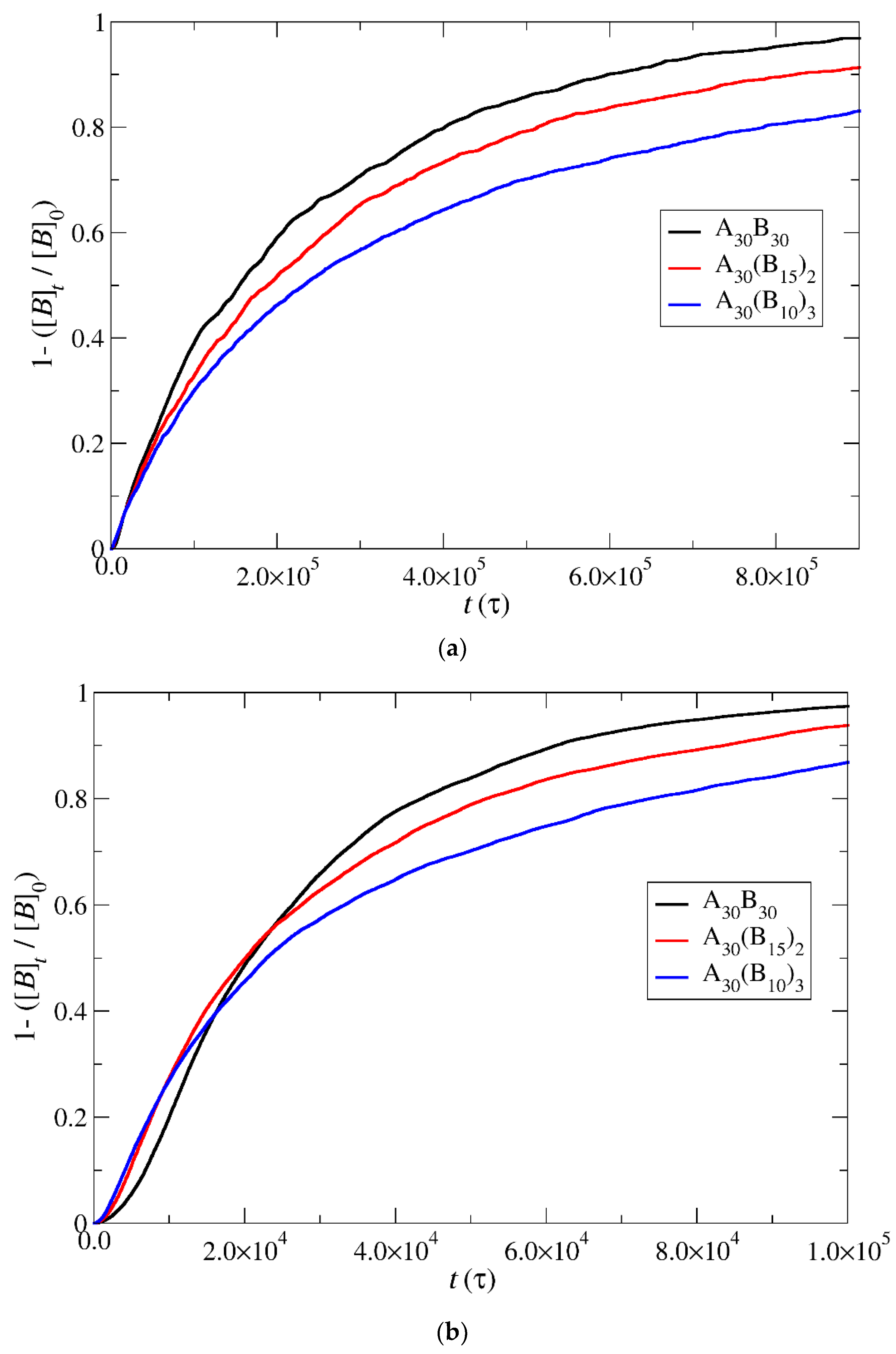
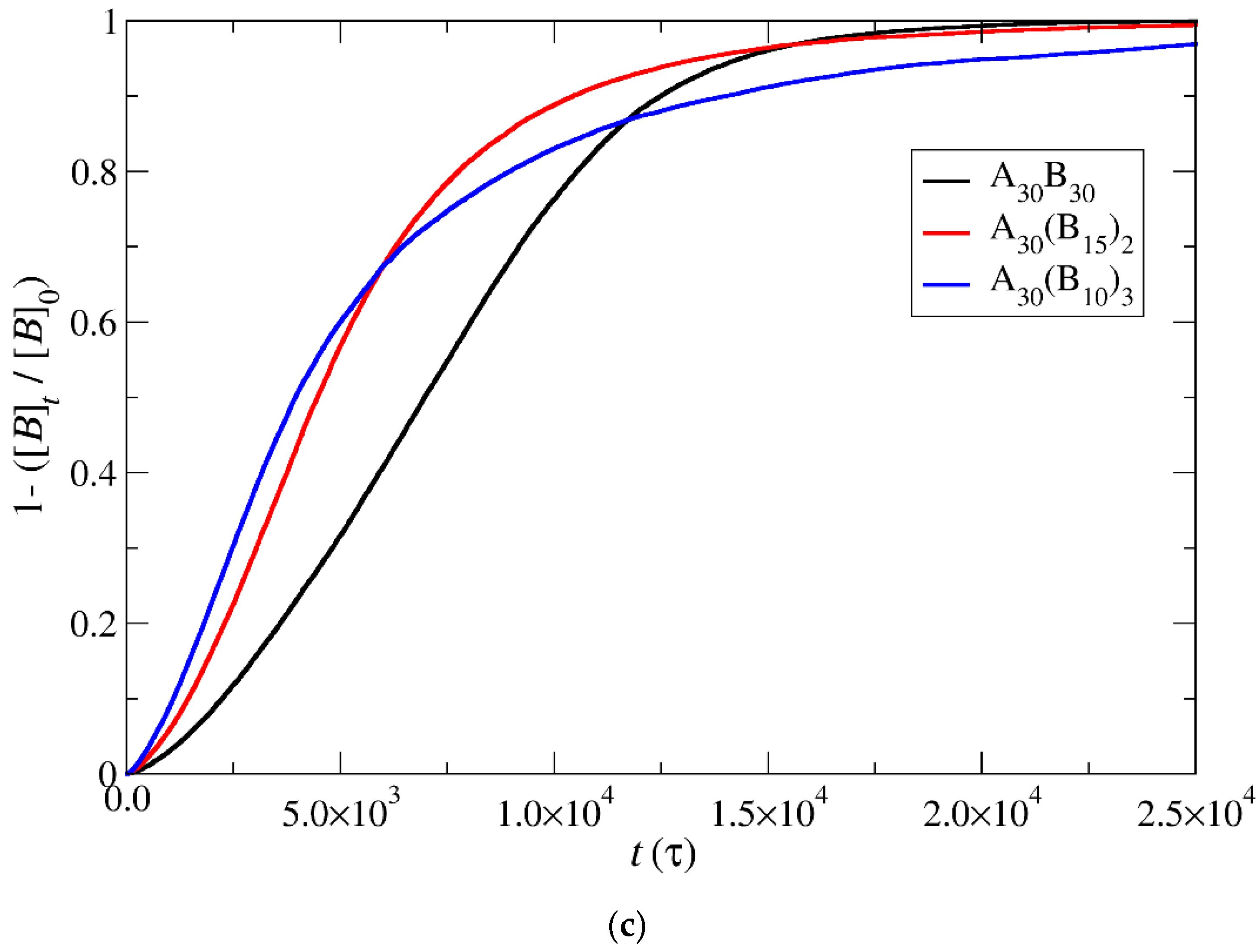
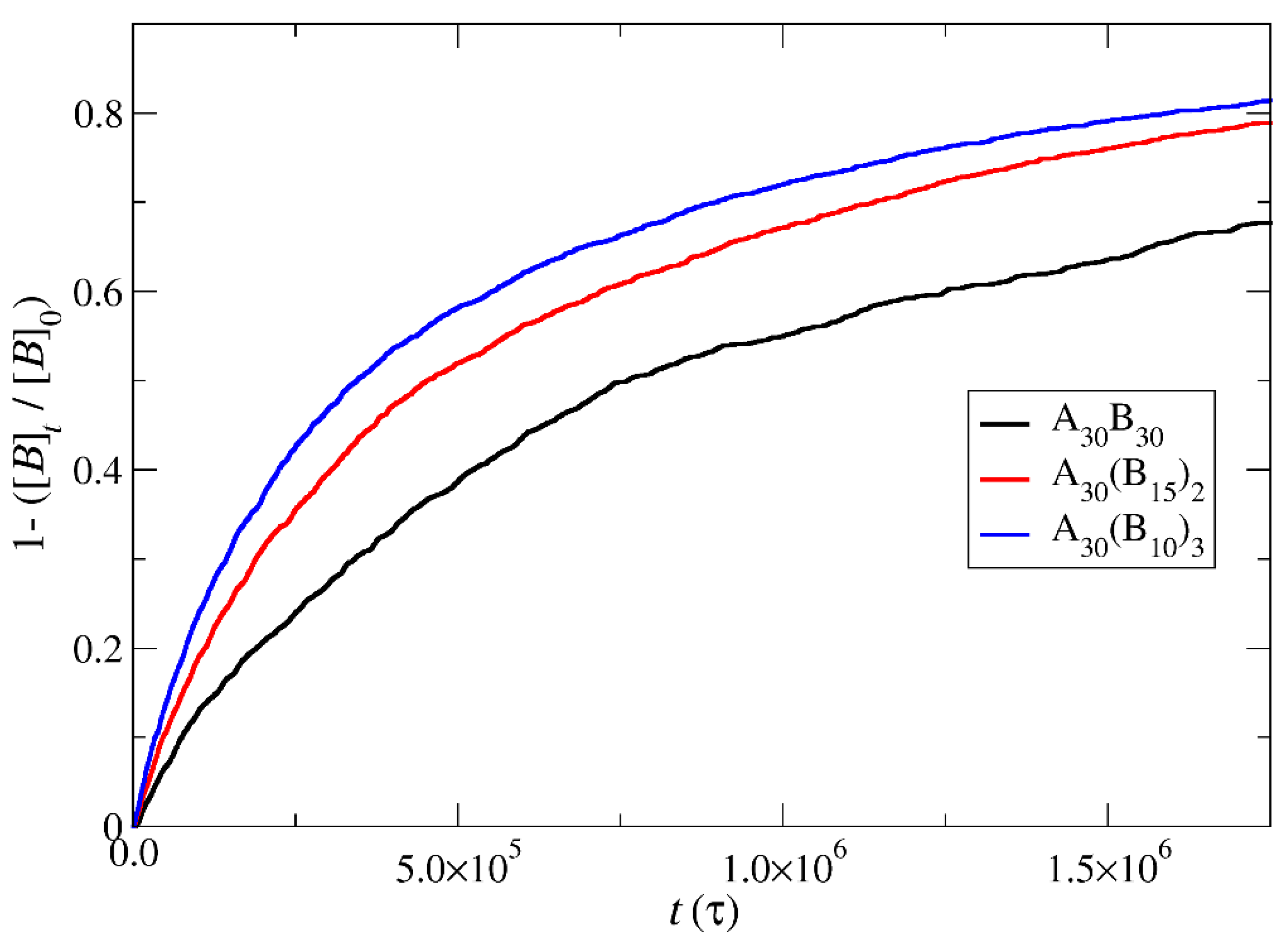
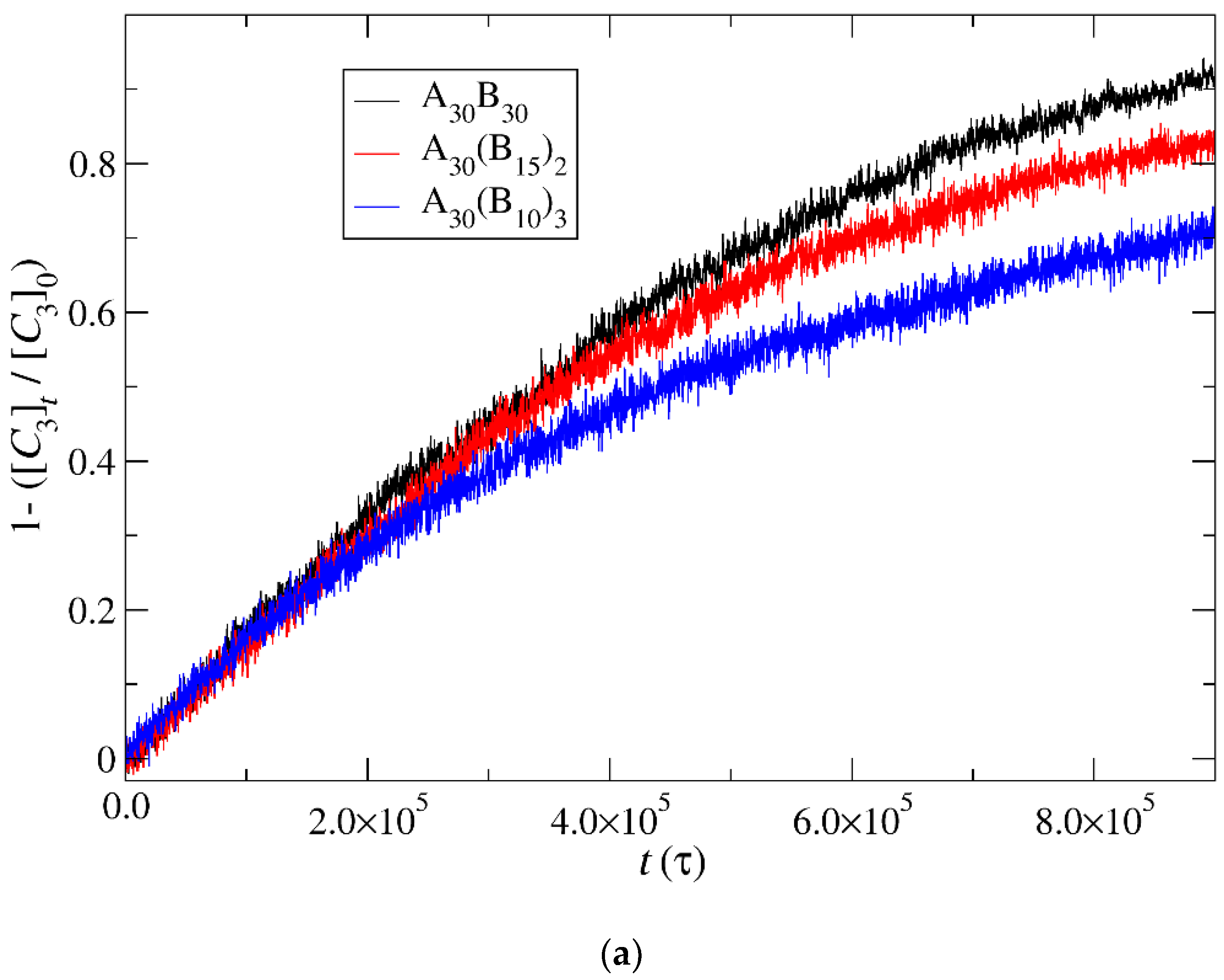
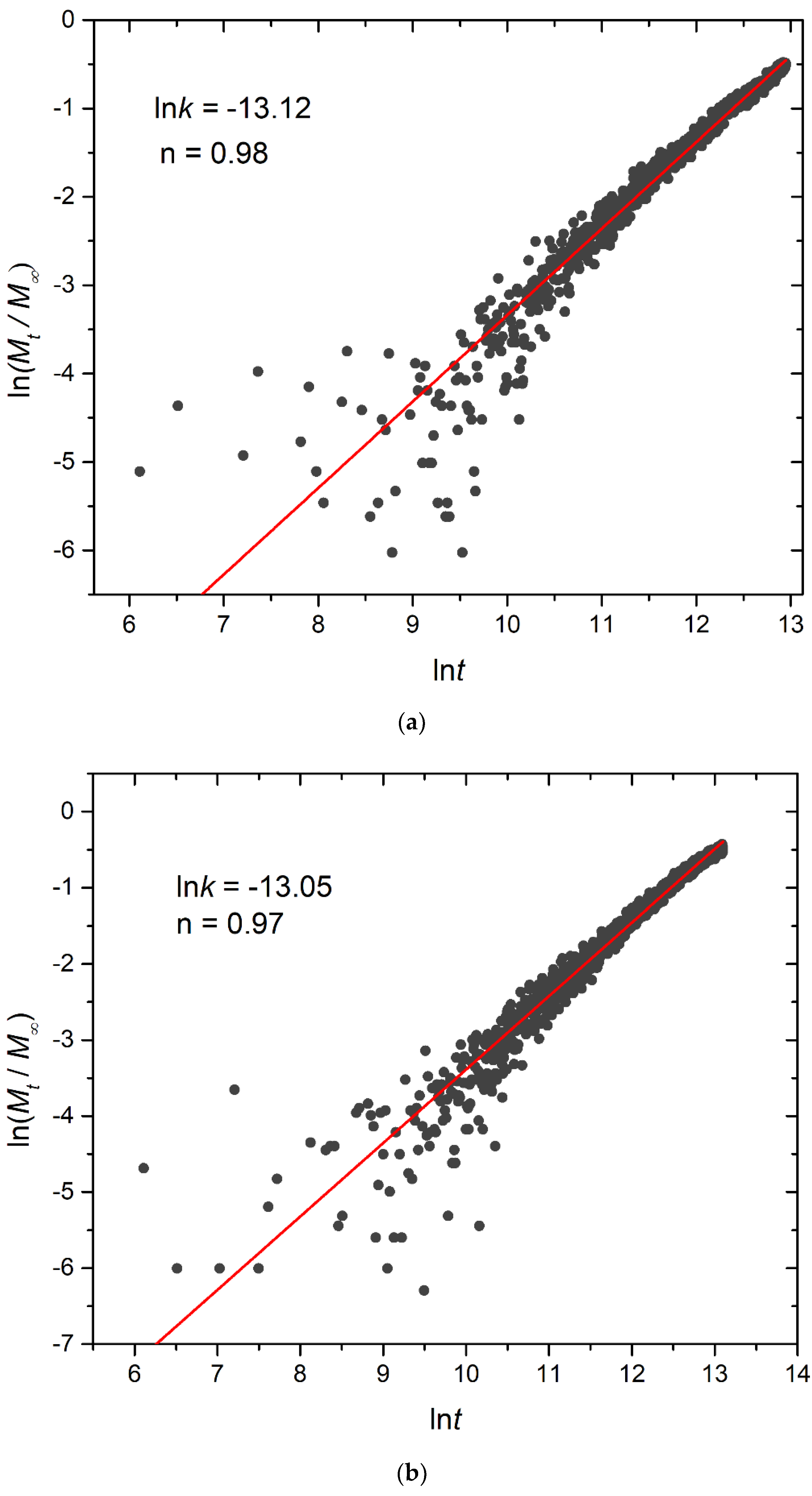
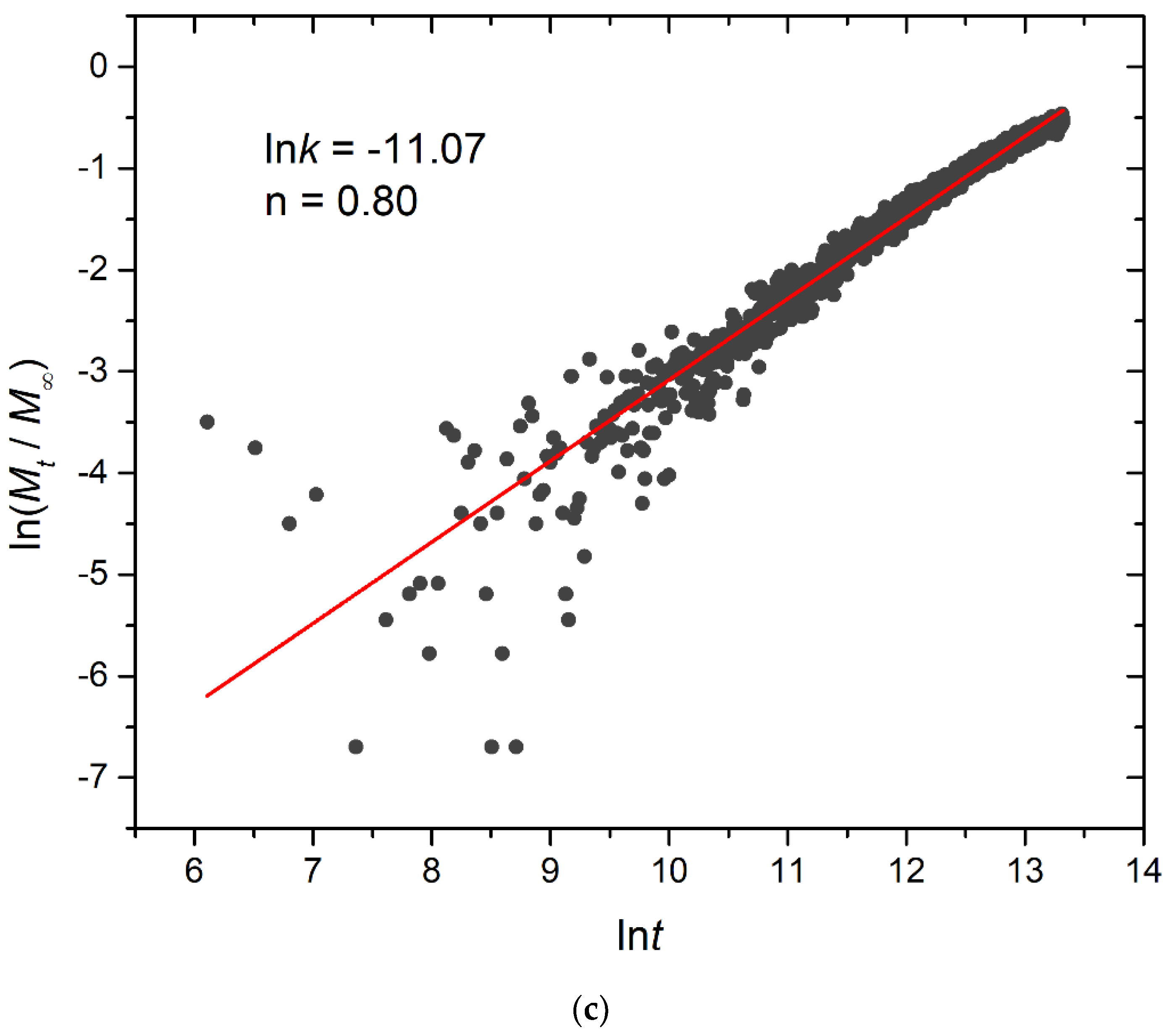
3.4. Kinetics of Cargo Release
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The Current Landscape of Nucleic Acid Therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Sinani, G.; Durgun, M.E.; Cevher, E.; Özsoy, Y. Polymeric-Micelle-Based Delivery Systems for Nucleic Acids. Pharmaceutics 2023, 15, 2021. [Google Scholar] [CrossRef] [PubMed]
- Tanbour, R.; Martins, A.M.; Pitt, W.G.; Husseini, G.A. Drug Delivery Systems Based on Polymeric Micelles and Ultrasound: A Review. Curr. Pharm. Des. 2016, 22, 2796–2807. [Google Scholar] [CrossRef] [PubMed]
- Negut, I.; Bita, B. Polymeric Micellar Systems—A Special Emphasis on “Smart” Drug Delivery. Pharmaceutics 2023, 15, 976. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, M.; Ke, L.; Wang, L.J.; Wu, C.; Li, C.; Li, Z.; Wu, Y.L. Flexible Polymeric Nanosized Micelles for Ophthalmic Drug Delivery: Research Progress in the Last Three Years. Nanoscale Adv. 2021, 3, 5240–5254. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Lu, J.; Wang, J.; Hao, P.; Li, C.; Qi, L.; Yang, L.; He, B.; Zhong, Z.; Hao, N. Redox-Sensitive Polymeric Micelles with Aggregation-Induced Emission for Bioimaging and Delivery of Anticancer Drugs. J. Nanobiotechnol. 2021, 19, 14. [Google Scholar] [CrossRef] [PubMed]
- Gioldasis, C.; Gergidis, L.N.; Vlahos, C. Micellization through Complexation of Oppositely Charged Diblock Copolymers: Effects of Composition, Polymer Architecture, Salt of Different Valency, and Thermoresponsive Block. J. Polym. Sci. 2021, 59, 191–204. [Google Scholar] [CrossRef]
- Suek, N.W.; Lamm, M.H. Computer Simulation of Architectural and Molecular Weight Effects on the Assembly of Amphiphilic Linear-Dendritic Block Copolymers in Solution. Langmuir 2008, 24, 3030–3036. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhao, Q.; Wang, L.; Huang, L.; Liu, Q.; Lu, X.; Cai, Y. Polymerization-Induced Self-Assembly Promoted by Liquid-Liquid Phase Separation. ACS Macro Lett. 2019, 8, 943–946. [Google Scholar] [CrossRef]
- György, C.; Armes, S.P. Recent Advances in Polymerization-Induced Self-Assembly (PISA) Syntheses in Non-Polar Media. Angew. Chem. Int. Ed. 2023, 62, e202308372. [Google Scholar] [CrossRef]
- Kim, J.H.; Li, Y.; Kim, M.S.; Kang, S.W.; Jeong, J.H.; Lee, D.S. Synthesis and Evaluation of Biotin-Conjugated PH-Responsive Polymeric Micelles as Drug Carriers. Int. J. Pharm. 2012, 427, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Ramesh, K.; Reddy, O.S.; Karthika, V.; Kumar, P.; Jo, S.H.; Yoo, S.I.I.; Park, S.H.; Lim, K.T. Redox-Responsive Comparison of Diselenide and Disulfide Core-Cross-Linked Micelles for Drug Delivery Application. Pharmaceutics 2023, 15, 1159. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lorenzo, C.; Bromberg, L.; Concheiro, A. Light-Sensitive Intelligent Drug Delivery Systems. Photochem. Photobiol. 2009, 85, 848–860. [Google Scholar] [CrossRef] [PubMed]
- DeWit, M.A.; Gillies, E.R. A Cascade Biodegradable Polymer Based on Alternating Cyclization and Elimination Reactions. J. Am. Chem. Soc. 2009, 131, 18327–18334. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.T.; Dilauro, A.M. Continuous Head-to-Tail Depolymerization: An Emerging Concept for Imparting Amplified Responses to Stimuli-Responsive Materials. ACS Macro Lett. 2014, 3, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.J.; Pessah, N.; Shamis, M.; Shabat, D. Self-Immolative Dendrimers. Angew. Chem.-Int. Ed. 2003, 42, 4494–4499. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jo, S.; Lee, Y.M.; Saravanakumar, G.; Lee, J.; Park, D.; Kim, W.J. Enzyme-Triggered Disassembly of Polymeric Micelles by Controlled Depolymerization via Cascade Cyclization for Anticancer Drug Delivery. ACS Appl. Mater. Interfaces 2021, 13, 8060–8070. [Google Scholar] [CrossRef]
- Sirianni, Q.E.A.; Liang, X.; Such, G.K.; Gillies, E.R. Polyglyoxylamides with a PH-Mediated Solubility and Depolymerization Switch. Macromolecules 2021, 54, 10547–10556. [Google Scholar] [CrossRef]
- Liu, G.; Wang, X.; Hu, J.; Zhang, G.; Liu, S. Self-Immolative Polymersomes for High-Efficiency Triggered Release and Programmed Enzymatic Reactions. J. Am. Chem. Soc. 2014, 136, 7492–7497. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, H.J.; Park, J.; Chae, J.A.; Song, H.W.; Choi, E.; Kim, H. Self-Immolative and Amphiphilic Poly(Benzyl Ether)-Based Copolymers: Synthesis and Triggered Demicellization via Head-to-Tail Depolymerization. Macromolecules 2022, 55, 6140–6149. [Google Scholar] [CrossRef]
- Murat, M.; Grest, G.S. Molecular Dynamics Study of Dendrimer Molecules in Solvents of Varying Quality. Macromolecules 1996, 29, 1278–1285. [Google Scholar] [CrossRef]
- Miliou, K.; Gergidis, L.N.; Vlahos, C. Polyelectrolyte Micelles in Salt-Free Solutions: Micelle Size and Electrostatic Potential. J. Polym. Sci. B Polym. Phys. 2018, 56, 924–934. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Stillinger, F.H. Rigorous Basis of the Frenkel-Band Theory of Association Equilibrium. J. Chem. Phys. 1963, 38, 1486–1494. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring Network Structure, Dynamics, and Function Using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy 2008), Pasadena, CA, USA, 19–24 August 2008. [Google Scholar]
- Cheng, L.; Cao, D. Effect of Tail Architecture on Self-Assembly of Amphiphiles for Polymeric Micelles. Langmuir 2009, 25, 2749–2756. [Google Scholar] [CrossRef]
- Georgiadis, C.; Moultos, O.; Gergidis, L.N.; Vlahos, C. Brownian Dynamics Simulations on the Self-Assembly Behavior of AB Hybrid Dendritic-Star Copolymers. Langmuir 2011, 27, 835–842. [Google Scholar] [CrossRef]
- Hwang, D.; Ramsey, J.D.; Kabanov, A.V. Polymeric Micelles for the Delivery of Poorly Soluble Drugs: From Nanoformulation to Clinical Approval. Adv. Drug Deliv. Rev. 2020, 156, 80–118. [Google Scholar] [CrossRef]
- Theodorou, D.N.; Suter, U.W. Shape of Unperturbed Linear Polymers: Polypropylene. Macromolecules 1985, 18, 1206–1214. [Google Scholar] [CrossRef]
- Peppas, N.A.; Sahlin, J.J. A Simple Equation for the Description of Solute Release. III. Coupling of Diffusion and Relaxation. Int. J. Pharm. 1989, 57, 169–172. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A Simple Equation for Description of Solute Release II. Fickian and Anomalous Release from Swellable Devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- He, E.; Yue, C.Y.; Tam, K.C. Binding and Release Studies of a Cationic Drug from a Star-Shaped Four-Arm Poly(Ethylene Oxide)-b-Poly(Methacrylic Acid). J. Pharm. Sci. 2010, 99, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Monticelli, L.; Melo, M.N.; Alessandri, R.; Tieleman, D.P.; Souza, P.C.T. Two Decades of Martini: Better Beads, Broader Scope. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2023, 13, e1620. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gioldasis, C.; Gkamas, A.; Vlahos, C. Impact of Copolymer Architecture on Demicellization and Cargo Release via Head-to-Tail Depolymerization of Hydrophobic Blocks or Branches. Polymers 2024, 16, 1127. https://doi.org/10.3390/polym16081127
Gioldasis C, Gkamas A, Vlahos C. Impact of Copolymer Architecture on Demicellization and Cargo Release via Head-to-Tail Depolymerization of Hydrophobic Blocks or Branches. Polymers. 2024; 16(8):1127. https://doi.org/10.3390/polym16081127
Chicago/Turabian StyleGioldasis, Christos, Apostolos Gkamas, and Costas Vlahos. 2024. "Impact of Copolymer Architecture on Demicellization and Cargo Release via Head-to-Tail Depolymerization of Hydrophobic Blocks or Branches" Polymers 16, no. 8: 1127. https://doi.org/10.3390/polym16081127