Preparation and Evaluation of Core–Shell Magnetic Molecularly Imprinted Polymers for Solid-Phase Extraction and Determination of Sterigmatocystin in Food

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Instrumentation
2.3. Synthesis of Silica Coated Fe3O4 Particles
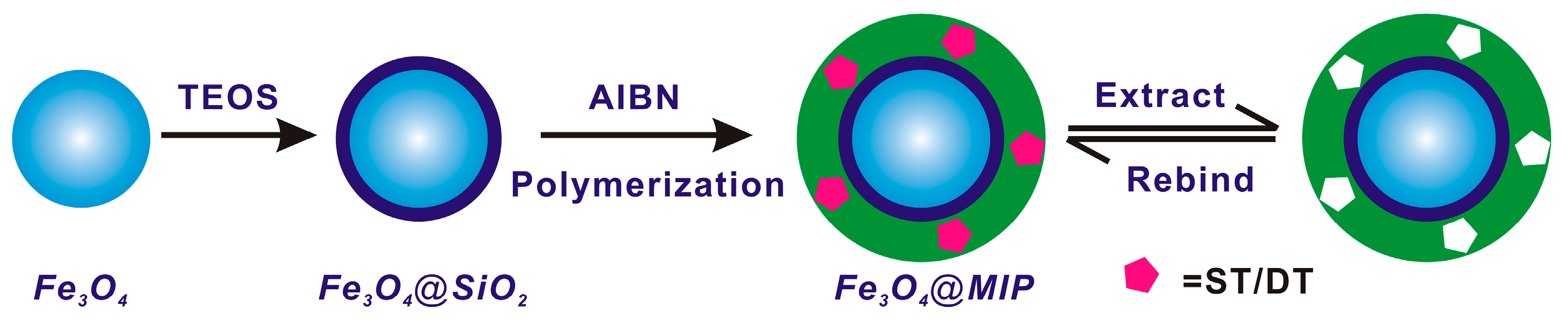
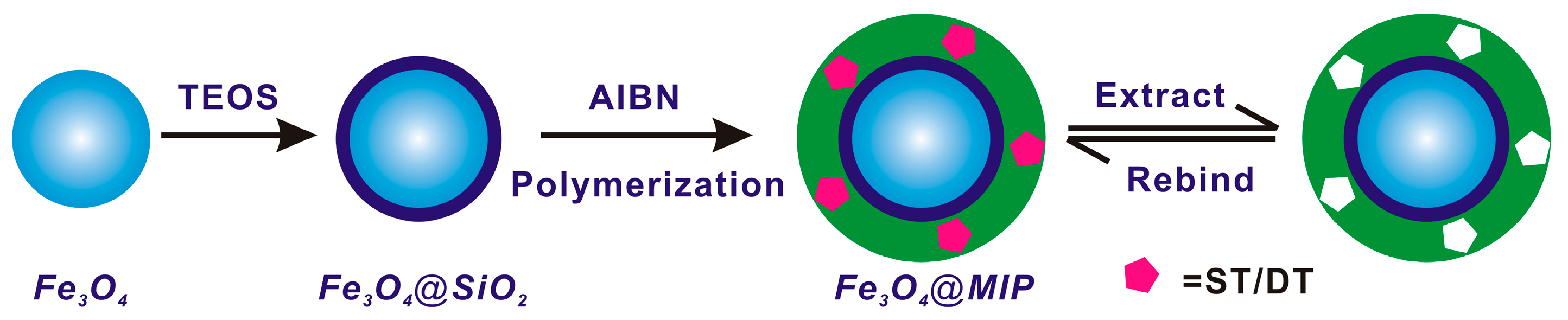
2.4. Preparation of Magnetic Core–Shell Fe3O4@MIPs and Fe3O4@NIPs Particles
2.5. Determination of Sterigmatocystin via Magnetic Fe3O4@MIPs
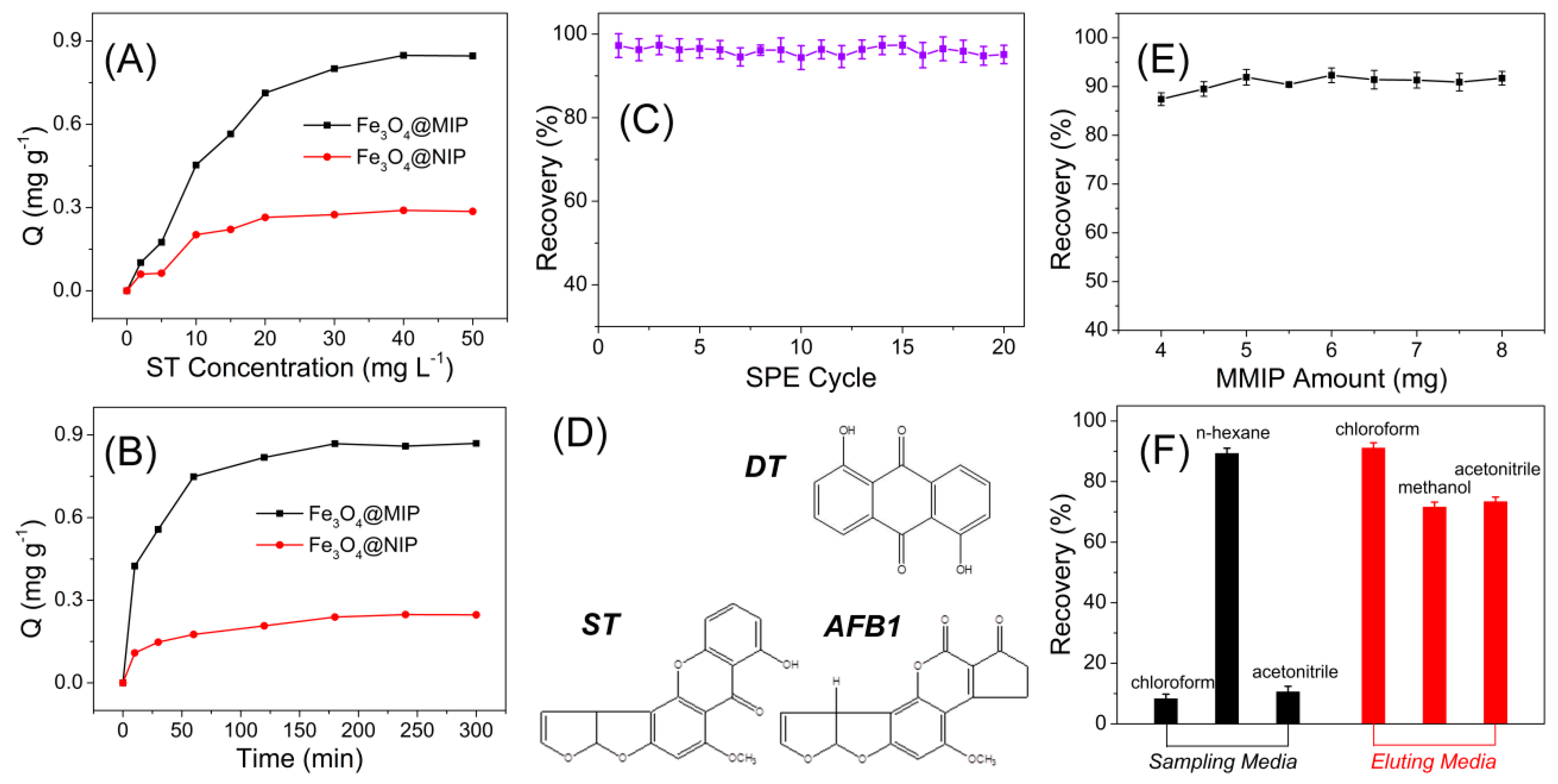
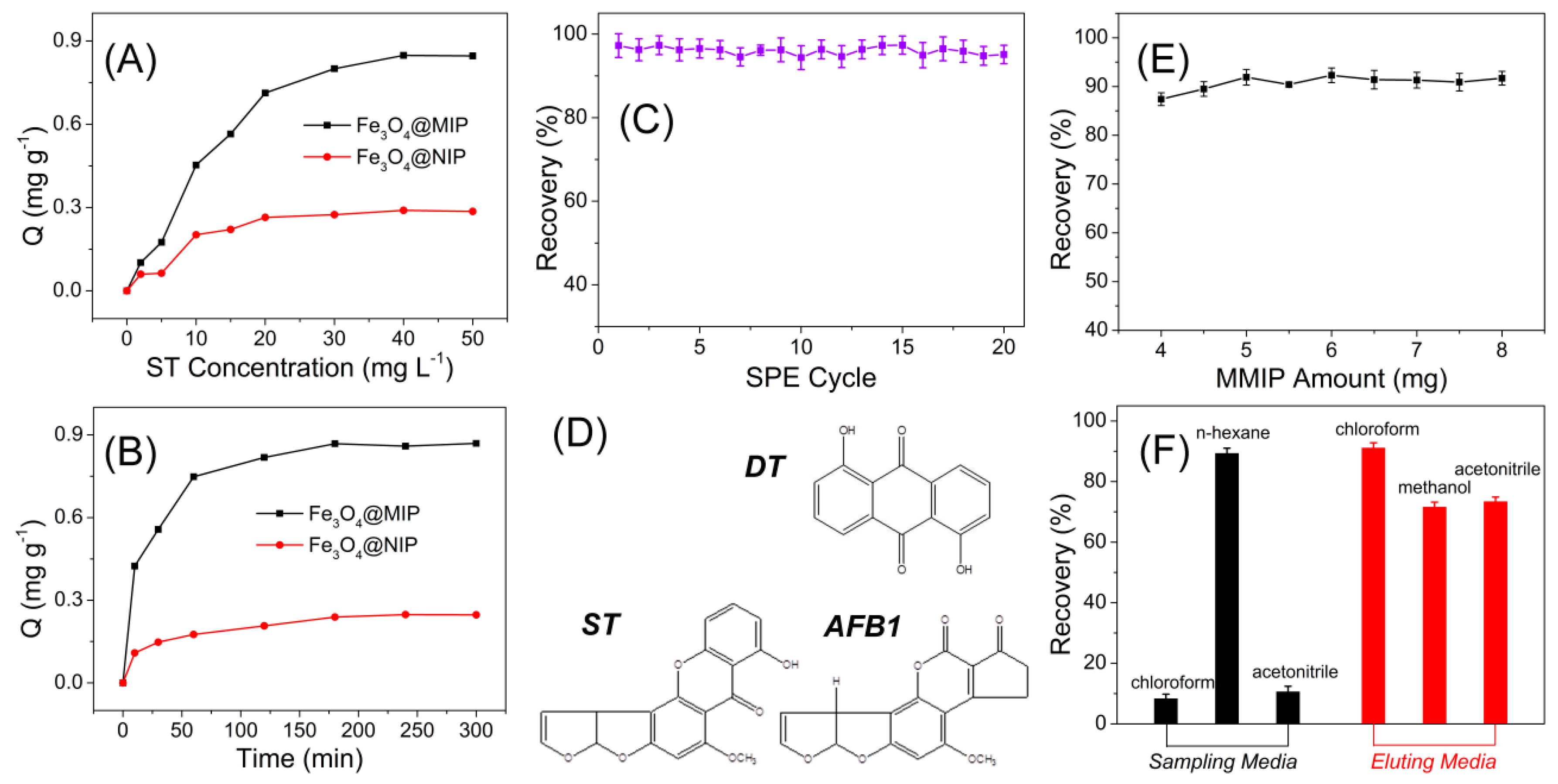
2.6. Adsorption Isotherms
2.7. Adsorption Kinetics Investigations
2.8. Real Sample Analysis
3. Results and Discussion
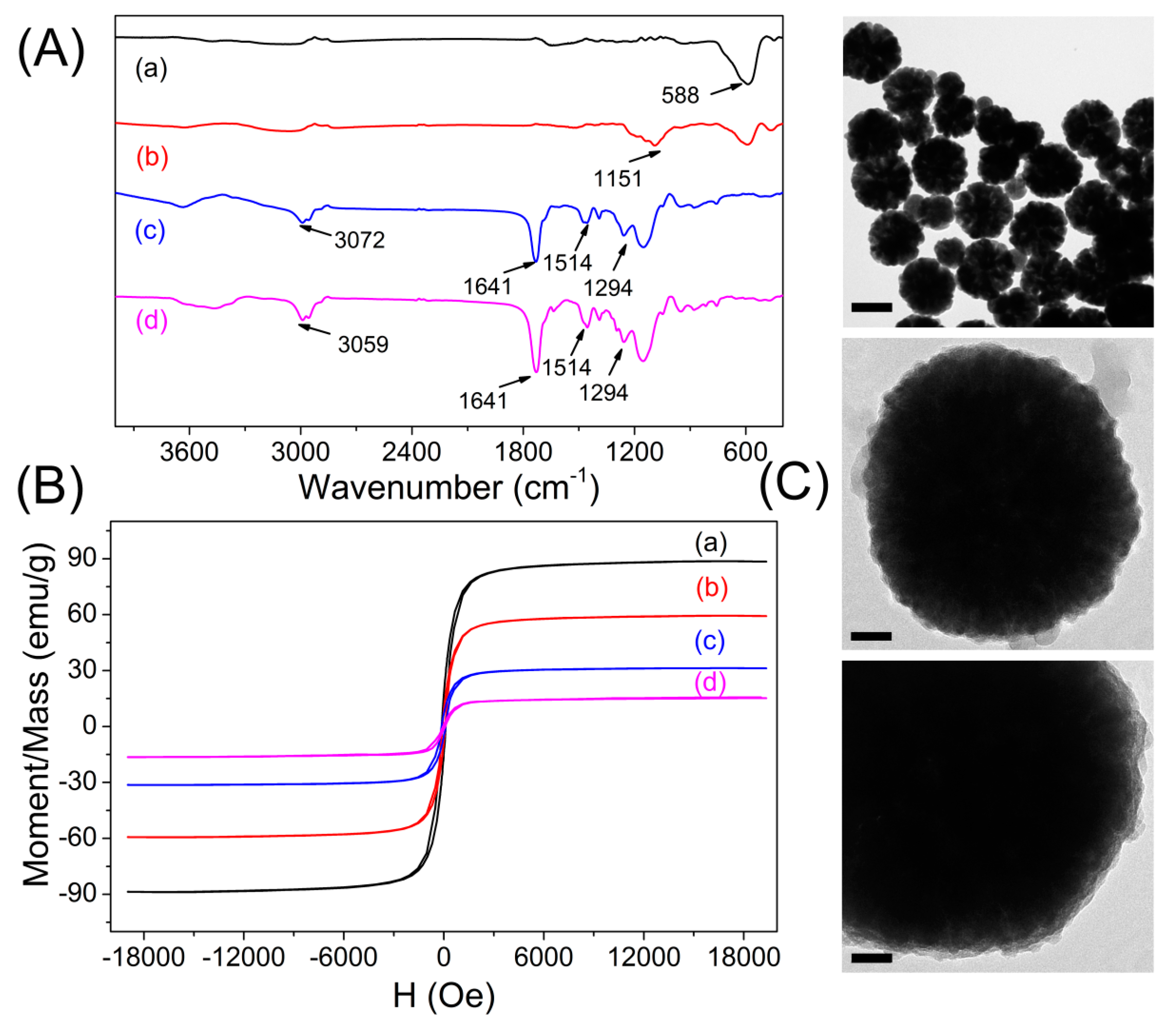
3.1. Preparation and Characterization of Imprinted Magnetic Nanoparticles
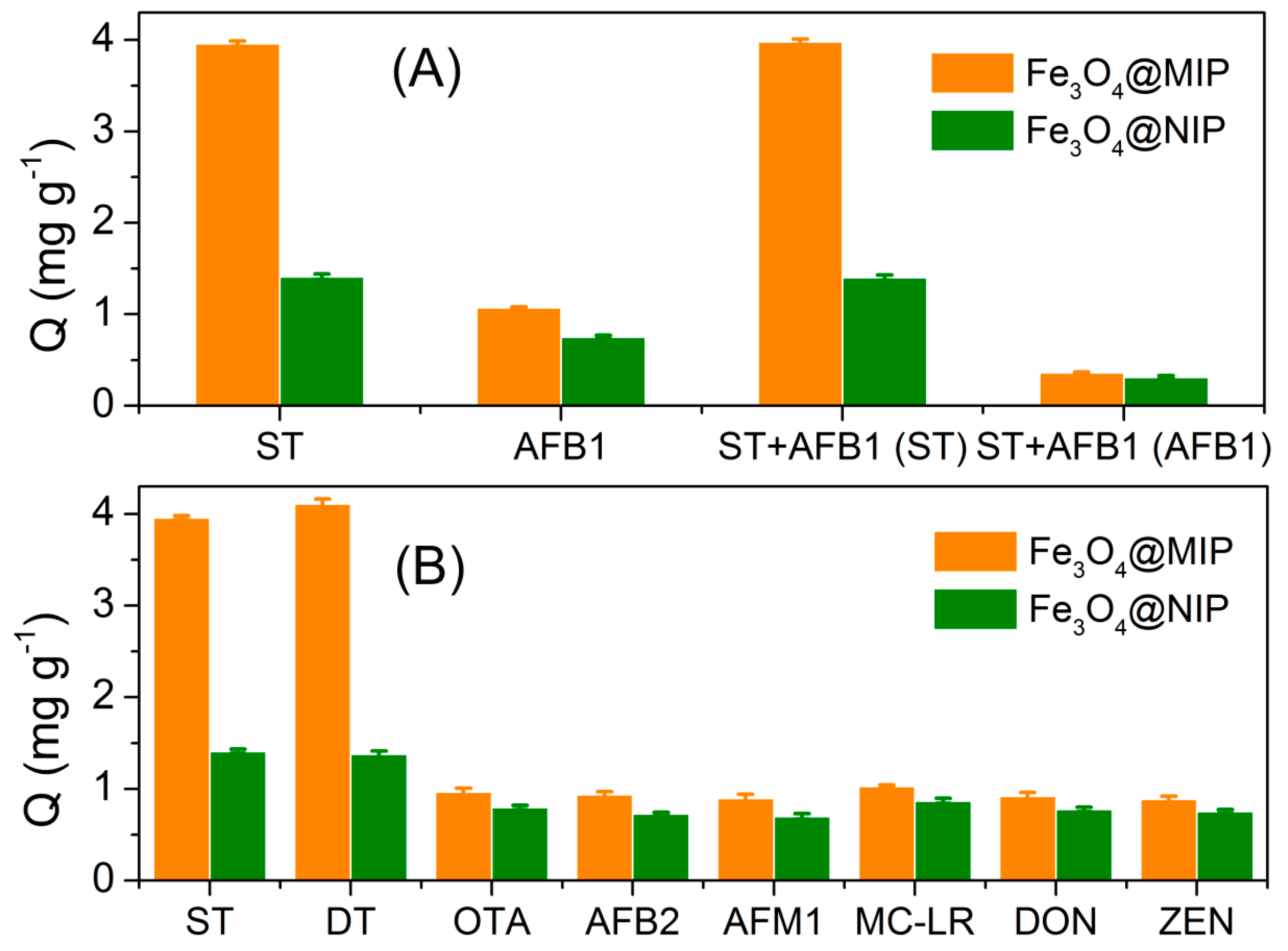
3.2. MIPs Recognition of Sterigmatocystin
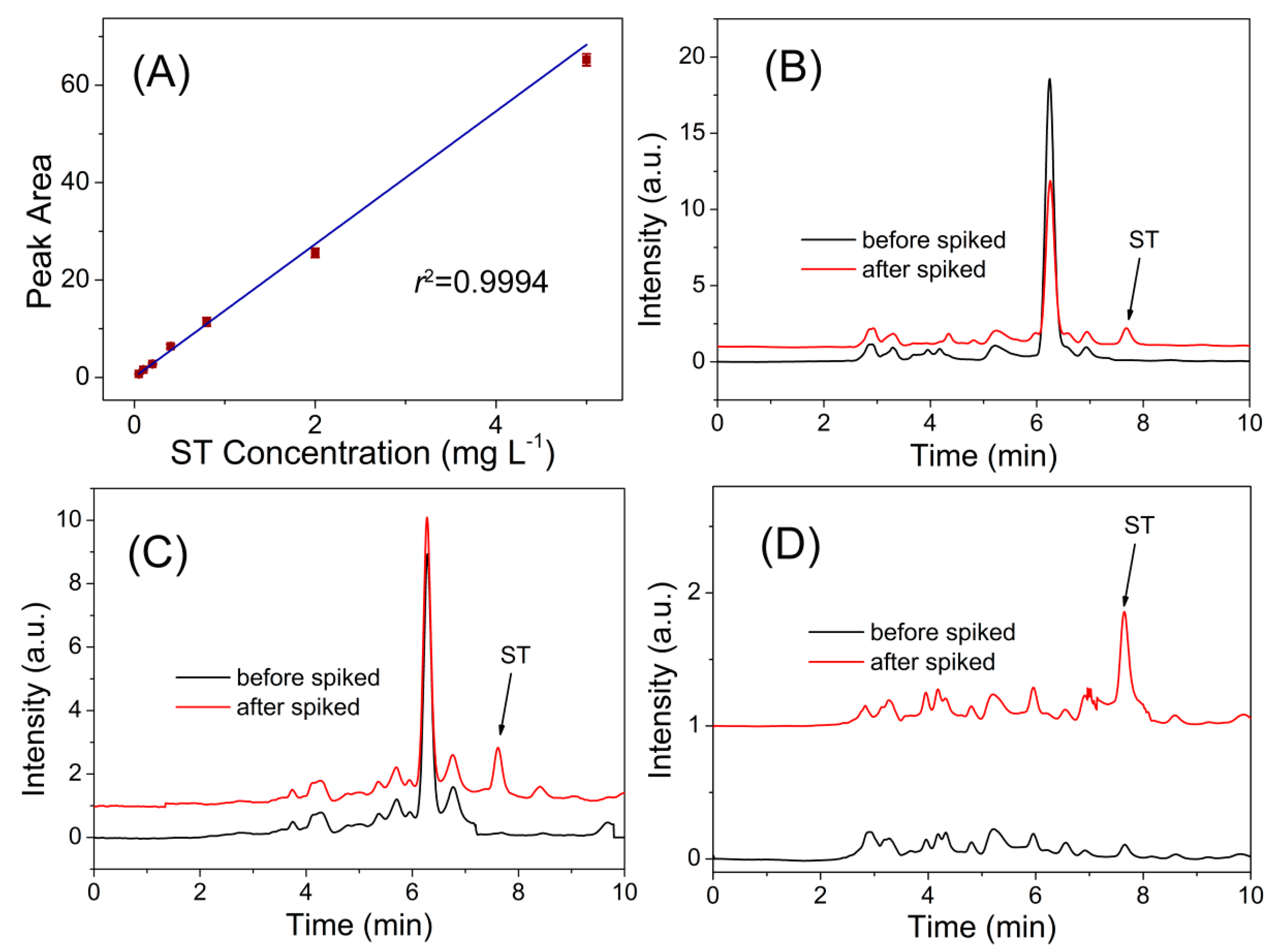
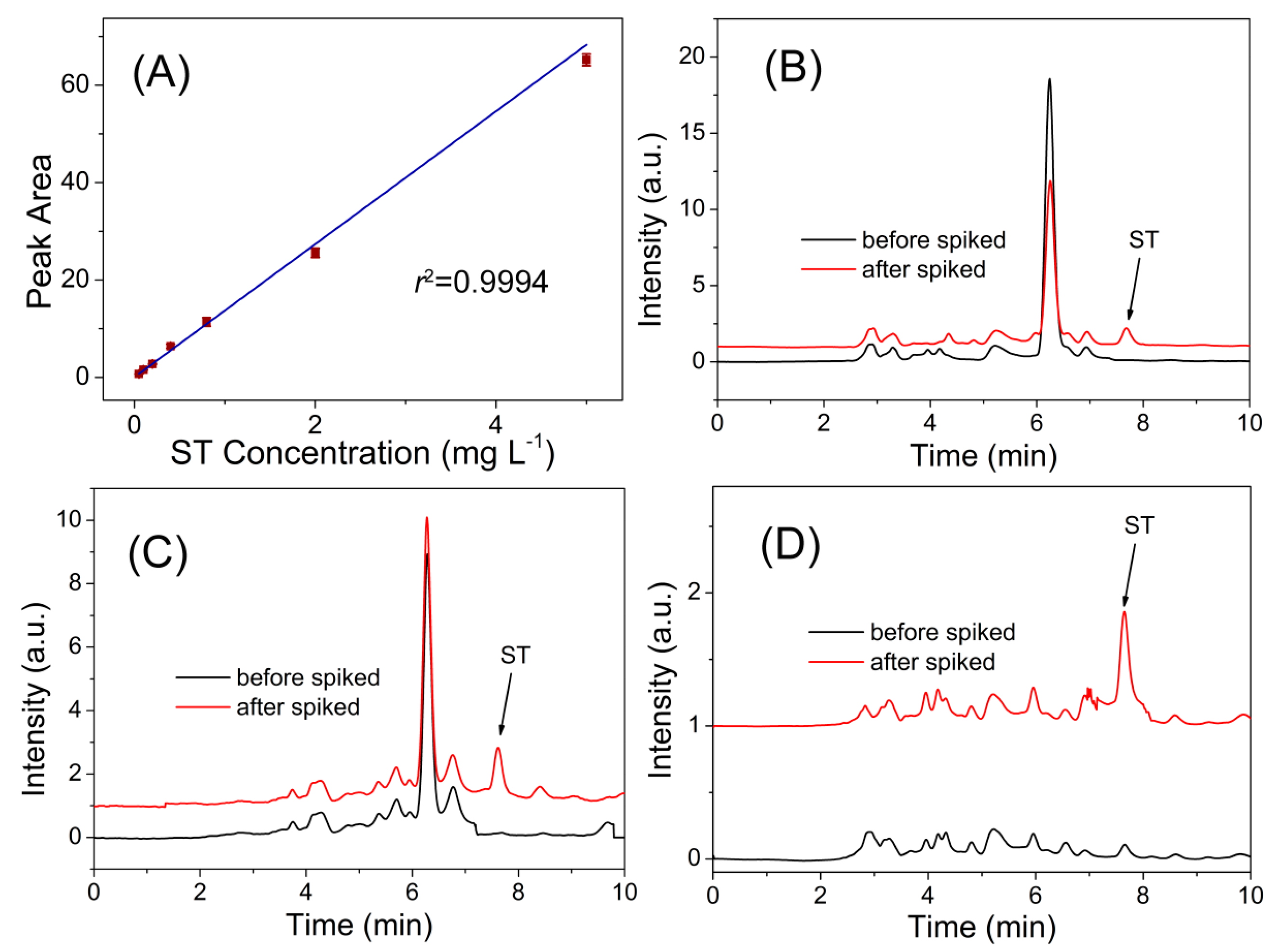
3.3. Real Sample Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zheng, R.; Xu, H.; Wang, W.; Zhan, R.; Chen, W. Simultaneous determination of aflatoxin B-1, B-2, G(1), G(2), ochratoxin A, and sterigmatocystin in traditional Chinese medicines by LC-MS-MS. Anal. Bioanal. Chem. 2014, 406, 3031–3039. [Google Scholar] [CrossRef] [PubMed]
- Wegner, S.; Bauer, J.I.; Dietrich, R.; Maertlbauer, E.; Usleber, E.; Gottschalk, C.; Gross, M. A highly specific competitive direct enzyme immunoassay for sterigmatocystin as a tool for rapid immunochemotaxonomic differentiation of mycotoxigenic Aspergillus species. Lett. Appl. Microbiol. 2017, 64, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.Z.; Goto, T. Determination of sterigmatocystin in grain using gas chromatography-mass spectrometry with an on-column injector. Mycotoxin Res. 2015, 31, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, Z.; Molnar, A.P.; Fejes, B.; Novak, L.; Karaffa, L.; Keller, N.P.; Fekete, E. Growth-phase sterigmatocystin formation on lactose is mediated via low specific growth rates in Aspergillus nidulans. Toxins 2016, 8, 354. [Google Scholar] [CrossRef] [PubMed]
- Ok, H.E.; Tian, F.; Hong, E.Y.; Paek, O.; Kim, S.-H.; Kim, D.; Chun, H.S. Harmonized collaborative validation of aflatoxins and sterigmatocystin in white rice and sorghum by liquid chromatography coupled to tandem mass spectrometry. Toxins 2016, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, P.; Wu, H.; Zhang, Q.; Ma, F.; Zhang, Z.; Ding, X.; Wang, H. An ultra-sensitive monoclonal antibody-based competitive enzyme immunoassay for sterigmatocystin in cereal and oil products. PLoS ONE 2014, 9, e106415. [Google Scholar] [CrossRef] [PubMed]
- Luan, L.; Chen, N.; Han, Z.; Liu, X.; Zheng, Y.; Wu, Y. Simultaneous determination of aflatoxin B-1, aflatoxin B-2, mycophenolic acid and sterigmatocystin in grape pomace by UHPLC-MS/MS. World Mycotoxin J. 2014, 7, 121–129. [Google Scholar] [CrossRef]
- Sharma, P.S.; D’Souza, F.; Kutner, W. Molecular imprinting for selective chemical sensing of hazardous compounds and drugs of abuse. TrAC Trends Anal. Chem. 2012, 34, 59–77. [Google Scholar] [CrossRef]
- Diaz-Diaz, G.; Antuna-Jimenez, D.; Carmen Blanco-Lopez, M.; Jesus Lobo-Castanon, M.; Miranda-Ordieres, A.J.; Tunon-Blanco, P. New materials for analytical biomimetic assays based on affinity and catalytic receptors prepared by molecular imprinting. TrAC Trends Anal. Chem. 2012, 33, 68–80. [Google Scholar] [CrossRef]
- Chen, L.; Xu, S.; Li, J. Recent advances in molecular imprinting technology: Current status, challenges and highlighted applications. Chem. Soc. Rev. 2011, 40, 2922–2942. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, M.; Wu, D.; Ma, X.; Wu, Z.; Xiang, S.; Chen, S. Molecularly imprinted nanofiber film for sensitive sensing 2,4,6-tribromophenol. Polymers 2016, 8, 222. [Google Scholar] [CrossRef]
- Asman, S.; Mohamad, S.; Sarih, N.M. Effects of RAFT agent on the selective approach of molecularly imprinted polymers. Polymers 2015, 7, 484–503. [Google Scholar] [CrossRef]
- Yusof, N.A.; Ab Rahman, S.K.; Hussein, M.Z.; Ibrahim, N.A. Preparation and characterization of molecularly imprinted polymer as SPE sorbent for melamine isolation. Polymers 2013, 5, 1215–1228. [Google Scholar] [CrossRef]
- Wang, X.Y.; Yu, J.L.; Kang, Q.; Shen, D.Z.; Li, J.H.; Chen, L.X. Molecular imprinting ratiometric fluorescence sensor for highly selective and sensitive detection of phycocyanin. Biosens. Bioelectron. 2016, 77, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Uzun, L.; Turner, A.P.F. Molecularly-imprinted polymer sensors: Realising their potential. Biosens. Bioelectron. 2016, 76, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, M.; Salinas, Y.; Beyazit, S.; Kunath, S.; Duma, L.; Prost, E.; Mayes, A.G.; Resmini, M.; Bui, B.T.S.; Haupt, K. Molecularly imprinted polymer coated quantum dots for multiplexed cell targeting and imaging. Angew. Chem. Int. Ed. 2016, 55, 8244–8248. [Google Scholar] [CrossRef] [PubMed]
- Baggiani, C.; Anfossi, L.; Giovannoli, C. Solid phase extraction of food contaminants using molecular imprinted polymers. Anal. Chim. Acta 2007, 591, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, V.B.; Ju, H.X. Molecular imprinting: A dynamic technique for diverse applications in analytical chemistry. Anal. Bioanal. Chem. 2004, 380, 587–605. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Yan, L.; Zhang, Z.; Wang, J.; Lv, P. Magnetic molecularly imprinted polymer preconcentration of 4-chlorophenol with determination by high-performance liquid chromatography. Anal. Lett. 2017, 50, 117–134. [Google Scholar] [CrossRef]
- Fang, G.Z.; Fan, C.; Liu, H.L.; Pan, M.F.; Zhu, H.D.; Wang, S. A novel molecularly imprinted polymer on CdSe/ZnS quantum dots for highly selective optosensing of mycotoxin zearalenone in cereal samples. RSC Adv. 2014, 4, 2764–2771. [Google Scholar] [CrossRef]
- Liu, H.L.; Fang, G.Z.; Zhu, H.D.; Wang, S. Application of molecularly imprinted polymer appended onto CdSe/ZnS quantum dots for optosensing of tocopherol in rice. Food Anal. Methods 2014, 7, 1443–1450. [Google Scholar] [CrossRef]
- Xu, L.H.; Fang, G.Z.; Pan, M.F.; Wang, X.F.; Wang, S. One-pot synthesis of carbon dots-embedded molecularly imprinted polymer for specific recognition of sterigmatocystin in grains. Biosens. Bioelectron. 2016, 77, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Fang, G.Z.; Wang, S. Highly sensitive and selective novel core-shell molecularly imprinted polymer based on NaYF4: Yb3+, Er3+ upconversion fluorescent nanorods. RSC Adv. 2013, 3, 3825–3828. [Google Scholar] [CrossRef]
- Liu, J.-M.; Cao, F.-Z.; Fang, G.-Z.; Wang, S. Upconversion nanophosphor-involved molecularly imprinted fluorescent polymers for sensitive and specific recognition of sterigmatocystin. Polymers 2017, 9, 299. [Google Scholar] [CrossRef]
- Machynakova, A.; Hrobonova, K. Preparation and application of magnetic molecularly imprinted polymers for the selective extraction of coumarins from food and plant samples. Anal. Methods 2017, 9, 2168–2176. [Google Scholar] [CrossRef]
- Shi, S.; Fan, D.; Xiang, H.; Li, H. Effective synthesis of magnetic porous molecularly imprinted polymers for efficient and selective extraction of cinnamic acid from apple juices. Food Chem. 2017, 237, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, X.; Li, J.; Wu, Z.; Wang, F.; Liu, L.; Tan, X.; Lei, F. Selective separation and determination of glucocorticoids in cosmetics using dual-template magnetic molecularly imprinted polymers and HPLC. J. Colloid Interface Sci. 2017, 504, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S. Application of magnetic molecularly imprinted polymer as a versatile and highly selective tool in food and environmental analysis: Recent developments and trends. TrAC Trends Anal. Chem. 2017, 90, 89–106. [Google Scholar] [CrossRef]
- Ji, W.; Sun, R.; Duan, W.; Wang, X.; Wang, T.; Mu, Y.; Guo, L. Selective solid phase extraction of chloroacetamide herbicides from environmental water samples by amphiphilic magnetic molecularly imprinted polymers. Talanta 2017, 170, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, P.; Ma, X.; Wang, H.; Lu, X.; Du, X. Preparation and evaluation of magnetic core-shell mesoporous molecularly imprinted polymers for selective adsorption of tetrabromobisphenol S. Talanta 2017, 166, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-M.; Yan, X.-P. Ultrasensitive, selective and simultaneous detection of cytochrome c and insulin based on immunoassay and aptamer-based bioassay in combination with Au/Ag nanoparticle tagging and ICP-MS detection. J. Anal. At. Spectrom. 2011, 26, 1191–1197. [Google Scholar] [CrossRef]
- Liu, Y.X.; Liu, L.; He, Y.H.; He, Q.H.; Ma, H. Quantum-dots-encoded-microbeads based molecularly imprinted polymer. Biosens. Bioelectron. 2016, 77, 886–893. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Freundlich Model | Langmuir Model | ||||
| KF | n | R2 | Qm | Rl | R2 |
| 3.12 | 1.67 | 0.9486 | 0.92 | 0.85 | 0.9948 |
| Pseudo-First-Order Kinetics | Pseudo-Second-Order Kinetics | ||||
| K1 (min−1) | qe (cal) (mg·g−1) | R2 | K2 (min−1) | qe (cal) (mg·g−1) | R2 |
| 0.045 | 0.53 | 0.9726 | 0.019 | 0.91 | 0.9923 |
| Intra-Particle Diffusion | Elovich | ||||
| kint (mg·g−1·min−1/2) | C (mg·g−1) | R2 | α (mg·g−1·min−1) | β (g·mg−1) | R2 |
| 0.2987 | 1.3782 | 0.8832 | 0.98 | 0.88 | 0.9529 |
| Method | Detection Limit (μg·L−1) | Linear Range (μg·L−1) | Reference |
|---|---|---|---|
| ELISA | 0.36 | NA | [6] |
| GC-MS | 3 | 10–150 | [3] |
| LC-MS | 3 | NA | [5] |
| LC-MS | 3 | NA | [7] |
| HPLC | 9 | 50–5000 | This Work |
| Fluorescence | 19 | 50–2000 | [22] |
| Fluorescence | 13 | 20–1000 | [24] |
| Samples | Spiked Amount (μg·kg−1) | Determined Amount (μg·kg−1, Mean ± SD) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| Wheat | 50 | 44.1 ± 0.9 | 88.1 ± 1.7 | 1.1 |
| 100 | 85.2 ± 4.8 | 85.2 ± 4.8 | 5.6 | |
| 200 | 174.4 ± 0.7 | 87.2 ± 0.4 | 0.4 | |
| Rice | 50 | 43.6 ± 1.9 | 87.2 ± 3.7 | 4.4 |
| 100 | 88.7 ± 1.1 | 88.7 ± 1.1 | 1.2 | |
| 200 | 161.2 ± 1.4 | 80.6 ± 0.7 | 0.9 | |
| Millet | 50 | 41.5 ± 1.7 | 82.9 ± 3.5 | 4.1 |
| 100 | 88.6 ± 1.4 | 88.6 ± 1.4 | 1.6 | |
| 200 | 170.8 ± 9.3 | 85.4 ± 4.6 | 5.4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.-M.; Wei, S.-Y.; Liu, H.-L.; Fang, G.-Z.; Wang, S. Preparation and Evaluation of Core–Shell Magnetic Molecularly Imprinted Polymers for Solid-Phase Extraction and Determination of Sterigmatocystin in Food. Polymers 2017, 9, 546. https://doi.org/10.3390/polym9100546
Liu J-M, Wei S-Y, Liu H-L, Fang G-Z, Wang S. Preparation and Evaluation of Core–Shell Magnetic Molecularly Imprinted Polymers for Solid-Phase Extraction and Determination of Sterigmatocystin in Food. Polymers. 2017; 9(10):546. https://doi.org/10.3390/polym9100546
Chicago/Turabian StyleLiu, Jing-Min, Shu-Yuan Wei, Hui-Lin Liu, Guo-Zhen Fang, and Shuo Wang. 2017. "Preparation and Evaluation of Core–Shell Magnetic Molecularly Imprinted Polymers for Solid-Phase Extraction and Determination of Sterigmatocystin in Food" Polymers 9, no. 10: 546. https://doi.org/10.3390/polym9100546
APA StyleLiu, J.-M., Wei, S.-Y., Liu, H.-L., Fang, G.-Z., & Wang, S. (2017). Preparation and Evaluation of Core–Shell Magnetic Molecularly Imprinted Polymers for Solid-Phase Extraction and Determination of Sterigmatocystin in Food. Polymers, 9(10), 546. https://doi.org/10.3390/polym9100546