Polyisoprene-Silica Nanoparticles Synthesized via RAFT Emulsifier-Free Emulsion Polymerization Using Water-Soluble Initiators


Abstract
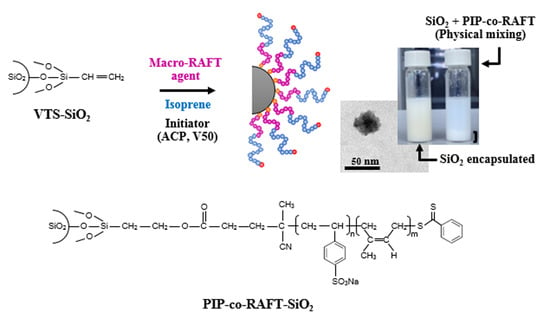
:
1. Introduction
2. Materials and Methods
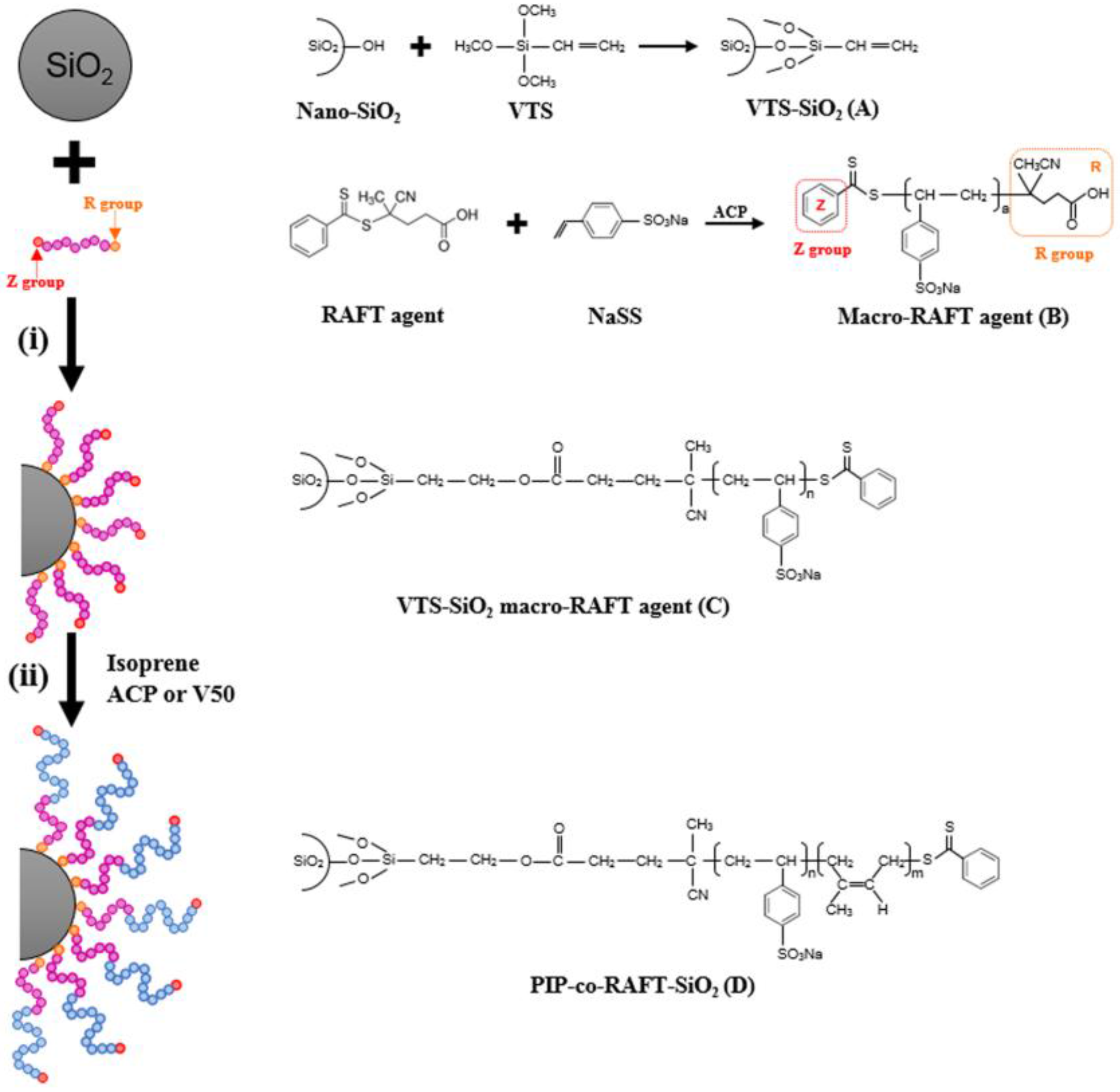
2.1. Materials
2.2. RAFT Emulsion Polymerization of Isoprene (PIP-co-RAFT)
2.3. Preparation of Polyisoprene-Silica Nanoparticles (PIP-co-RAFT-SiO2)
2.4. Characterization of Nanoparticles
2.5. Preparation and Properties of NR/PIP-R-SiO2 Nanocomposites
3. Results and Discussion
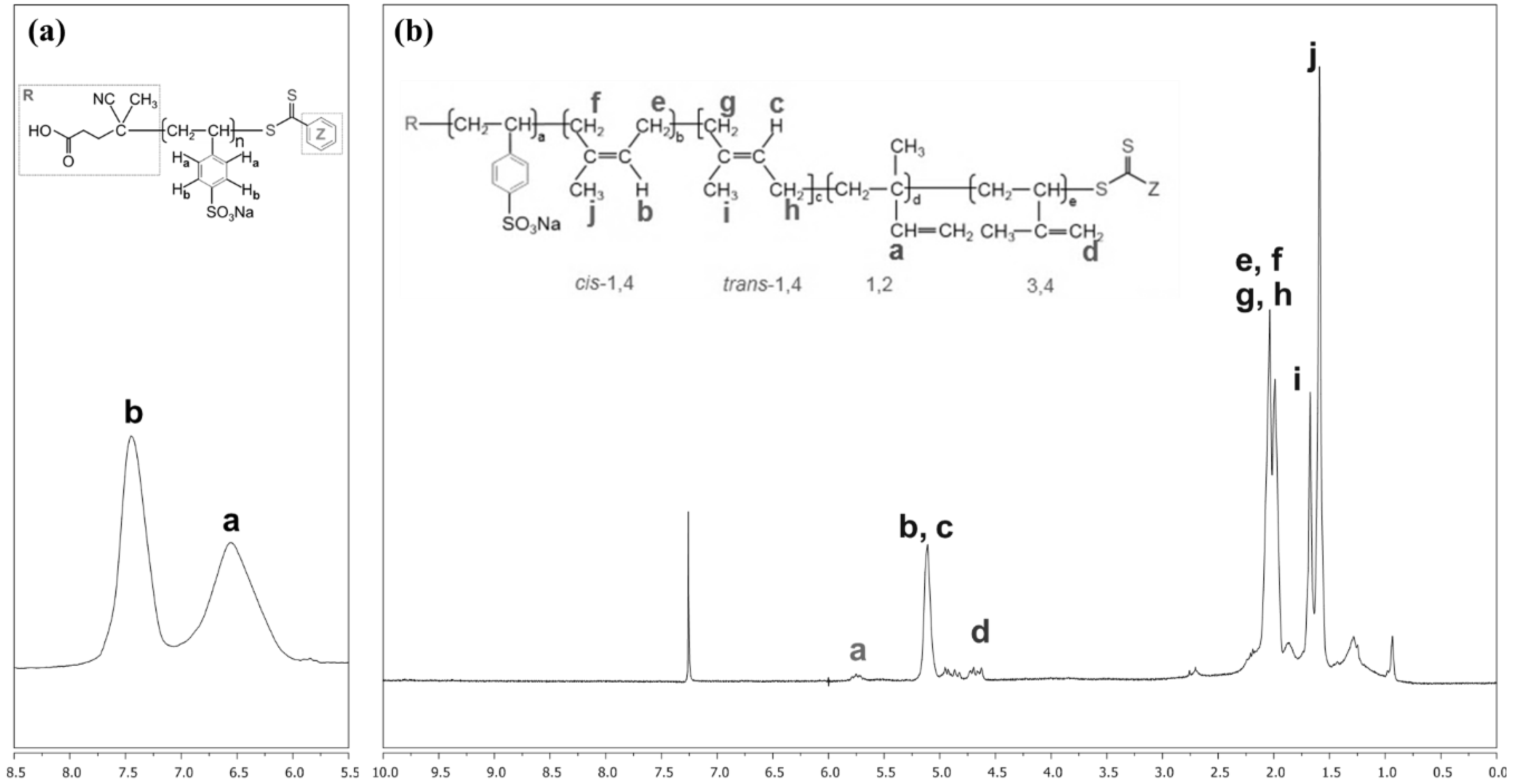
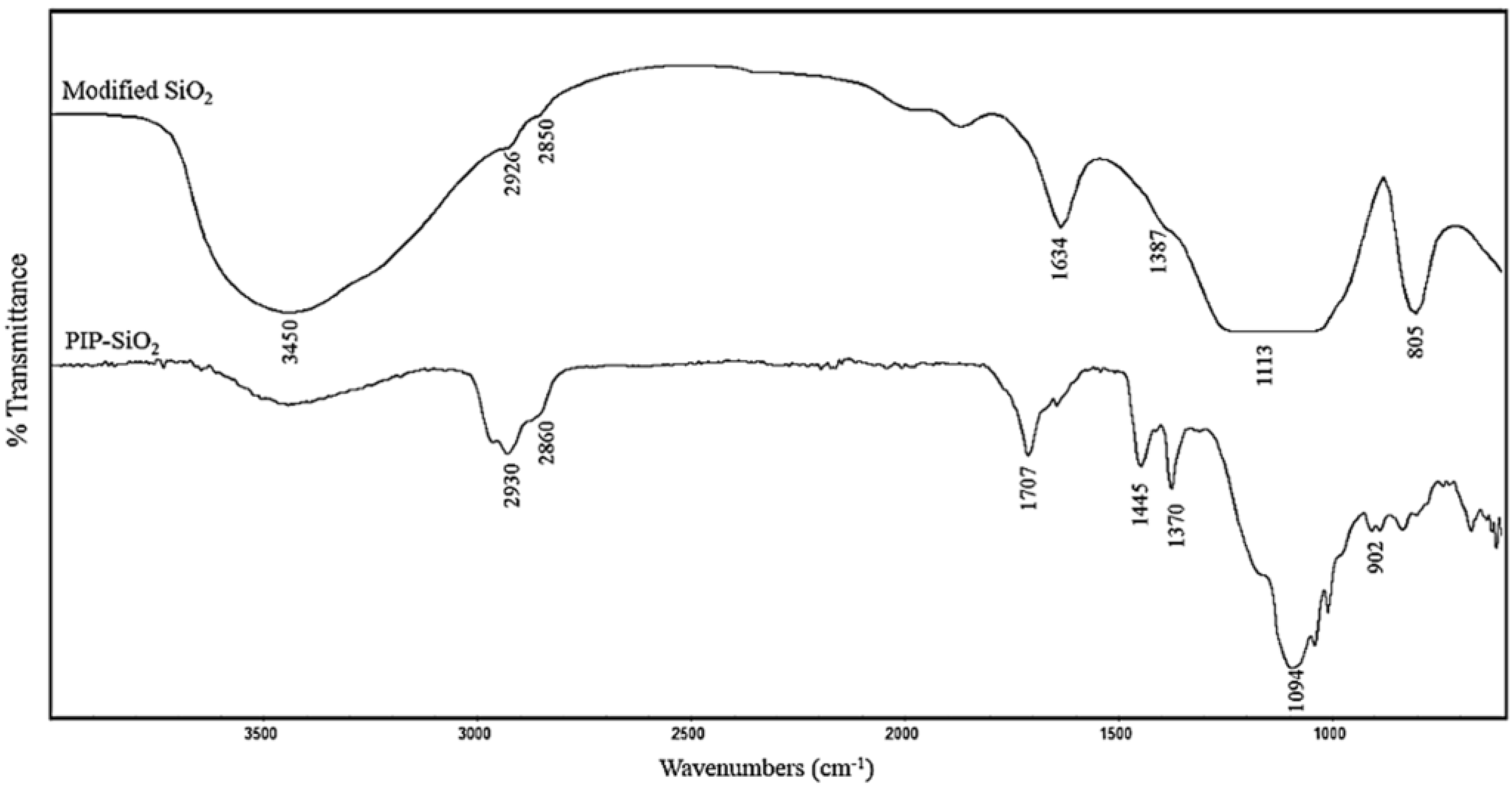
3.1. Characterization of PIP-co-RAFT and PIP-co-RAFT-SiO2 Nanoparticles
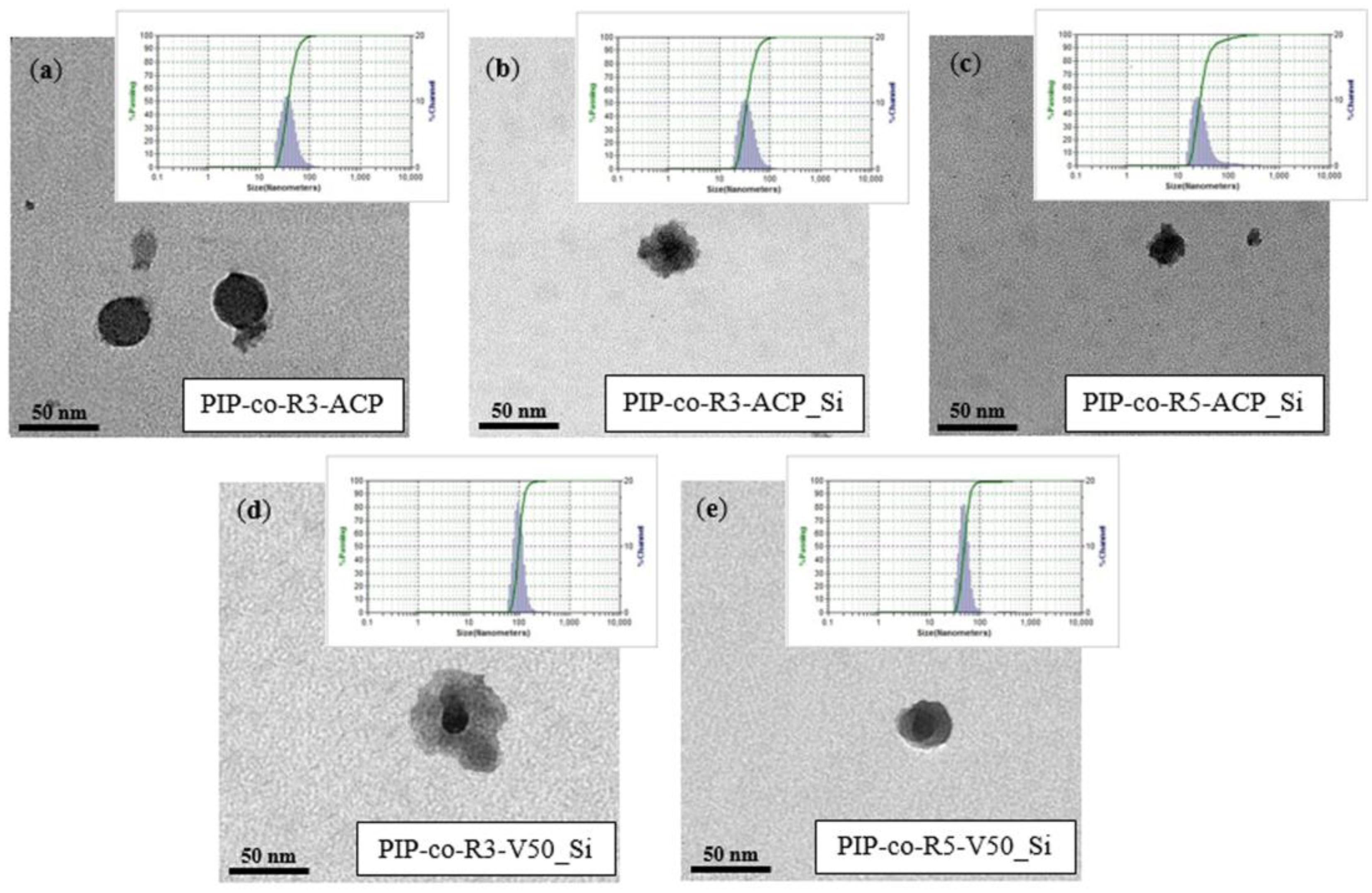
3.2. Morphology of PIP-co-RAFT and PIP-co-RAFT-SiO2 Nanoparticles
3.3. PIP-co-RAFT Nanoparticles: Effect of Initiator
3.4. PIP-co-RAFT-SiO2 Nanoparticles
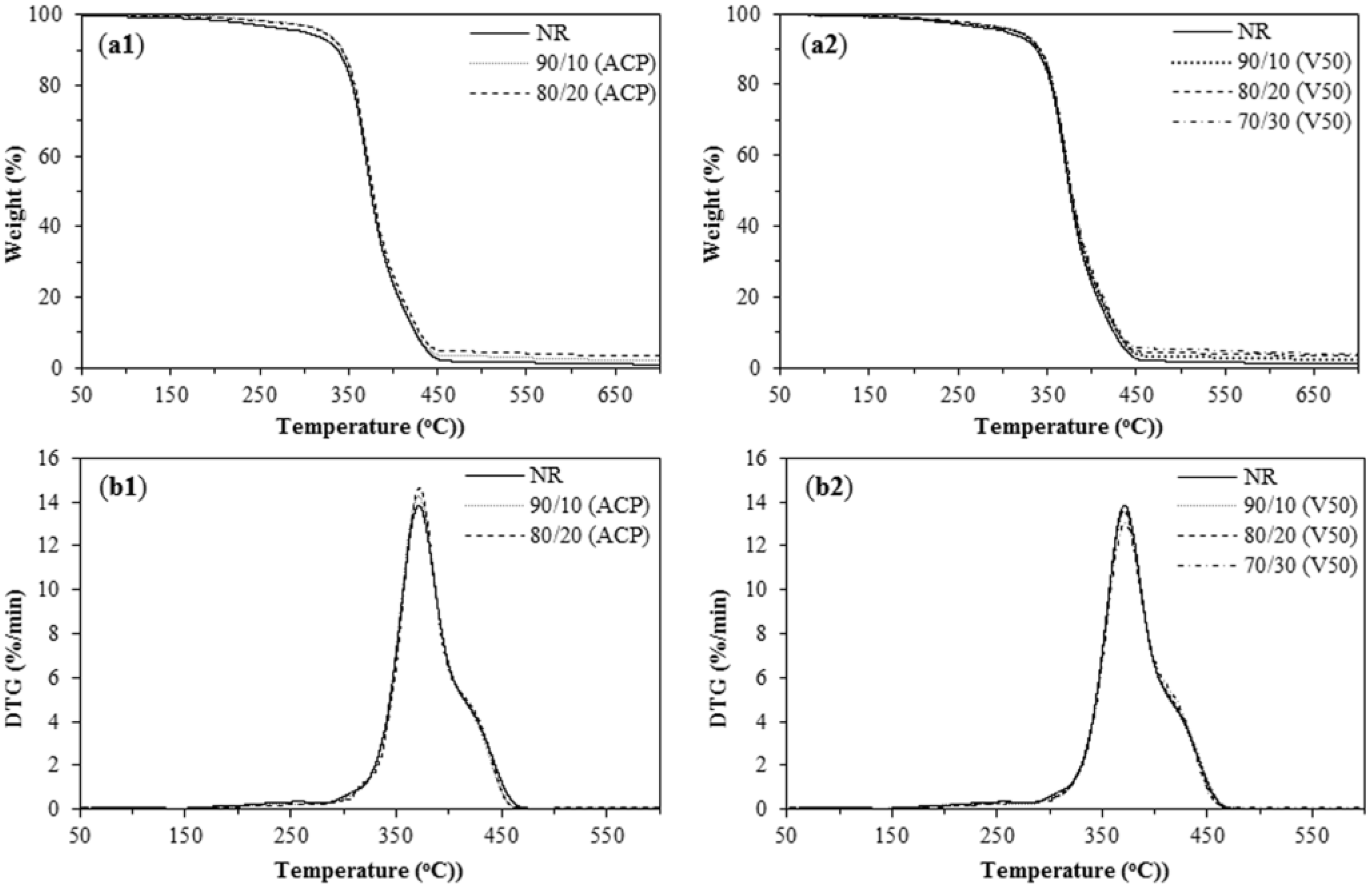
3.5. Mechanical and Thermal Properties of NR/PIP-R-SiO2 Nanocomposites
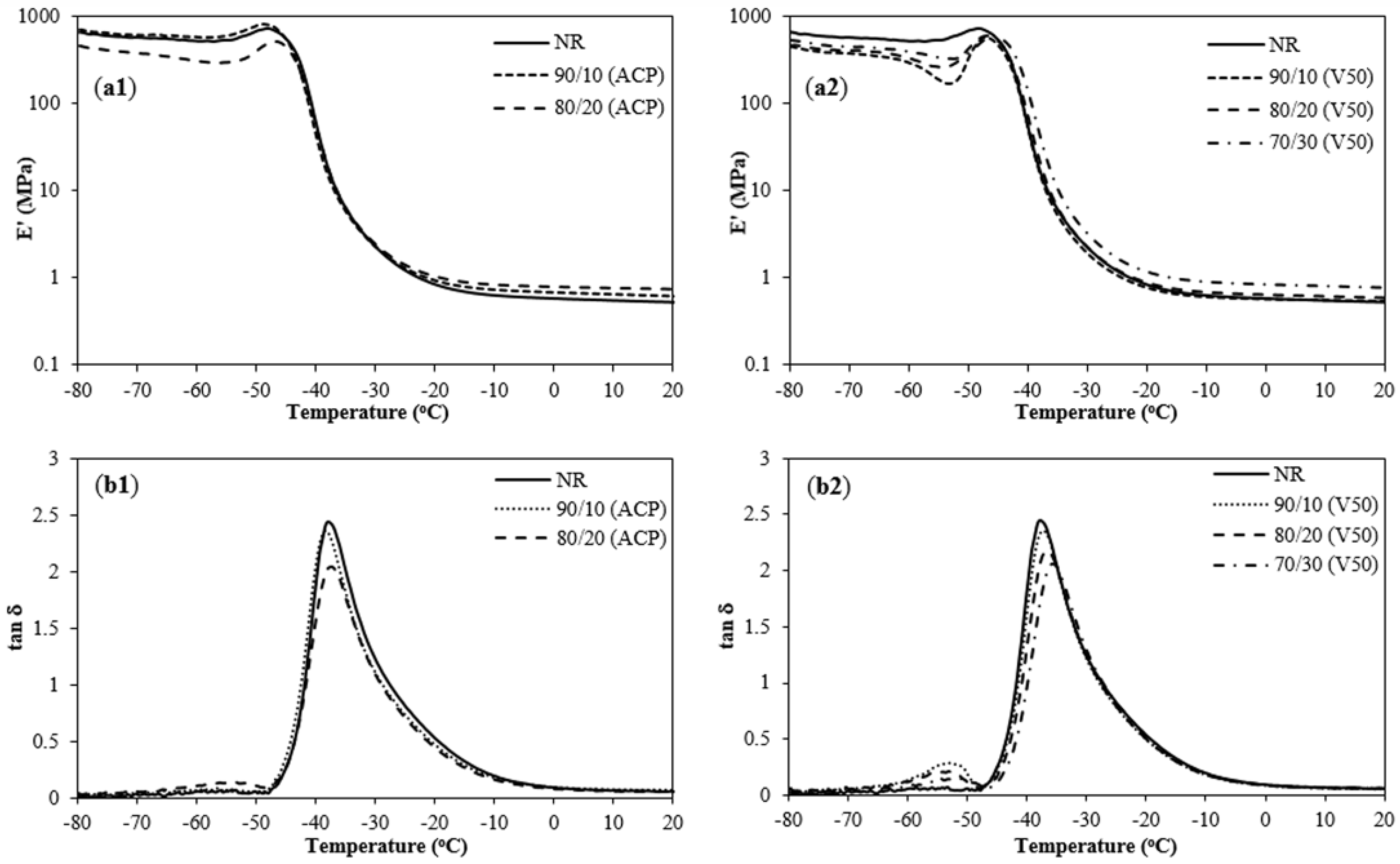
3.6. Dynamic Mechanical Properties of NR/PIP-R-SiO2 Nanocomposites
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cunningham, M.F. Controlled/living radical polymerization in aqueous dispersed systems. Prog. Polym. Sci. 2008, 33, 365–398. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Van Steenberge, P.H.M.; Reyniers, M.-F.; Marin, G.B. The strength of multi-scale modeling to unveil the complexity of radical polymerization. Prog. Polym. Sci. 2016, 58, 59–89. [Google Scholar] [CrossRef]
- Asua, J.M. Challenges in polymerization in dispersed media. In Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2017; pp. 1–22. [Google Scholar]
- Wei, R.; Luo, Y.; Li, Z. Synthesis of structured nanoparticles of styrene/butadiene block copolymers via RAFT seeded emulsion polymerization. Polymer 2010, 51, 3879–3886. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, W.; Wang, Y.; Sun, C.; Han, M.; Zhang, C. Synthesis and self-assembly of amphiphilic gradient copolymer via RAFT emulsifier-free emulsion polymerization. J. Colloid Interface Sci. 2012, 369, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Benicewicz, B.C. Functionalization of silica nanoparticles via the combination of surface-initiated RAFT polymerization and click reactions. Macromolecules 2008, 41, 7986–7992. [Google Scholar] [CrossRef]
- Bar-Nes, G.; Hall, R.; Sharma, V.; Gaborieau, M.; Lucas, D.; Castignolles, P.; Gilbert, R.G. Controlled/living radical polymerization of isoprene and butadiene in emulsion. Eur. Polym. J. 2009, 45, 3149–3163. [Google Scholar] [CrossRef]
- Yao, L.; Yuan, Y.C.; Rong, M.Z.; Zhang, M.Q. Self-healing linear polymers based on raft polymerization. Polymer 2011, 52, 3137–3145. [Google Scholar] [CrossRef]
- Keddie, D.J.; Moad, G.; Rizzardo, E.; Thang, S.H. RAFT agent design and synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar] [CrossRef]
- Ferguson, C.J.; Hughes, R.J.; Pham, B.T.T.; Hawkett, B.S.; Gilbert, R.G.; Serelis, A.K.; Such, C.H. Effective ab initio emulsion polymerization under RAFT control. Macromolecules 2002, 35, 9243–9245. [Google Scholar] [CrossRef]
- Chaduc, I.; Crepet, A.; Boyron, O.; Charleux, B.; D’Agosto, F.; Lansalot, M. Effect of the pH on the RAFT polymerization of acrylic acid in water. Application to the synthesis of poly(acrylic acid)-stabilized polystyrene particles by RAFT emulsion polymerization. Macromolecules 2013, 46, 6013–6023. [Google Scholar] [CrossRef]
- Zhou, J.; He, R.; Ma, J. RAFT-mediated polymerization-induced self-assembly of poly(acrylic acid)-b-poly(hexafluorobutyl acrylate): Effect of the pH on the synthesis of self-stabilized particles. Polymers 2016, 8, 207. [Google Scholar] [CrossRef]
- Rieger, J.; Stoffelbach, F.; Bui, C.; Alaimo, D.; Jérôme, C.; Charleux, B. Amphiphilic poly(ethylene oxide) macromolecular RAFT agent as a stabilizer and control agent in ab initio batch emulsion polymerization. Macromolecules 2008, 41, 4065–4068. [Google Scholar] [CrossRef]
- Rieger, J.; Osterwinter, G.; Bui, C.; Stoffelbach, F.; Charleux, B. Surfactant-free controlled/living radical emulsion (co)polymerization of n-butyl acrylate and methyl methacrylate via RAFT using amphiphilic poly(ethylene oxide)-based trithiocarbonate chain transfer agents. Macromolecules 2009, 42, 5518–5525. [Google Scholar] [CrossRef]
- Fréal-Saison, S.; Save, M.; Bui, C.; Charleux, B.; Magnet, S. Emulsifier-free controlled free-radical emulsion polymerization of styrene via RAFT using dibenzyltrithiocarbonate as a chain transfer agent and acrylic acid as an ionogenic comonomer: Batch and spontaneous phase inversion processes. Macromolecules 2006, 39, 8632–8638. [Google Scholar] [CrossRef]
- Mitsukami, Y.; Donovan, M.S.; Lowe, A.B.; McCormick, C.L. Water-soluble polymers. 81. Direct synthesis of hydrophilic styrenic-based homopolymers and block copolymers in aqueous solution via RAFT. Macromolecules 2001, 34, 2248–2256. [Google Scholar] [CrossRef]
- Yeole, N.; Hundiwale, D. Effect of hydrophilic macro-RAFT agent in surfactant-free emulsion polymerization. Colloids Surf. A Physicochem. Eng. Asp. 2011, 392, 329–334. [Google Scholar] [CrossRef]
- Ji, J.; Yan, L.; Xie, D. Surfactant-free synthesis of amphiphilic diblock copolymer in aqueous phase by a self-stability process. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3098–3107. [Google Scholar] [CrossRef]
- Shibuya, K.; Nagao, D.; Ishii, H.; Konno, M. Advanced soap-free emulsion polymerization for highly pure, micron-sized, monodisperse polymer particles. Polymer 2014, 55, 535–539. [Google Scholar] [CrossRef]
- Samakande, A.; Sanderson, R.D.; Hartmann, P.C. RAFT-mediated polystyrene-clay nanocomposites prepared by making use of initiator-bound MMT clay. Eur. Polym. J. 2009, 45, 649–657. [Google Scholar] [CrossRef]
- Ngo, V.G.; Bressy, C.; Leroux, C.; Margaillan, A. Synthesis of hybrid TiO2 nanoparticles with well-defined poly(methyl methacrylate) and poly(tert-butyldimethylsilyl methacrylate) via the RAFT process. Polymer 2009, 50, 3095–3102. [Google Scholar] [CrossRef]
- Moradi, R.; Karimi-Sabet, J.; Shariaty-Niassar, M.; Koochaki, M. Preparation and characterization of polyvinylidene fluoride/graphene superhydrophobic fibrous films. Polymers 2015, 7, 1444. [Google Scholar] [CrossRef]
- Kim, H.; Abdala, A.A.; Macosko, C.W. Graphene/polymer nanocomposites. Macromolecules 2010, 43, 6515–6530. [Google Scholar] [CrossRef]
- Zou, H.; Wu, S.; Shen, J. Polymer/silica nanocomposites: Preparation, characterization, properties, and applications. Chem. Rev. 2008, 108, 3893–3957. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, T.; Baleizão, C.; Farinha, J. Functional films from silica/polymer nanoparticles. Materials 2014, 7, 3881. [Google Scholar] [CrossRef] [PubMed]
- Moraes, J.; Ohno, K.; Maschmeyer, T.; Perrier, S. Synthesis of silica-polymer core-shell nanoparticles by reversible addition-fragmentation chain transfer polymerization. Chem. Commun. 2013, 49, 9077–9088. [Google Scholar] [CrossRef] [PubMed]
- Jankiewicz, B.J.; Jamiola, D.; Choma, J.; Jaroniec, M. Silica-metal core-shell nanostructures. Adv. Colloid Interface Sci. 2012, 170, 28–47. [Google Scholar] [CrossRef] [PubMed]
- Salami-Kalajahi, M.; Haddadi-Asl, V.; Rahimi-Razin, S.; Behboodi-Sadabad, F.; Najafi, M.; Roghani-Mamaqani, H. A study on the properties of PMMA/silica nanocomposites prepared via RAFT polymerization. J. Polym. Res. 2012, 19, 9793. [Google Scholar] [CrossRef]
- Buhin, Z.; Blagojević, S.L.; Leskovac, M. In situ emulsion polymerization and characterization of poly(butyl acrylate-co-methyl methacrylate)/silica nanosystems. Polym. Eng. Sci. 2013, 53, 2292–2298. [Google Scholar] [CrossRef]
- Tumnantong, D.; Rempel, G.L.; Prasassarakich, P. Synthesis of polystyrene-silica nanoparticles via RAFT emulsifier-free emulsion polymerization. Eur. Polym. J. 2016, 80, 145–157. [Google Scholar] [CrossRef]
- Sun, D.; Li, B.B.; Xu, Z.L. Pervaporation of ethanol/water mixture by organophilic nano-silica filled PDMS composite membranes. Desalination 2013, 322, 159–166. [Google Scholar] [CrossRef]
- Kongsinlark, A.; Rempel, G.L.; Prasassarakich, P. Synthesis of monodispersed polyisoprene-silica nanoparticles via differential microemulsion polymerization and mechanical properties of polyisoprene nanocomposite. Chem. Eng. J. 2012, 193, 215–226. [Google Scholar] [CrossRef]
- Tancharernrat, T.; Rempel, G.L.; Prasassarakich, P. Preparation of styrene butadiene copolymer–silica nanocomposites via differential microemulsion polymerization and NR/SBR–SiO2 membranes for pervaporation of water–ethanol mixtures. Chem. Eng. J. 2014, 258, 290–300. [Google Scholar] [CrossRef]
- Tancharernrat, T.; Rempel, G.L.; Prasassarakich, P. Synthesis of polybutadiene-silica nanoparticles via differential microemulsion polymerization and their hydrogenated nanoparticles by diimide reduction. Polym. Degrad. Stab. 2015, 118, 69–81. [Google Scholar] [CrossRef]
- Liu, C.H.; Pan, C.Y. Grafting polystyrene onto silica nanoparticles via RAFT polymerization. Polymer 2007, 48, 3679–3685. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, P.; Zhang, C.; Zhao, Y. Facile synthesis of highly pure block copolymers by combination of RAFT polymerization, click reaction and de-grafting process. Polym. Chem. 2012, 3, 1803–1812. [Google Scholar] [CrossRef]
- Perkowski, A.J.; Nicewicz, D.A. Direct catalytic anti-Markovnikov addition of carboxylic acids to alkenes. J. Am. Chem. Soc. 2013, 135, 10334–10337. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Zheng, S.; Chen, H.; Yao, M.; Zhang, K.; Jia, X.; Mou, J.; Xu, H.; Wu, R.; Shi, J. A combined “RAFT” and “graft from” polymerization strategy for surface modification of mesoporous silica nanoparticles: Towards enhanced tumor accumulation and cancer therapy efficacy. J. Mater. Chem. B 2014, 2, 5828–5836. [Google Scholar] [CrossRef]
- Water Soluble Azo Initiators. Available online: http://www.wako-chem.co.jp/kaseihin_en/waterazo/index.htm (accessed on 20 October 2017).
- Chonkaew, W.; Minghvanish, W.; Kungliean, U.; Rochanawipart, N.; Brostow, W. Vulcanization characteristics and dynamic mechanical behavior of natural rubber reinforced with silane modified silica. J. Nanosci. Nanotechnol. 2011, 11, 2018–2024. [Google Scholar] [CrossRef] [PubMed]
- Piya-areetham, P.; Prasassarakich, P.; Rempel, G.L. Organic solvent-free hydrogenation of natural rubber latex and synthetic polyisoprene emulsion catalyzed by water-soluble rhodium complexes. J. Mol. Catal. A Chem. 2013, 372, 151–159. [Google Scholar] [CrossRef]
- Baboo, M.; Dixit, M.; Sharma, K.; Saxena, N.S. Mechanical and thermal characterization of cis-polyisoprene and trans-polyisoprene blends. Polym. Bull. 2011, 66, 661–672. [Google Scholar] [CrossRef]
- Kader, M.A.; Kim, W.D.; Kaang, S.; Nah, C. Morphology and dynamic mechanical properties of natural rubber/nitrile rubber blends containing trans-polyoctylene rubber as a compatibilizer. Polym. Int. 2005, 54, 120–129. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | [R]:[I] a | Particle size (nm) | PSD | % Total solid content | Monomer conversion (%) |
|---|---|---|---|---|---|
| ACP initiator | |||||
| PIP-co-R1-ACP | 1:1 | 87.5 | 1.2 | 16.1 | 72.9 |
| PIP-co-R2-ACP | 2:1 | 54.0 | 1.3 | 17.0 | 77.3 |
| PIP-co-R3-ACP | 3:1 | 37.4 | 1.3 | 16.3 | 74.5 |
| PIP-co-R4-ACP | 4:1 | 36.3 | 1.4 | 17.8 | 81.0 |
| PIP-co-R5-ACP | 5:1 | 28.0 | 1.4 | 18.8 | 86.2 |
| V50 initiator | |||||
| PIP-co-R1-V50 | 1:1 | - b | - b | - b | - b |
| PIP-co-R2-V50 | 2:1 | 184.3 | 1.1 | 7.4 | 31.8 |
| PIP-co-R3-V50 | 3:1 | 165.9 | 1.1 | 10.0 | 44.4 |
| PIP-co-R4-V50 | 4:1 | 61.5 | 1.2 | 19.8 | 85.5 |
| PIP-co-R5-V50 | 5:1 | 58.6 | 1.1 | 18.6 | 81.8 |
| Name | [R]:[I] a | Particle size (nm) | PSD | % Total solid content | Monomer conversion (%) | % GE | % Si Encap eff |
|---|---|---|---|---|---|---|---|
| ACP initiator | |||||||
| PIP-co-R1-ACP-Si | 1:1 | 73.1 | 1.5 | 14.8 | 64.0 | 86.4 | 18.3 |
| PIP-co-R2-ACP-Si | 2:1 | 44.1 | 1.5 | 17.1 | 77.1 | 90.4 | 50.4 |
| PIP-co-R3-ACP-Si | 3:1 | 37.5 | 1.2 | 19.2 | 84.2 | 82.5 | 55.0 |
| PIP-co-R4-ACP-Si | 4:1 | 27.2 | 1.5 | 21.0 | 91.0 | 89.1 | 73.5 |
| PIP-co-R5-ACP-Si | 5:1 | 24.8 | 1.5 | 19.0 | 86.8 | 89.2 | 61.8 |
| V50 initiator | |||||||
| PIP-co-R1-V50-Si | 1:1 | - b | - b | - b | - b | - b | - b |
| PIP-co-R2-V50-Si | 2:1 | - b | - b | - b | - b | - b | - b |
| PIP-co-R3-V50-Si | 3:1 | 90.9 | 1.5 | 19.5 | 80.4 | 82.2 | 66.5 |
| PIP-co-R4-V50-Si | 4:1 | 66.1 | 1.4 | 22.1 | 96.0 | 91.4 | 56.6 |
| PIP-co-R5-V50-Si | 5:1 | 48.2 | 1.2 | 16.8 | 48.2 | 96.2 | 33.1 |
| Properties | NR | NR/PIP-R-ACP-SiO2 (ACP initiator) a | NR/PIP-R-V50-SiO2 (V50 initiator) b | |||
|---|---|---|---|---|---|---|
| NR/PIP-R-SiO2 | 100 | 90/10 | 80/20 | 90/10 | 80/20 | 70/30 |
| SiO2 content c (wt %) | - | 1.0 | 2.0 | 1.0 | 2.0 | 3.0 |
| Mechanical properties | ||||||
| Tensile strength (MPa) | 17.9 ± 0.7 | 24.2 ± 2.1 | 23.5 ± 1.0 | 18.4 ± 1.6 | 16.0 ± 1.2 | 15.1 ± 2.8 |
| 300% Modulus (MPa) | 1.11 ± 0.02 | 1.19 ± 0.02 | 1.24 ± 0.01 | 1.09 ± 0.01 | 1.19 ± 0.01 | 1.37 ± 0.08 |
| Elongation at break (%) | 871 ± 32 | 947 ± 08 | 972 ± 15 | 889 ± 39 | 891 ± 34 | 935 ± 77 |
| Thermal properties | ||||||
| Tid (°C) | 349.4 | 348.7 | 348.8 | 348.9 | 348.6 | 348.8 |
| Tmax (°C) | 376.7 | 375.6 | 376.7 | 377.0 | 376.6 | 377.3 |
| NR/PIP-co-R-SiO2 (wt/wt) | SiO2 Content c (wt %) | E′max (MPa) | E′20°C (MPa) | tan δ | Tg (°C) |
|---|---|---|---|---|---|
| 100/0 | - | 732 | 0.53 | 2.44 | −39.8 |
| NR/PIP-R-ACP-SiO2 (ACP Initiator) a | |||||
| 90/10 | 1.0 | 814 | 0.62 | 2.36 | −38.8 |
| 80/20 | 2.0 | 517 | 0.74 | 2.04 | −37.6 |
| NR/PIP-R-V50-SiO2 (V50 Initiator) b | |||||
| 90/10 | 1.0 | 584 | 0.55 | 2.36 | −37.5 |
| 80/20 | 2.0 | 608 | 0.60 | 2.17 | −36.9 |
| 70/30 | 3.0 | 591 | 0.79 | 2.06 | −36.0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tumnantong, D.; Rempel, G.L.; Prasassarakich, P. Polyisoprene-Silica Nanoparticles Synthesized via RAFT Emulsifier-Free Emulsion Polymerization Using Water-Soluble Initiators. Polymers 2017, 9, 637. https://doi.org/10.3390/polym9110637
Tumnantong D, Rempel GL, Prasassarakich P. Polyisoprene-Silica Nanoparticles Synthesized via RAFT Emulsifier-Free Emulsion Polymerization Using Water-Soluble Initiators. Polymers. 2017; 9(11):637. https://doi.org/10.3390/polym9110637
Chicago/Turabian StyleTumnantong, Dusadee, Garry L. Rempel, and Pattarapan Prasassarakich. 2017. "Polyisoprene-Silica Nanoparticles Synthesized via RAFT Emulsifier-Free Emulsion Polymerization Using Water-Soluble Initiators" Polymers 9, no. 11: 637. https://doi.org/10.3390/polym9110637
APA StyleTumnantong, D., Rempel, G. L., & Prasassarakich, P. (2017). Polyisoprene-Silica Nanoparticles Synthesized via RAFT Emulsifier-Free Emulsion Polymerization Using Water-Soluble Initiators. Polymers, 9(11), 637. https://doi.org/10.3390/polym9110637