Tillage Practice Impacts on the Carbon Sequestration Potential of Topsoil Microbial Communities in an Agricultural Field


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site and Soil Characteristics
2.2. Experiment Design
2.3. Microbial Analysis
2.4. Calculation and Statistics
3. Results
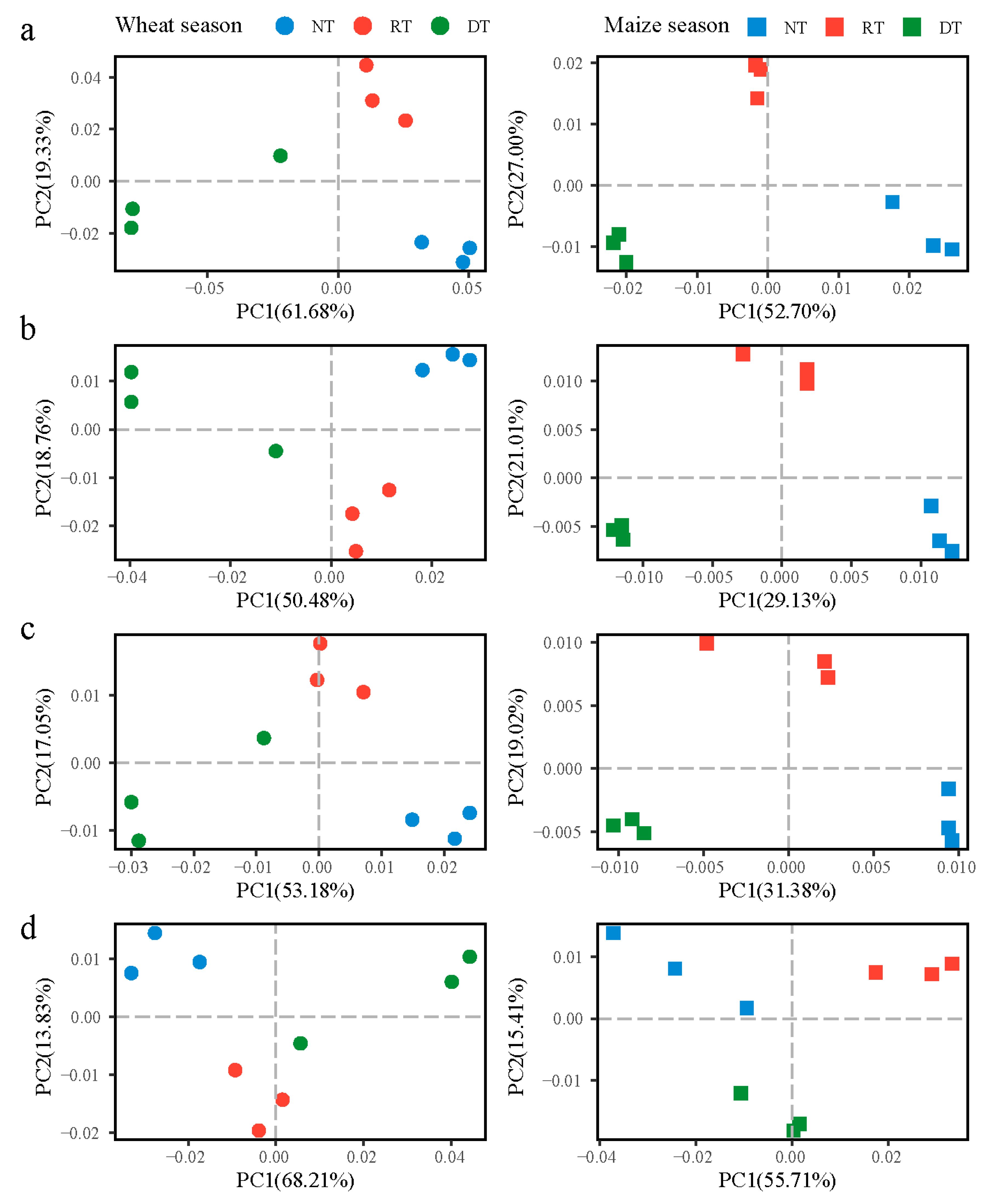
3.1. Diversity and Abundance between Treatments
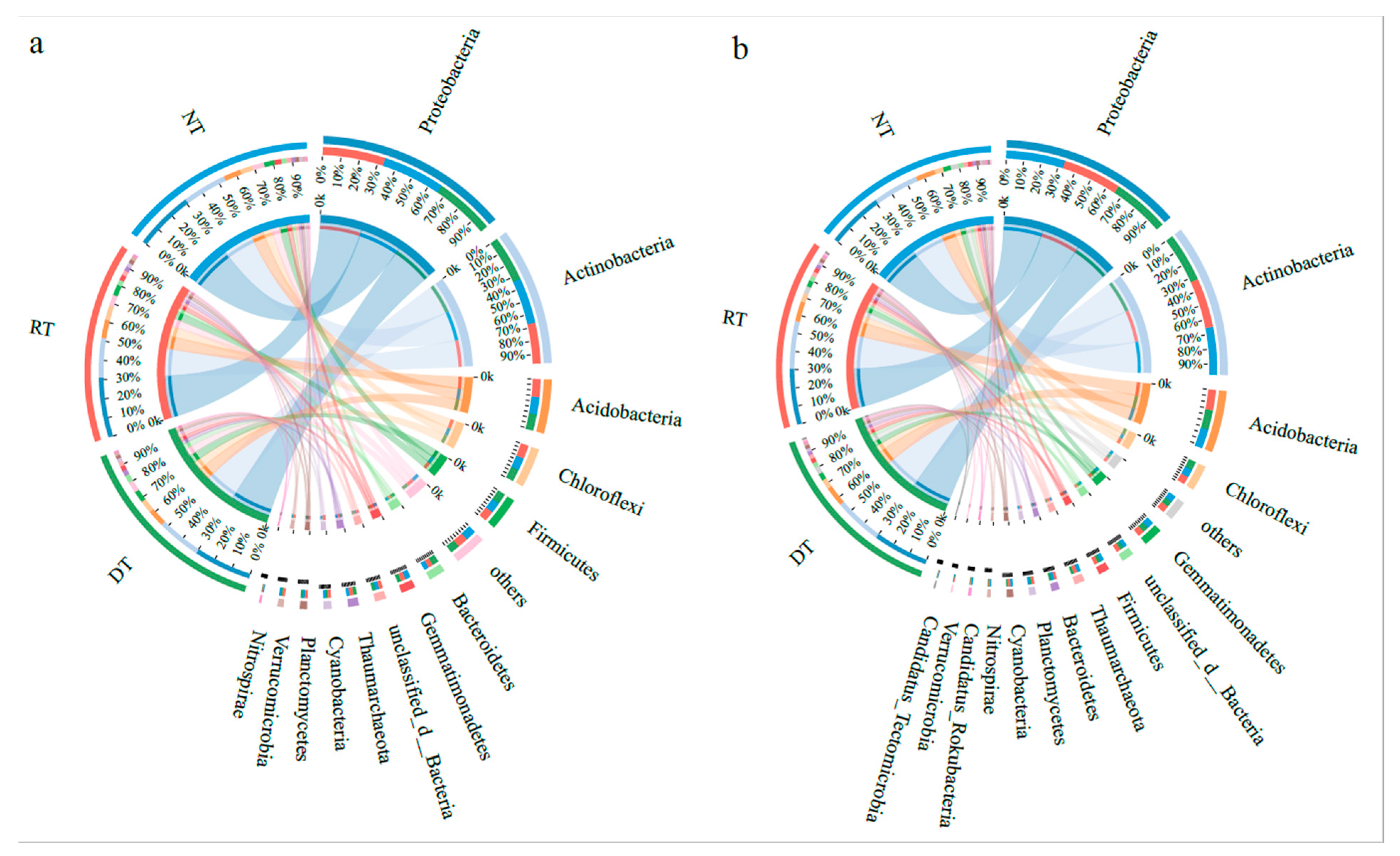
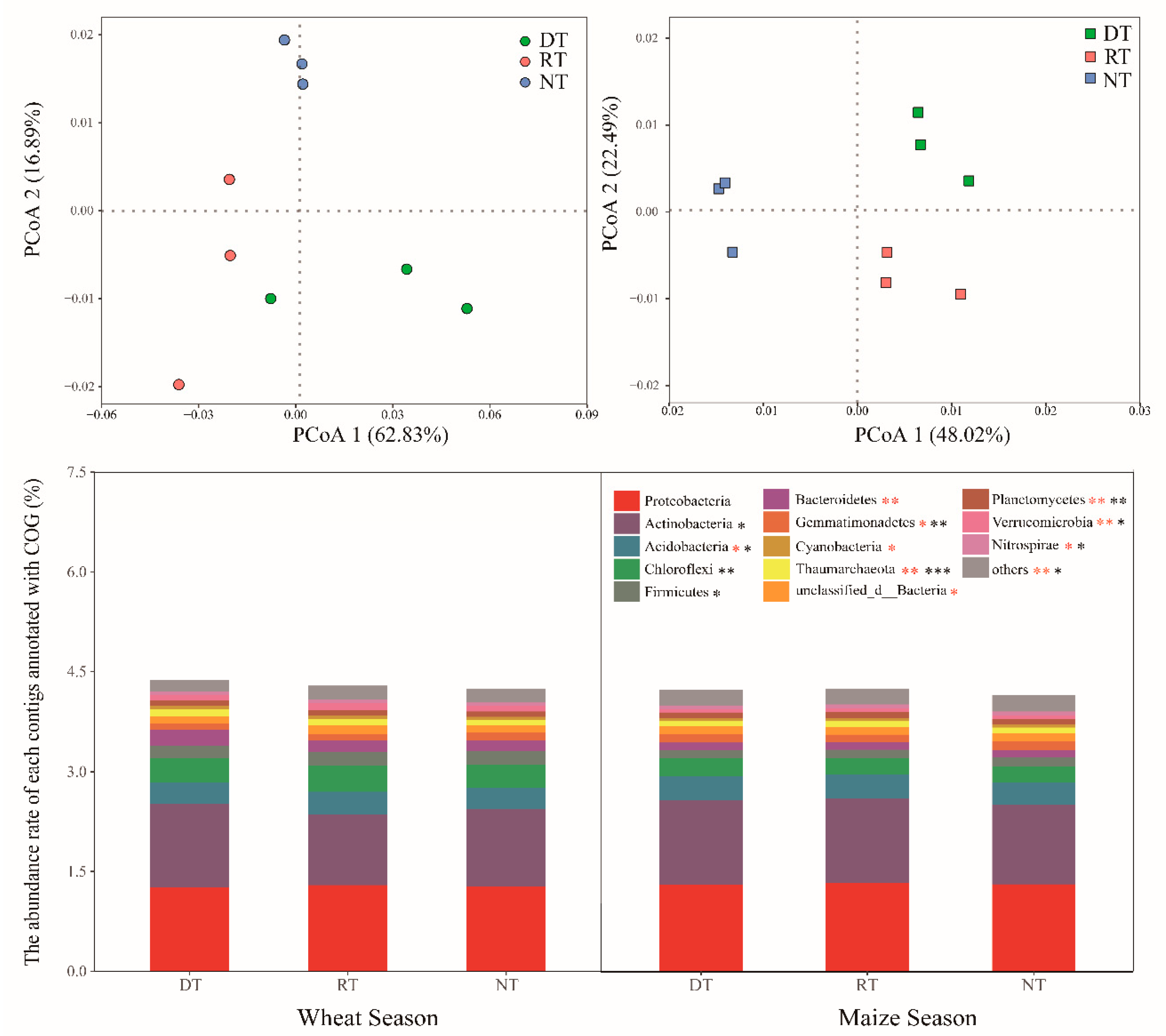
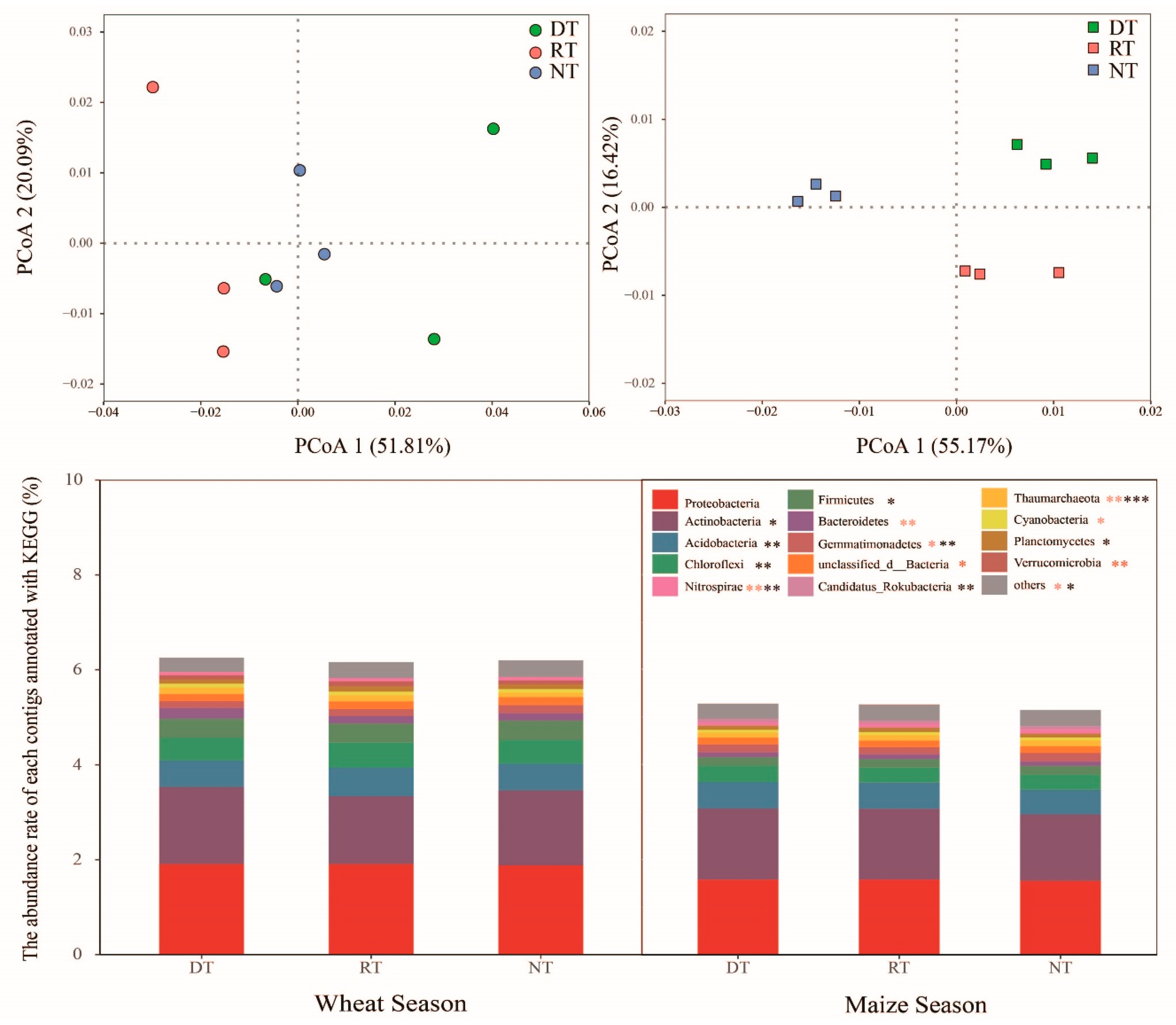
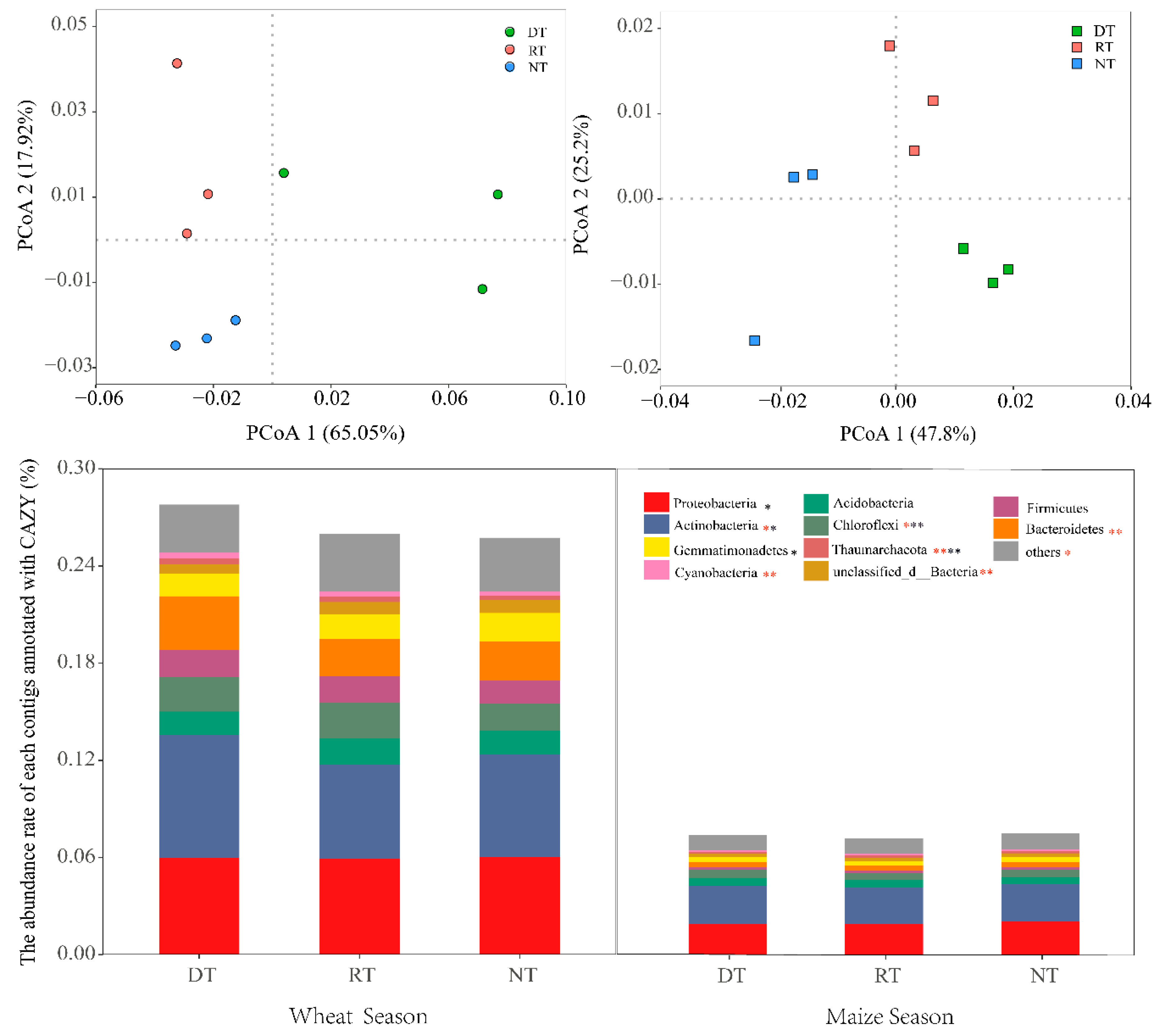
3.2. Microbial Community and Function Structure
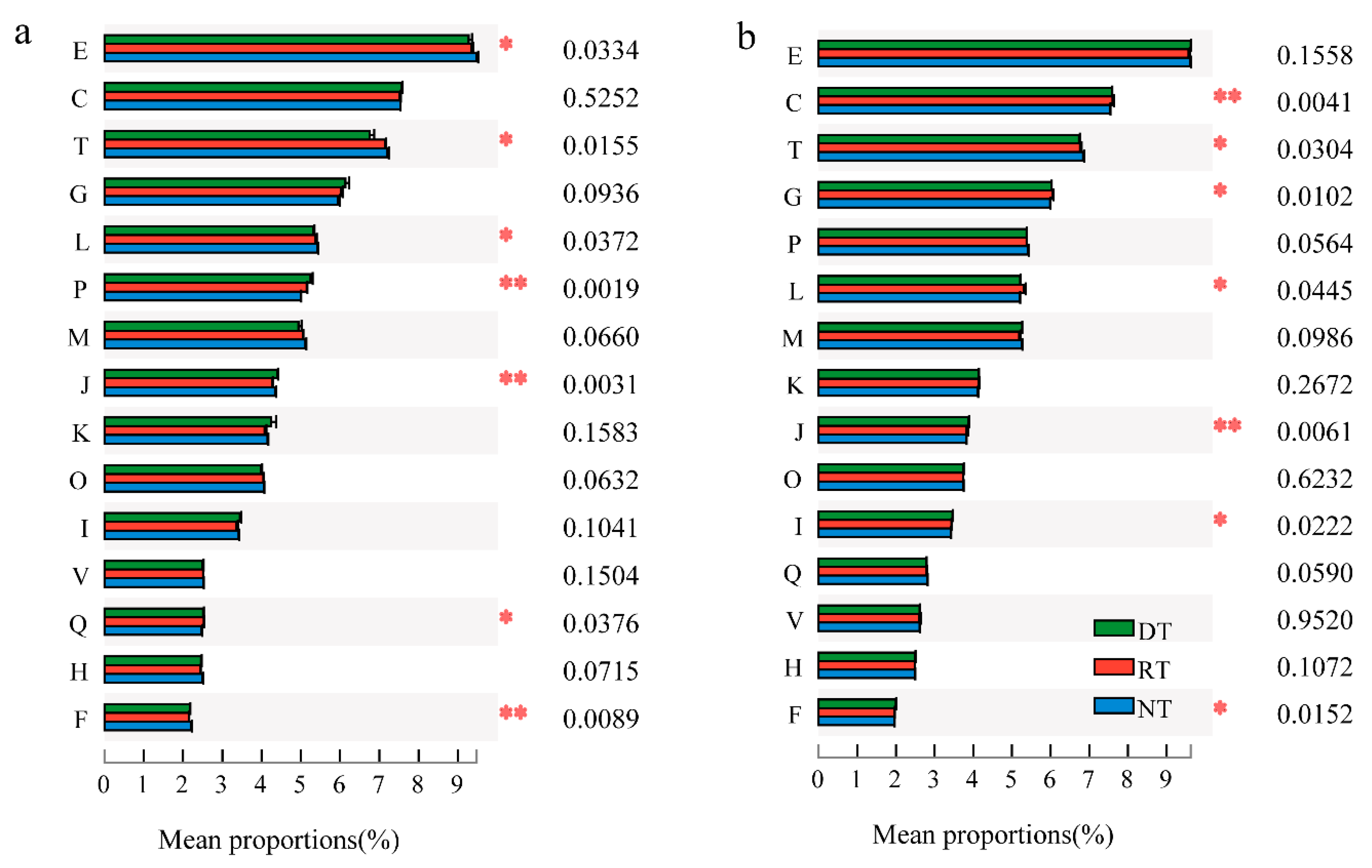
3.3. COG
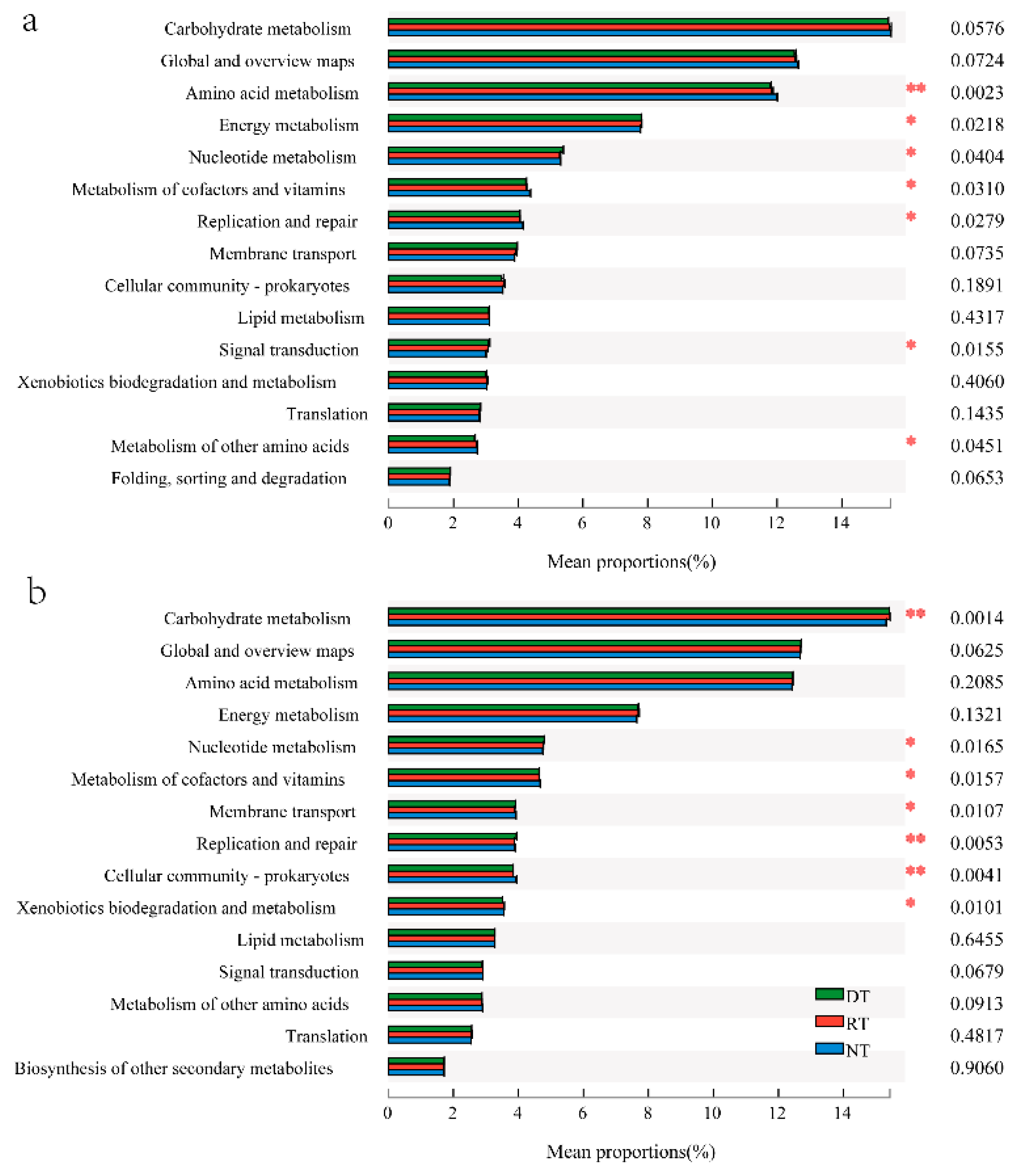
3.4. KEGG
3.5. CAZy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, M.; Shen, J.; Yuan, L.; Jiang, R.; Chen, X.; Davies, W.J.; Zhang, F. Improving crop productivity and resource use efficiency to ensure food security and environmental quality in China. J. Exp. Bot. 2012, 63, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.L.N.; Aparna, K.; Mohanty, S.R. Microbiology and biochemistry of soil organic matter, carbon sequestration and soil health. Indian J. Fertil. 2019, 15, 124–138. [Google Scholar]
- Zang, H.; Blagodatskaya, E.; Wen, Y.; Xu, X.; Kuzyakov, Y. Carbon sequestration and turnover in soil under the energy crop Miscanthus: Repeated 13C natural abundance approach and literature synthesis. GCB Bioenergy 2018, 4, 262–271. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Sun, L.; Lal, R.; Bol, R.; Wang, Y.; Gao, X.; Ding, F.; Liang, S.; Li, S.; Wang, J. Microbial assimilation dynamics differs but total mineralization from added root and shoot residues is similar in agricultural Alfisols. Soil Biol. Biochem. 2020, 9, 107901. [Google Scholar] [CrossRef]
- Sun, R.; Li, W.; Dong, W.; Tian, Y.; Hu, C.; Liu, B. Tillage changes vertical distribution of soil bacterial and fungal communities. Front. Microbiol. 2018, 9, 699. [Google Scholar] [CrossRef]
- Li, S.; Lu, J.; Liang, G.; Wu, X.; Zhang, M.; Plougonven, E.; Wang, Y.; Gao, L.; Abdelrhman, A.A.; Song, X.; et al. Factors governing soil water repellency under tillage management: The role of pore structure and hydrophobic substances. Land Degrad. Dev. 2020, 1–14. [Google Scholar] [CrossRef]
- Gao, L.; Wang, B.; Li, S.; Han, Y.; Zhang, X.; Gong, D.; Ma, M.; Liang, G.; Wu, H.; Wu, X.; et al. Effects of different long-term tillage systems on the composition of organic matter by 13C CP/TOSS NMR in physical fractions in the Loess Plateau of China. Soil Till. Res. 2019, 194, 1–13. [Google Scholar] [CrossRef]
- Gao, L.; Becker, E.; Liang, G.; Houssou, A.A.; Wu, H.; Wu, X.; Cai, D.; Degré, A. Effect of different tillage systems on aggregate structure and inner distribution of organic carbon. Geoderma 2017, 288, 97–104. [Google Scholar] [CrossRef]
- Li, S.; Wu, X.; Liang, G.; Gao, L.; Wang, B.; Lu, J.; Abdelrhman, A.A.; Song, X.; Zhang, M.; Zheng, F.; et al. Is least limiting water range a useful indicator of the impact of tillage management on maize yield? Soil Till. Res. 2020, 199, 1–12. [Google Scholar] [CrossRef]
- Wang, B.; Gao, L.; Yu, W.; Wei, X.; Li, J.; Li, S.; Song, X.; Liang, G.; Cai, D.; Wu, X. Distribution of soil aggregates and organic carbon in deep soil under long-term conservation tillage with residual retention in dryland. J. Arid Land 2019, 11, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Wang, B.; Li, S.; Wu, H.; Wu, X.; Liang, G.; Gong, D.; Zhang, X.; Cai, D.; Degré, A. Soil wet aggregate distribution and pore size distribution under different tillage systems after 16 years in the Loess Plateau of China. Catena 2019, 173, 38–47. [Google Scholar] [CrossRef]
- Tian, S.; Ning, T.; Wang, Y.; Liu, Z.; Li, G.; Li, Z.; Lal, R. Crop yield and soil carbon responses to tillage method changes in North China. Soil Till. Res. 2016, 163, 207–213. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, L.; Chen, Q.; Wen, X.; Liao, Y. Conservation tillage increases soil bacterial diversity in the dryland of northern China. Agron. Sustain. Dev. 2016, 36, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L.; Owens, S.; Gilbert, J.A.; Wall, D.H.; Caporaso, J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, A.; Dick, W.A. Bacterial community diversity in soil under two tillage practices as determined by pyrosequencing. Microb. Ecol. 2015, 70, 853–859. [Google Scholar] [CrossRef]
- Lijbert, B. Soil Ecology and Ecosystem Services; Oxford University Press: Oxford, UK, 2012; pp. 45–58. [Google Scholar]
- Wen, Y.; Freeman, B.; Ma, Q.; Evans, C.; Chadwick, D.; Zang, H.; Jones, D. Raising the groundwater table in the non-growing season can reduce greenhouse gas emissions and maintain crop productivity in cultivated fen peats. J. Clean. Prod. 2020, 262, 121179. [Google Scholar] [CrossRef]
- Wen, Y.; Zang, H.; Freeman, B.; Musarika, S.; Evans, C.; Chadwick, D.; Jones, D. Microbial utilization of low molecular weight organic carbon substrates in cultivated peats in response to warming and soil degradation. Soil Biol. Biochem. 2019, 139, 107629. [Google Scholar] [CrossRef]
- Essel, E.; Li, L.; Deng, C.; Xie, J.; Zhang, R.; Luo, Z.; Cai, L. Evaluation of bacterial and fungal diversity in a long-term spring wheat–field pea rotation field under different tillage practices. Can. J. Soil Sci. 2018, 98, 619–637. [Google Scholar] [CrossRef]
- Ceja-Navarro, J.A.; Rivera-Ordu, A.F.N.; Patiño-Zúñiga, L.; Vila-Sanjurjo, A.N.; Crossa, J.; Govaerts, B.; Dendooven, L. Phylogenetic and multivariate analyses to determine the effects of different tillage and residue management practices on soil bacterial communities. Appl. Environ. Microb. 2010, 76, 3685–3691. [Google Scholar] [CrossRef] [Green Version]
- Pastorelli, R.; Vignozzi, N.; Landi, S.; Piccolo, R.; Orsini, R.; Seddaiu, G.; Roggero, P.P.; Pagliai, M. Consequences on macroporosity and bacterial diversity of adopting a no-tillage farming system in a clayish soil of Central Italy. Soil Biol. Biochem. 2013, 66, 78–93. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Cui, S.; Liang, G.; Zhang, Q. Microbial-derived carbon components are critical for enhancing soil organic carbon in no-tillage croplands: A global perspective. Soil Till. Res. 2021, 205, 104758. [Google Scholar] [CrossRef]
- Anderson, C.; Beare, M.; Buckley, H.L.; Lear, G. Bacterial and fungal communities respond differently to varying tillage depth in agricultural soils. Peer J. 2017, 5, e3930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoharan, L.; Kushwaha, S.K.; Ahrén, D.; Hedlund, K. Agricultural land use determines functional genetic diversity of soil microbial communities. Soil Biol. Biochem. 2017, 115, 423–432. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Q.; Sun, X.; Chen, D.; Insam, H.; Koide, R.; Zhang, S. Effects of mixed-species litter on bacterial and fungal lignocellulose degradation functions during litter decomposition. Soil Biol. Biochem. 2020, 141, 107690. [Google Scholar] [CrossRef]
- Chang, H.; Haudenshield, J.S.; Bowen, C.R.; Hartman, G.L. Metagenome-wide association study and machine learning prediction of bulk soil microbiome and crop productivity. Front. Microbiol. 2017, 8, 519. [Google Scholar] [CrossRef]
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci. 2014, 5, 209. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Barber, Ã.N.A.; Laughlin, D.C. Seeing the forest for the genes: Using metagenomics to infer the aggregated traits of microbial communities. Front. Microbiol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, J.; Cao, H.; Tian, R.; Cai, L.; Ding, W.; Qian, P. Post-translational modifications are enriched within protein functional groups important to bacterial adaptation within a deep-sea hydrothermal vent environment. Microbiome 2016, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Kolmeder, C.A.; Ritari, J.; Verdam, F.J.; de Vos, W.M. Colonic metaproteomic signatures of active bacteria and the host in obesity. Proteomics 2015, 15, 3544–3552. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Lombard, V.; Ramulu, G.H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2013, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Gao, Y.; Wang, S.; Xu, D.; Yu, H.; Wu, L.; Lin, Q.; Hu, Y.; Li, X.; He, Z.; et al. The microbial gene diversity along an elevation gradient of the Tibetan grassland. ISME J. 2014, 8, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.; Luo, R.; Sadakane, K.; Lam, T. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, H.; Park, J.; Takagi, T. MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006, 34, 5623–5630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Jensen, L.J.; Julien, P.; Kuhn, M.; Von, M.C.; Muller, J.; Doerks, T.; Bork, P. eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2007, 36, D250–D254. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Gougoulias, C.; Clark, J.M.; Shaw, L.J. The role of soil microbes in the global carbon cycle: Tracking the below-ground microbial processing of plant-derived carbon for manipulating carbon dynamics in agricultural systems. J. Sci. Food Agric. 2014, 94, 2362–2371. [Google Scholar] [CrossRef]
- Helgason, B.L.; Walley, F.L.; Germida, J.J. Fungal and bacterial abundance in long-term no-till and intensive-till soils of the northern great plains. Soil Sci. Soc. Am. J. 2009, 73, 120–127. [Google Scholar] [CrossRef]
- Raaijmakers, J.M.; Paulitz, T.C.; Steinberg, C.; Alabouvette, C.; Moënne-Loccoz, Y. The rhizosphere: A playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 2009, 321, 341–361. [Google Scholar] [CrossRef] [Green Version]
- Kölbl, A.; Kögel-Knabner, I. Content and composition of free and occluded particulate organic matter in a differently textured arable Cambisol as revealed by solid-state 13C NMR spectroscopy. J. Soil Sci. Plant Nut. 2004, 167, 45–53. [Google Scholar] [CrossRef]
- Robertson, S.A.; Mason, S.L.; Hack, E.; Abbott, G.D. A comparison of lignin oxidation, enzymatic activity and fungal growth during white-rot decay of wheat straw. Org. Geochem. 2008, 39, 945–951. [Google Scholar] [CrossRef]
- Chen, X.; Hu, Y.; Feng, S.; Rui, Y.; Zhang, Z.; He, H.; He, X.; Ge, T.; Wu, J.; Su, Y. Lignin and cellulose dynamics with straw incorporation in two contrasting cropping soils. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.J.; Cao, C.G.; Zhang, Z.S.; Liu, T.Q.; Li, C.F. Short-term effects of tillage practices and wheat-straw returned to rice fields on topsoil microbial community structure and microbial diversity in central China. J. Agro-Environ. Sci. 2013, 32, 1577–1584. (In Chinese) [Google Scholar]
- Malik, A.A.; Chowdhury, S.; Schlager, V.; Oliver, A.; Puissant, J.; Vazquez, P.G.M.; Jehmlich, N.B.; Martin, G.; Robert, I.; Gleixner, G. Soil fungal: Bacterial ratios are linked to altered carbon cycling. Front. Microbiol. 2016, 7, 1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H. Effect of Long-Term Application of Fertilizations on Microbial Community Structure and Organic Carbon Turnover and Accumulation in Sandy Loam Soils; University of Chinese Academy of Science: Nanjing, China, 2013. [Google Scholar]
- Ananyeva, N.D.; Susyan, E.A.; Chernova, O.V.; Chernov, I.Y.; Makarova, O.L. The ratio of fungi and bacteria in the biomass of different types of soil determined by selective inhibition. Microbiology 2006, 75, 702–707. [Google Scholar] [CrossRef]
- Six, J.; Frey, S.D.; Thiet, R.K.; Batten, K.M. Bacterial and fungal contributions to carbon sequestration in agroecosystems. Soil Sci. Soc. Am. J. 2006, 70, 555–569. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.Y.; Zhang, X.Y.; Wang, P.P.; Tian, J.; Song, J.L. Current advances in the structure, applications of CBM, and the interactions between CBM and polysaccharides. J. Cell. Sci. Technol. 2019, 27, 66–73. (In Chinese) [Google Scholar]
- Wang, C.; Dong, D.; Wang, H.; Müller, K.; Qin, Y.; Wang, H.; Wu, W. Metagenomic analysis of microbial consortia enriched from compost: New insights into the role of Actinobacteria in lignocellulose decomposition. Biotechnol. Biofuels 2016, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Větrovský, T.; Steffen, K.T.; Baldrian, P. Potential of cometabolic transformation of polysaccharides and lignin in lignocellulose by soil Actinobacteria. PLoS ONE 2014, 9, e89108. [Google Scholar] [CrossRef] [PubMed]
- Xun, W.; Zhao, J.; Xue, C.; Zhang, G.; Ran, W.; Wang, B.; Shen, Q.; Zhang, R. Significant alteration of soil bacterial communities and organic carbon decomposition by different long-term fertilization management conditions of extremely low-productivity arable soil in South China. Environ. Microbiol. 2016, 18, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Herbert, S.J.; Hashemi, A.M.; Zhang, X.; Ding, G. Effects of agricultural management on soil organic matter and carbon transformation-a review. Plant Soil Environ. 2006, 52, 531. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Zhang, H.; Xue, Y.; Gao, Y.; Qian, X.; Zhao, H.; Cheng, H.; Li, Z.; Liu, C. Response of fungal community and function to different tillage and straw returning methods. Sci. Agric. Sin. 2019, 52, 2280–2294. (In Chinese) [Google Scholar]
- Kirkby, E.; Kirkegaard, J.A.; Strong, M.; Richardson, A.E. Microorganisms and nutrient stoichiometry as mediators of soil organic matter dynamics. Nutr. Cycl. Agroecosyst. 2020, 117, 273–298. [Google Scholar]
- Jansson, J.K.; Hofmockel, K.S. Soil microbiomes and climate change. Nat. Rev. Microbiol. 2020, 18, 35–46. [Google Scholar] [CrossRef]
- Liang, C.; Amelung, W.; Lehmann, J.; Kästner, M. Quantitative assessment of microbial necromass contribution to soil organic matter. Glob. Chang. Biol. 2019, 25, 3578–3590. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Wheat Season | Maize Season | ||||
|---|---|---|---|---|---|---|
| NT | RT | DT | NT | RT | DT | |
| Bacteria | 95.96 ± 0.12 a | 95.31 ± 0.29 b | 94.79 ± 0.01 c | 96.07 ± 0.31 | 96.06 ± 0.24 | 95.94 ± 0.23 |
| Archaea | 2.83 ± 0.09 b | 3.46 ± 0.19 a | 3.73 ± 0.14 a | 3.43 ± 0.33 | 3.42 ± 0.22 | 3.56 ± 0.26 |
| Fungi | 1.21 ± 0.06 b | 1.23 ± 0.11 b | 1.47 ± 0.15 a | 0.50 ± 0.02 | 0.52 ± 0.05 | 0.50 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, H.; Zhang, H.; Li, Z.; Liu, K.; Zamanian, K. Tillage Practice Impacts on the Carbon Sequestration Potential of Topsoil Microbial Communities in an Agricultural Field. Agronomy 2021, 11, 60. https://doi.org/10.3390/agronomy11010060
Dai H, Zhang H, Li Z, Liu K, Zamanian K. Tillage Practice Impacts on the Carbon Sequestration Potential of Topsoil Microbial Communities in an Agricultural Field. Agronomy. 2021; 11(1):60. https://doi.org/10.3390/agronomy11010060
Chicago/Turabian StyleDai, Hongcui, Hui Zhang, Zongxin Li, Kaichang Liu, and Kazem Zamanian. 2021. "Tillage Practice Impacts on the Carbon Sequestration Potential of Topsoil Microbial Communities in an Agricultural Field" Agronomy 11, no. 1: 60. https://doi.org/10.3390/agronomy11010060