
Analysis of Cytosine Methylation in Genomic DNA of Solanum × michoacanum (+) S. tuberosum Somatic Hybrids

 ,
,  ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. DNA Isolation
2.3. Conversion of DArT Markers and PCR Analysis
2.4. Methylation-Sensitive Amplified Polymorphism Analysis
| mchparent | tbrparent | somatic hybrid | methylation changes in somatic hybrid |
| A | A | A | No change |
| A | A | B | Hyper-methylation of the somatic hybrid |
| A | B | A | Methylation pattern of the mch parent |
| B | A | A | Methylation pattern of the tbr parent |
| B | B | B | No change |
| B | B | A | Hypo-methylation of the somatic hybrid |
| B | A | B | Methylation pattern of the mch parent |
| A | B | B | Methylation pattern of the tbr parent |
2.5. High-Performance Liquid Chromatography Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orczyk, W.; Przetakiewicz, J.; Nadolska-Orczyk, A. Somatic hybrids of Solanum tuberosum—Application to genetics and breeding. Plant Cell Tissue Organ. Cult. 2003, 74, 1–13. [Google Scholar] [CrossRef]
- Liu, S.; Li, F.; Kong, L.; Sun, Y.; Qin, L.; Chen, S.; Cui, H.; Huang, Y.; Xia, G. Genetic and epigenetic changes in somatic hybrid introgression lines between wheat and tall wheat grass. Genetics 2015, 199, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Roychoudhury, A. Epigenetic regulation during salinity and drought stress in plants: Histone modification and DNA methylation. Plant Gene 2017, 11, 199–204. [Google Scholar] [CrossRef]
- Chwialkowska, K.; Korotko, U.; Kosinska, J.; Szarejko, I.; Kwasniewski, M. Methylation sensitive amplification polymorphism sequencing (MSAP-Seq)—A method for high—Throughput analysis of differentially methylated CCGG sites in plants with large genomes. Front. Plant Sci. 2017, 8, 2056. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chinnusamy, V.; Mohapatra, T. Epigenetics of Modified DNA Bases:5-Methylcytosine and Beyond. Front. Genet. 2018, 9, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, C.; Pérez, R.; Bazaga, P.; Herrera, C.M. Global DNA cytosine methylation as an evolving trait: Phylogenetic signal and correlated evolution with genome size in angiosperms. Front. Genet. 2015, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Singroha, G.; Sharma, P. Epigenetic Modifications in Plants under Abiotic Stress, Epigenetics; Meccariello, R., Ed.; IntechOpen: London, UK, 2019; Available online: https://www.intechopen.com/books/epigenetics/epigenetic-modifications-in-plants-under-abiotic-stress (accessed on 26 April 2021). [CrossRef] [Green Version]
- Sapna, H.; Ashwini, N.; Ramesh, S.; Nataraja, K. Assessment of DNA methylation pattern under drought stress using methylation-sensitive randomly amplified polymorphism analysis in rice. Plant Genet. Resour. 2020, 18, 222–230. [Google Scholar] [CrossRef]
- Garg, R.; Chevala, V.V.S.N.; Shankar, R.; Jain, M. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci. Rep. 2015, 14922. [Google Scholar] [CrossRef] [Green Version]
- Steward, N.; Kusano, T.; Sano, H. Expression of ZmMET1, a gene encoding a DNA methyltransferase from maize, is associated not only with DNA replication in actively proliferating cells, but also with altered DNA methylation status in cold-stressed quiescent cells. Nucleic Acids Res. 2000, 28, 3250–3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, X.; Wang, X.; Yang, G.; Wu, Y.; Su, S.; Li, S.; Liu, H.; Yuan, Y. Analysis of the DNA methylation of maize (Zea mays L.) in response to cold stress based on methylation- sensitive amplified polymorphisms. J. Plant Biol. 2013, 56, 32–38. [Google Scholar] [CrossRef]
- Xin, C.; Hou, R.; Wu, F.; Zhao, Y.; Xiao, H.; Si, W.; Ali, M.E.; Cai, L.; Guo, J. Analysis of cytosine methylation status in potato by methylation-sensitive amplified polymorphisms under low-temperature stress. J. Plant Biol. 2015, 58, 383–390. [Google Scholar] [CrossRef]
- Gao, W.; Li, S.; Huang, Y.; Deng, C.; Lu, L. Detection of genome DNA methylation change in spinach induced by 5-azaC. Mol. Cell Probes 2014, 28, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Dann, A.L.; Wilson, C.R. Comparative assessment of genetic and epigenetic variation among regenerants of potato (Solanum tuberosum) derived from long-term nodal tissue-culture and cell selection. Plant Cell Rep. 2011, 30, 631–639. [Google Scholar] [CrossRef]
- Tiwari, J.K.; Saurabh, S.; Chandel, P.; Singh, B.P.; Bhardwaj, V. Analysis of genetic and epigenetic variation in in vitro propagated potato somatic hybrid by AFLP and MSAP markers. Electron. J. Biotechnol. 2013, 16, 5. [Google Scholar] [CrossRef]
- Da, K.; Nowak, J.; Flinn, B. Potato cytosine methylation and gene expression changes induced by a beneficial bacterial endophyte, Burkholderia phytofirmans strain PsJN. Plant Physiol. Biochem. 2012, 50, 24–34. [Google Scholar] [CrossRef]
- Le, T.N.; Schumann, U.; Smith, N.A.; Tiwari, S.; Au, P.C.K.; Zhu, Q.H.; Taylor, J.M.; Kazan, K.; Llewellyn, D.J.; Zhang, R.; et al. DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol. 2014, 15, 458. [Google Scholar] [CrossRef] [PubMed]
- Tirnaz, S.; Batley, J. DNA methylation: Toward crop disease resistance improvement. Trends Plant Sci. 2019, 12, 1149. [Google Scholar] [CrossRef]
- Pikaard, C.S.; Scheid, O.M. Epigenetic regulation in plants. Cold Spring Harb. Perspect. Biol. 2014, 6, a019315. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.F.; An, Y.C.; et al. Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettell, R.I.S.; Dennis, E.S. Reactivation of a silent Ac following tissue culture is associated with heritable alterations in its methylation pattern. Mol. Gen. Genet. 1991, 229, 365–372. [Google Scholar] [CrossRef]
- Smulders, M.J.M.; de Klerk, G.J. Epigenetics in plant tissue culture. Plant Growth Regul. 2011, 63, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.; Chen, Z.J. Genomic and expression plasticity of polyploidy. Curr. Opin. Plant Biol. 2010, 13, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Laird, P.W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Kurdyukov, S.; Bullock, M. DNA methylation analysis: Choosing the right method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Marconi, G.; Capomaccio, S.; Comino, C.; Acquadro, A.; Portis, E.; Porceddu, A.; Albertini, E. Methylation content sensitive enzyme ddRAD (MCSeEd): A reference-free, whole genome profiling system to address cytosine/adenine methylation changes. Sci. Rep. 2019, 9, 14864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.J.; Palanca-Ballester, C.; Urtasun, R.; Alemany-Cosme, E.; Lahoz, A.; Sandoval, J. Methods for analysis of specific DNA methylation status. Methods 2021, 187, 3–12. [Google Scholar] [CrossRef]
- Li, X.L.; Lin, Z.X.; Nie, Y.C.; Guo, X.P.; Zhang, X.L. Methylation-sensitive amplification polymorphism of epigenetic changes in cotton under salt stress. Acta Agron. Sin. 2009, 35, 588–596. [Google Scholar] [CrossRef]
- Marconi, G.; Pace, R.; Traini, A.; Raggi, L.; Lutts, S.; Chiusano, M.; Guiducci, M.; Falcinelli, M.; Benincasa, P.; Albertini, E. Use of MSAP markers to analyse the effects of salt stress on DNA methylation in rapeseed (Brassica napus var. oleifera). PLoS ONE 2013, 8, e75597. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhang, M.; Fu, R.; Qian, X.; Rong, P.; Zhang, Y.; Jiang, P.; Wang, J.; Lu, X.; Wang, D.; et al. Epigenetic mechanisms of salt tolerance and heterosis in Upland cotton (Gossypium hirsutum L.) revealed by methylation-sensitive amplified polymorphism analysis. Euphytica 2016, 208, 477–491. [Google Scholar] [CrossRef]
- Gautam, M.; Dang, Y.; Ge, X.; Shao, Y.; Li, Z. Genetic and epigenetic changes in oilseed rape (Brassica napus L.) extracted from intergeneric allopolyploid and additions with Orychophragmus. Front. Plant Sci. 2016, 7, 438. [Google Scholar] [CrossRef] [Green Version]
- Abid, G.; Mingeot, D.; Muhovski, Y.; Mergeai, G.; Aouida, M.; Abdelkarim, S.; Aroua, I.; El Ayed, M.; Mhamdi, M.; Sassi, K.; et al. Analysis of DNA methylation patterns associated with drought stress response in faba bean (Vicia faba L.) using methylation-sensitive amplification polymorphism (MSAP). Environ. Exp. Bot. 2017, 142, 34–44. [Google Scholar] [CrossRef]
- Li, S.; Xia, Q.; Wang, F.; Yu, X.; Ma, J.; Kou, H.; Lin, X.; Gao, X.; Liu, B. Laser irradiation-induced DNA methylation changes are heritable and accompanied with transpositional activation of mPing in rice. Front. Plant Sci. 2017, 8, 363. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Qin, L.; Xie, C.; Li, W.; Yuan, J.; Kong, L.; Yu, W.; Xia, G.; Liu, S. Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol. 2014, 55, 1354–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Rong, H.; Xie, T.; Jiang, J.; Wu, J.; Wang, Y. Comparison of DNA methylation in the developing seeds of yellow- and black-seeded Brassica napus through MSAP analysis. Euphytica 2016, 209, 157–169. [Google Scholar] [CrossRef]
- Marfil, C.F.; Camadro, E.L.; Masuelli, R.W. Phenotypic instability and epigenetic variability in a diploid potato of hybrid origin, Solanum ruiz-lealii. BMC Plant Biol. 2009, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aversano, R.; Caruso, I.; Aronne, G.; De Micco, V.; Scognamiglio, N.; Carputo, D. Stochastic changes affect Solanum wild species following autopolyploidization. J. Exp. Bot. 2013, 64, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Fulneček, J.; Kovařík, A. How to interpret Methylation Sensitive Amplified Polymorphism (MSAP) profiles? BMC Genet. 2014, 15, 2. [Google Scholar] [CrossRef] [Green Version]
- González-Benito, M.E.; Ibáñez, A.; Pirredda, M.; Mira, S.; Martin, C. Application of the MSAP technique to evaluate epigenetic changes in plant conservation. Int. J. Mol. Sci. 2020, 21, 7459. [Google Scholar] [CrossRef]
- Duan, H.; Li, J.; Zhu, Y.; Jia, W.; Wang, H.; Jiang, L.; Zhou, Y. Responsive changes of DNA methylation in wheat (Triticum aestivum) under water deficit. Sci. Rep. 2020, 10, 7938. [Google Scholar] [CrossRef]
- Smyda, P.; Jakuczun, H.; Dębski, K.; Śliwka, J.; Thieme, R.; Nachtigall, M.; Wasilewicz-Flis, I.; Zimnoch-Guzowska, E. Development of somatic hybrids Solanum × michoacanum Bitter. (Rydb.) (+) S. tuberosum L. and autofused 4x S. × michoacanum plants as potential sources of late blight resistance for potato breeding. Plant Cell Rep. 2013, 32, 1231–1241. [Google Scholar] [CrossRef] [Green Version]
- Smyda-Dajmund, P.; Śliwka, J.; Wasilewicz-Flis, I.; Jakuczun, H.; Zimnoch-Guzowska, E. Genetic composition of interspecific potato somatic hybrids and autofused 4x plants evaluated by DArT and cytoplasmic DNA markers. Plant Cell Rep. 2016, 35, 1345–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Albertini, E.; Marconi, G. Methylation-Sensitive Amplified Polymorphism (MSAP) Marker to investigate drought-stress response in Montepulciano and Sangiovese grape cultivars. In Plant Epigenetics and Epigenomics; Spillane, C., McKeown, P., Eds.; Humana Press: Totowa, NJ, USA, 2014; Volume 1112, pp. 151–164. [Google Scholar] [CrossRef]
- Havliš, J.; Trbušek, M. 5-Methylcytosine as a marker for the monitoring of DNA methylation. J. Chromatogr. A 2002, 781, 373–392. [Google Scholar] [CrossRef]
- Johnston, J.W.; Harding, K. HPLC analysis of plant DNA methylation: A study of critical methodological factors. Plant Physiol. Biochem. 2005, 43, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, P.T.; Orłowska, R. Plant tissue culture environment as a switch-key of (epi)genetic changes. Plant Cell Tissue Cult. 2019, 140, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ma, Y.; Chen, F.; Song, W.; Zhang, L. Analysis of DNA methylation patterns of PLBs derived from Cymbidium hybridium based on MSAP. Plant Cell Tissue Organ. Cult. 2009, 98, 67–77. [Google Scholar] [CrossRef]
- Gao, X.; Yang, D.; Cao, D.; Ao, M.; Sui, X.; Wang, Q.; Kimatu, J.N.; Wang, L. In Vitro Micropropagation of Freesia hybrida and the Assessment of Genetic and Epigenetic Stability in Regenerated Plantlets. J. Plant Growth Regul. 2010, 29, 257–267. [Google Scholar] [CrossRef]
- Li, X.; Yu, X.; Wang, N.; Feng, Q.; Dong, Z.; Liu, L.; Shen, J.; Liu, B. Genetic and epigenetic instabilities induced by tissue culture in wild barley (Hordeum brevisubulatum (Trin.) Link). Plant Cell Tissue Organ. Cult. 2007, 90, 153–168. [Google Scholar] [CrossRef]
- Phillips, R.L.; Kaeppler, S.; Olhoft, P. Genetic instability of plant tissue cultures: Breakdown of normal controls. Proc. Natl. Acad. Sci. USA 1994, 91, 5222–5226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, J.K.; Saurabh, S.; Chandel, P.; Devi, S.; Ali, N.; Bist, C.M.; Singh, B.P. Analysis of genetic and epigenetic changes in potato somatic hybrids between Solanum tuberosum and S. etuberosum by AFLP and MSAP markers. Agric. Res. 2015, 4, 339–346. [Google Scholar] [CrossRef]
- Sheng, X.G.; Zhao, Z.Q.; Yu, H.F.; Wang, J.S.; Gu, H.H. Rapid alterations of DNA sequence and cytosine methylation induced by somatic hybridization between Brassica oleracea L. var. italica and Brassica nigra (L.) Koch. Plant Cell Tissue Organ. Cult. 2013, 115, 395–405. [Google Scholar] [CrossRef]
- Xu, S.X.; Cai, D.F.; Tan, F.Q.; Fang, Y.N.; Xie, K.D.; Grosser, J.W.; Guo, W.W. Citrus somatic hybrid: An alternative system to study rapid structural and epigenetic reorganization in allotetraploid genomes. Plant Cell Tissue Organ. Cult. 2014, 119, 511–522. [Google Scholar] [CrossRef]
- Cai, Y.F.; Xiang, F.N.; Zhi, D.Y.; Liu, H.; Xia, G.M. Genotyping of somatic hybrids between Festuca arundinacea Schreb. and Triticum aestivum L. Plant Cell Rep. 2007, 26, 1809–1819. [Google Scholar] [CrossRef]
- Jia, L.; Zhai, H.; He, S.; Yang, Y.; Liu, Q. Analysis of drought tolerance and genetic and epigenetic variations in a somatic hybrid between Ipomoea batatas (L.) Lam. and I. triloba L. J. Integr. Agric. 2017, 16, 36–46. [Google Scholar] [CrossRef]
- Jackson, S.A. Epigenomics: Dissecting hybridization and polyploidization. Genome Biol. 2017, 18, 117. [Google Scholar] [CrossRef] [Green Version]
- Aversano, R.; Scarano, M.T.; Aronne, G.; Caruso, I.; D’Amelia, V.; De Micco, V.; Fasano, C.; Termolino, P.; Carputo, D. Genotype-specific changes associated to early synthesis of autotetraploids in wild potato species. Euphytica 2015, 202, 307–316. [Google Scholar] [CrossRef]
- Wenzl, P.; Carling, J.; Kudrna, D.; Jaccoud, D.; Huttner, E.; Kleinhofs, A.; Kilian, A. Diversity Arrays Technology (DArT) for whole-genome profiling of barley. Proc. Natl. Acad. Sci. USA 2004, 101, 9915–9920. [Google Scholar] [CrossRef] [Green Version]
- Pootakham, W.; Sonthirod, C.; Naktang, C.; Jomchai, N.; Sangsrakru, D.; Tangphatsornruang, S. Effects of methylation-sensitive enzymes on the enrichment of genic SNPs and the degree of genome complexity reduction in a two-enzyme genotyping-by-sequencing (GBS) approach: A case study in oil palm (Elaeis guineensis). Mol. Breed. 2016, 36, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaccoud, D.; Peng, K.; Feinstein, D.; Kilian, A. Diversity Arrays: A solid-state technology for sequence information independent genotyping. Nucleic Acids Res. 2001, 29, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Vaillancourt, R.; Mendham, N.; Zhou, M. Comparative mapping of quantitative trait loci associated with waterlogging tolerance in barley (Hordeum vulgare L.). BMC Genom. 2008, 9, 401. [Google Scholar] [CrossRef] [Green Version]
- Crossa, J.; Burgueño, J.; Dreisigacker, S.; Vargas, M.; Herrera-Foessel, S.A.; Lillemo, M.; Singh, R.P.; Trethowan, R.; Warburton, M.; Franco, J.; et al. Association analysis of historical bread wheat germplasm using additive genetic covariance of relatives and population structure. Genetics 2007, 177, 1889–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sołtys-Kalina, D.; Szajko, K.; Sierocka, I.; Śliwka, J.; Strzelczyk-Żyta, D.; Wasilewicz-Flis, I.; Jakuczun, H.; Szweykowska-Kulinska, Z.; Marczewski, W. Novel candidate genes AuxRP and Hsp90 influence the chip color of potato tubers. Mol. Breed. 2015, 35, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Śliwka, J.; Sołtys-Kalina, D.; Szajko, K.; Wasilewicz-Flis, I.; Strzelczyk-Żyta, D.; Zimnoch-Guzowska, E.; Jakuczun, H.; Marczewski, W. Mapping of quantitative trait loci for tuber starch and leaf sucrose contents in diploid potato. Theor. Appl. Genet. 2016, 129, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Thieme, R.; Darsow, U.; Gavrilenko, T.; Dorokhov, D.; Tiemann, H. Production of somatic hybrids between S. tuberosum L. and late blight resistant Mexican wild potato species. Euphytica 1997, 97, 189–200. [Google Scholar] [CrossRef]
- Szczerbakowa, A.; Tarwacka, J.; Oskiera, M.; Jakuczun, H.; Wielgat, B. Somatic hybridization between the diploids of S. x michoacanum and S. tuberosum. Acta Physiol. Plant 2010, 32, 867–873. [Google Scholar] [CrossRef]
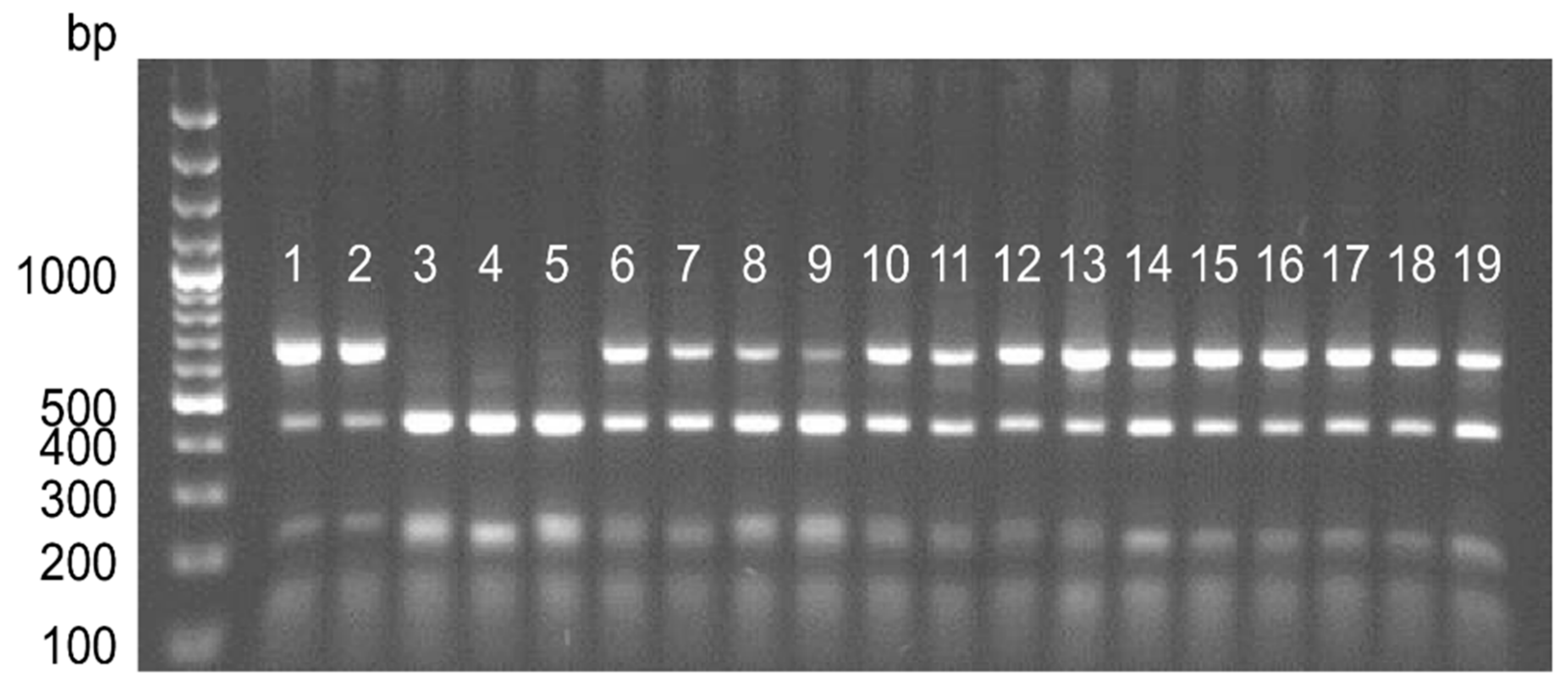

{kind=link}
| Name of Combination | Primer Combination | No. of Bands Scored | Band Size Range (bp) | No. of Methylated CCGG Sites * | % of Methylated CCGG Sites |
|---|---|---|---|---|---|
| EHM_2 | fam_EcoRI+3_CCA (+) HpaII/MspI+3_AAC | 137 | 52–592 | 58 | 42.3 |
| EHM_3 | fam_EcoRI+3_CCA (+) HpaII/MspI+3_ACA | 75 | 54–584 | 36 | 48 |
| EHM_8 | fam_EcoRI+3_CCA (+) HpaII/MspI+3_ACT | 86 | 54–567 | 44 | 51.1 |
| EHM_9 | fam_EcoRI+3_CCA (+) HpaII/MspI+3_AGC | 85 | 51–588 | 48 | 56.4 |
| EHM_12 | fam_EcoRI+3_CAA (+) HpaII/MspI+3_AGC | 120 | 51–600 | 46 | 38.3 |
| EHM_13 | fam_EcoRI+3_CAA (+) HpaII/MspI+3_AGG | 119 | 51–630 | 63 | 52.9 |
| Total | 622 | 295 |
| MSAP | HPLC | ||||
|---|---|---|---|---|---|
| Genotype | Total no. of Bands | Unmethylated CCGG Sites (%) | Methylated CCGG Sites (%) | % of Cytosine DNA Methylation | Range of % of Single Readings |
| mch/8 | 190 | 123 (64.7) | 67 (35.3) | 15.8 | 15.7–15.9 |
| mch/39 | 148 | 105 (70.9) | 43 (29.1) | 15.5 | 15.4–15.6 |
| DG 81-68 | 177 | 90 (50.8) | 87 (49.2) | 15.9 | 15.9–16.0 |
| dHBard | 186 | 85 (45.7) | 101 (54.3) | 15.7 | 15.6–15.9 |
| cv. Rywal | 213 | 121 (56.8) | 92 (43.2) | 16.9 | 16.7–17.0 |
| Mean | 182.8 | 104.8 | 78 | ||
| MSAP | HPLC | |||||
|---|---|---|---|---|---|---|
| Fusion Combination | Number of Individuals | Total No. of Analysed Bands | Mean Unmethylated CCGG Sites Per Individual (%) | Mean Methylated CCGG Sites Per Individual (%) | Mean % of Cytosine DNA Methylation Per Individual | Mean Range of % of Cytosine Methylation Per Individual |
| mch/8 (+) dHBard | 4 | 261 | 140.5 (54.9) | 118 (45.1) | 18.8 | 16.1–21.5 |
| mch/8 (+) cv. Rywal | 2 | 272 | 134.5 (52) | 130.5 (48) | 18.3 | 17.5–19.0 |
| mch/39 (+) DG 81-68 | 38 | 236 | 117.7 (50.6) | 116.6 (49.4) | 19.4 | 3.9–22.5 |
| mch/39 (+) dHBard | 44 | 240 | 127.3 (53) | 112.7 (47) | 17.3 | 13.1–19.7 |
| mch/39 (+) cv. Rywal | 8 | 251 | 117.4 (46.8) | 133.6 (53.2) | 15.9 | 14.0–19.2 |
| Total | 96 | |||||
| Mean Percentage of the Analyzed Bands in the Fusion Combinations | ||||||||
|---|---|---|---|---|---|---|---|---|
| Parent mch | Parent tbr | Somatic hybrids | mch/8 (+) dHBard | mch/8 (+) cv. Rywal | mch/39 (+) DG 81-68 | mch/39 (+) dHBard | mch/39 (+) cv. Rywal | Mean |
| No changes | ||||||||
| A | A | A | 10.4 | 15.5 | 7.0 | 6.1 | 8.4 | 9.5 |
| B | B | B | 20.5 | 24.5 | 18.5 | 17.5 | 18.9 | 20.0 |
| 14.7 | ||||||||
| Changes in somatic hybrids | ||||||||
| A | A | B | 5.7 | 5.9 | 5.7 | 5.6 | 7.6 | 6.1 |
| B | B | A | 17.0 | 7.2 | 17.3 | 19.5 | 12.9 | 14.8 |
| 10.4 | ||||||||
| Parent-like banding patterns | ||||||||
| A | B | A | 19.4 | 11.8 | 12.2 | 15.1 | 10.8 | 13.9 |
| B | A | B | 7.9 | 4.2 | 12.4 | 11.1 | 15.0 | 10.1 |
| B | A | A | 8.2 | 18.3 | 12.8 | 12.2 | 14.8 | 13.3 |
| A | B | B | 10.8 | 12.5 | 14.2 | 12.8 | 11.6 | 12.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smyda-Dajmund, P.; Śliwka, J.; Villano, C.; Janiszewska, M.; Aversano, R.; Bednarek, P.T.; Carputo, D.; Zimnoch-Guzowska, E. Analysis of Cytosine Methylation in Genomic DNA of Solanum × michoacanum (+) S. tuberosum Somatic Hybrids. Agronomy 2021, 11, 845. https://doi.org/10.3390/agronomy11050845
Smyda-Dajmund P, Śliwka J, Villano C, Janiszewska M, Aversano R, Bednarek PT, Carputo D, Zimnoch-Guzowska E. Analysis of Cytosine Methylation in Genomic DNA of Solanum × michoacanum (+) S. tuberosum Somatic Hybrids. Agronomy. 2021; 11(5):845. https://doi.org/10.3390/agronomy11050845
Chicago/Turabian StyleSmyda-Dajmund, Paulina, Jadwiga Śliwka, Clizia Villano, Marta Janiszewska, Riccardo Aversano, Piotr Tomasz Bednarek, Domenico Carputo, and Ewa Zimnoch-Guzowska. 2021. "Analysis of Cytosine Methylation in Genomic DNA of Solanum × michoacanum (+) S. tuberosum Somatic Hybrids" Agronomy 11, no. 5: 845. https://doi.org/10.3390/agronomy11050845
APA StyleSmyda-Dajmund, P., Śliwka, J., Villano, C., Janiszewska, M., Aversano, R., Bednarek, P. T., Carputo, D., & Zimnoch-Guzowska, E. (2021). Analysis of Cytosine Methylation in Genomic DNA of Solanum × michoacanum (+) S. tuberosum Somatic Hybrids. Agronomy, 11(5), 845. https://doi.org/10.3390/agronomy11050845