1. Introduction
Rice (
Oryza sativa L.) is one of the most important crops worldwide, as it feeds more than 50% of the world’s population. Biotic stresses caused by plant pathogens are the primary limiting factors affecting rice production [
1]. Sheath blight disease (ShB), which is caused by
Rhizoctonia solani AG1-1A [
2,
3], is considered to be the second most important disease in rice (
Oryza sativa L.) and causes yield losses ranging from 20 to 50% [
4,
5]. ShB is notably difficult to control due to its wide host range and high genetic variability [
3]. Reducing the threat of ShB has long been a major issue facing rice pathologists and breeders [
3,
4,
6,
7,
8,
9,
10,
11,
12].
In addition, ShB in rice is thought to be a typical quantitative trait controlled by a number of genes, and more than 50 quantitative trait loci (QTLs) for rice ShB resistance, distributed over all 12 rice chromosomes, have been identified [
3,
6,
8,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24]. Most QTL mapping in rice ShB was performed by using conventional linkage mapping methods in the segregating populations derived from the crossing between typical ShB-tolerant and ShB-sensitive rice varieties that was performed in previous studies [
3]. Genome-wide association studies (GWASs) could overcome the limitations (i.e., high cost, poor mapping resolution, and only two alleles that can be studied) of linkage mapping [
25] and enables researchers to use the postgenomic data to exploit natural genetic diversity and identify elite genes in the genome [
26]. Moreover, GWAS has been used to identify natural variations for ShB resistance in maize [
27]. To the best of our knowledge, at least four GWAS have been performed on rice ShB. The first GWAS on rice ShB was performed in 217 sub-core rice entries with 155 markers and demonstrated a significant association of ten marker loci [
28]. Moreover, the second rice ShB GWAS detected 11 single-nucleotide polymorphism (SNP) loci significantly associated with ShB resistance using 299 diverse rice varieties and 44,000 SNP markers [
29]. The third rice ShB GWAS was performed using 2,977,750 SNPs to analyze 563 rice accessions [
30]. Finally, the fourth study used 700,000 SNPs to perform GWAS on the phenotype of 228 rice accessions [
31]. The phenotyping of the above four GWASs on rice ShB resistance was performed for only one year. Furthermore, compared with the above four studies, no higher-resolution GWAS on rice ShB resistance within natural populations has been performed.
Ting’s rice collection, which is one of the earliest rice collections in China, consists of 150 varieties constructed from 2262 of 7128 original landraces [
32]. Ting’s core collection has been used for association mapping of rice agronomic traits and aluminum tolerance, and abundant genetic diversity has been identified [
33,
34,
35,
36]. Therefore, Ting’s core collection may be an appropriate population for GWASs on rice ShB resistance.
In the present study, a GWAS for rice ShB resistance based on lesion length/plant height (LL/PH) was performed using Ting’s core collection with more than 5.1 million high-quality SNPs. Candidate regions identified by GWAS were compared with regions identified as QTLs in previous studies and with ShB-resistant mutants and/or candidate genes. This study provides important information regarding candidate genes for ShB resistance improvement in rice.
4. Discussion
Ting’s core collection may represent a useful reservoir of rice genotypes and a potential source of beneficial alleles for ShB resistance in rice breeding because the abundant genetic variations of agronomic traits and aluminum tolerance in this collection have been reported in our previous studies [
32,
33,
34,
35,
36]. Although the population size of Ting’s core collection is smaller than that of the other two studies examining ShB resistance GWAS [
29,
30], the resolution in the present study is higher than that of the above two studies. Moreover, our previous study [
36] observed that the phenotypic diversity of several agronomic traits was comparable to that of populations with larger population sizes [
48,
49,
50], or even higher for some agronomic traits. Ting’s core collection consists of rice landraces that are considerably easier to utilize than wild rice in rice breeding because they have more abundant genetic diversity and remain in an intermediate stage between wild rice and cultivars in their domestication histories [
37]. Ting’s core collection shows a less complicated population structure, a shorter LD decay distance, and a lower kinship value than other populations utilized in rice GWAS [
33]. Thus, Ting’s core collection may be a suitable population for rice ShB resistance. Furthermore, we compared the significant loci between four GWAS and our study in rice ShB resistance. We detected one significant SNP (8,675,283) on chromosome 7 in 2016 which was very close to SAL ID
L14 (chr. 7, 8,569,372,
Figure 3A) identified in GWAS research about rice ShB resistance [
31]. In addition, there was a peak with consecutive loci detected in 2016 which are below the significant threshold (
p ≤ 1.93 × 10
−8) located in the region of
qSB-6 on chromosome 6 (
Figure 3A) [
29], while there was no identical region associated with ShB resistance between the study of Jia et al. [
28] and our study, as well as between the study of Oreiro et al. [
31] and our study. The reasons for few loci being identical in different GWAS research might be (i) differential rice population, or (ii) differential traits representing rice ShB resistance.
Approximately 5.1 million SNPs were employed in the present study, which might be the highest resolution in rice GWAS. A mixed linear model was performed by EMMAX software, and this software’s strong points have been described in a previous study [
51]. The abovementioned information elucidates and supports the results obtained through ShB GWAS.
In the present study, ShB resistance was estimated by one trait, i.e., lesion length/plant height (LL/pH), which had been utilized in previous studies [
20,
30,
37]. It appears that LL/pH is the most applicable method for phenotyping ShB resistance within Ting’s core collection because pH can affect the development of ShB pathogens in rice plants and influence eventual phenotypic outcomes [
14]. Moreover, pH was frequently reported to be closely linked with ShB resistance [
6,
8,
21]. Furthermore, genetic diversity in Ting’s core collection was identified as abundant, and pH in this collection varied from 60 cm to 230 cm [
34]. LL might not reflect the real resistance to ShB; for instance, a value of 16.7% LL/pH cannot be compared to an 8.8% one for evaluating rice ShB resistance, which does not mean that the ShB resistance of the variety with 60 cm pH is stronger than that with 230 cm pH. Thus, we did not invest time estimating ShB resistance by other traits in the present study.
As hypothesized, there was a wide range of phenotypic variation for ShB resistance in Ting’s core collection in both 2016 and 2017. The varieties in Ting’s core collection, which were identified with small LL/pH values, could be employed as donor parents to introgress ShB resistance in elite rice varieties. ShB resistance was estimated in both the
indica and
japonica subgroups. Our results showed that the ShB resistance of
indica was stronger than that of
japonica, which was in keeping with the findings obtained in two previous studies [
29,
30].
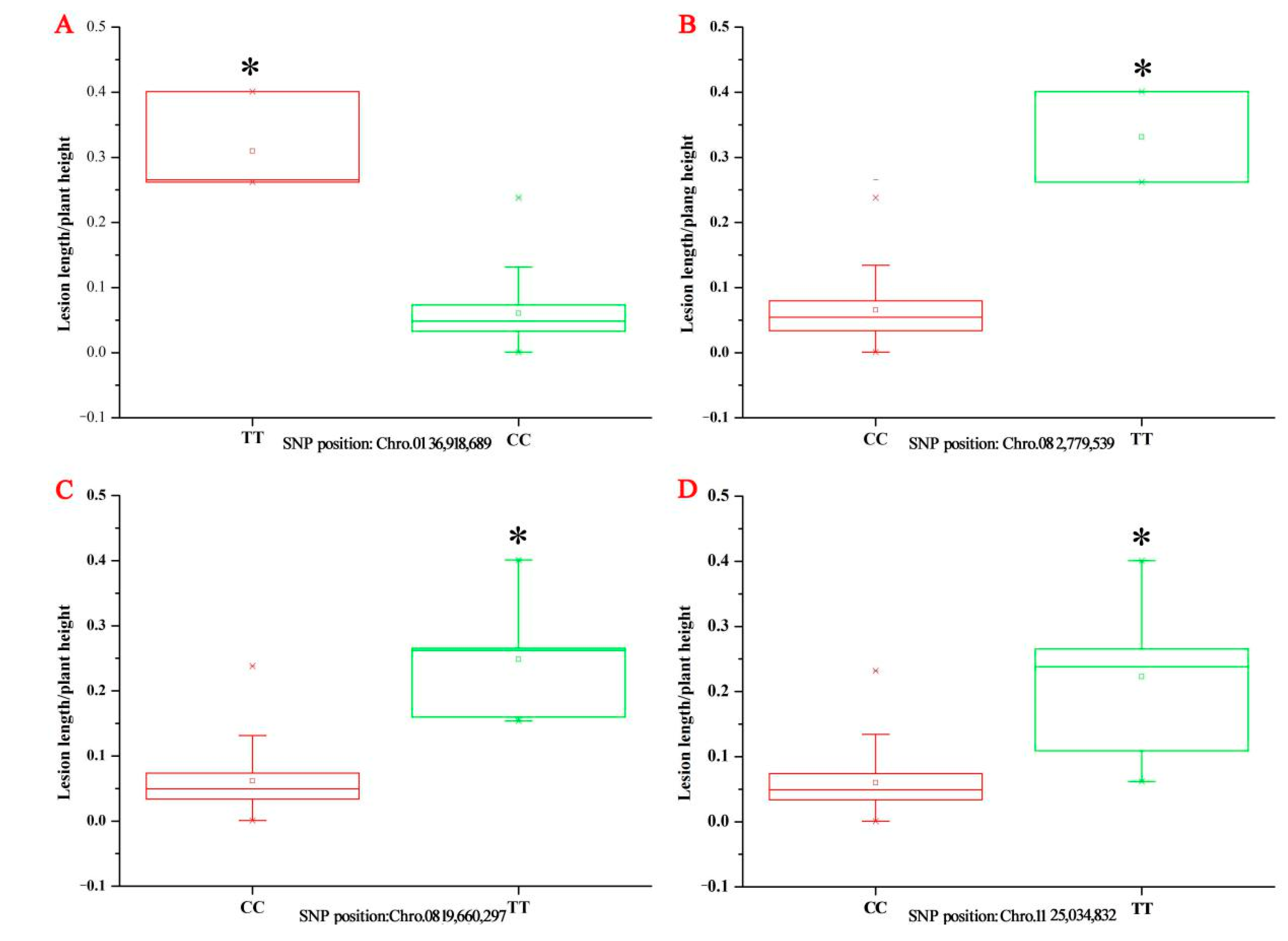
There was no identical significant SNP in 2016 and 2017, but identical SNPs located in previous major QTLs confirmed in many previous studies, such as
qShB9-2 and
qSBR11-1, could be detected in two years in our study if a lower significant threshold value was set (
p < 0.00001,
Figure 3). Previous QTL regions were sufficiently wide such that they could be detected in different years and multiple locations in previous studies (QTLs were summarized in a review by Molla et al. (2020)) [
3]. It was indicated that the environment had more influence than the genotype on ShB resistance in the field, which might explain the lack of identical significant SNPs found in the two years [
37]. Due to the wide range in QTL regions for rice ShB resistance, 13 gene-based SNPs that were significantly related to ShB resistance were supported by previous studies. The above 13 candidate genes could be hypothesized to defend against ShB (
Table 2). For instance,
LOC_Os02g42310, which encodes the putative serine carboxypeptidase homolog and serine carboxypeptidase, has been reported to be related to disease resistance in oats [
51].
LOC_Os11g40780 is annotated as encoding a disease-resistance protein.
LOC_Os11g05700 encodes an ABC transporter family protein, while an ABC transporter family protein has been reported to be related to disease resistance in barley [
52].
More than 50 QTLs for rice ShB resistance distributed on all of the 12 chromosomes were identified, but no single gene was cloned. The reason for the above finding might be that (i) rice ShB resistance is controlled by a number of QTLs, and there are no major QTLs responsible for ShB resistance, and (ii) rice ShB is influenced more easily by the environment than genotype, which leads to the determination of a gene’s function. It appears that mining the critical genes in possible pathways of ShB resistance is more important than locating or cloning a major QTL. Thus, we performed GO and KEGG analysis for the genes located at a region within 10 kb of the significant SNPs in two different years.
Although no identical SNPs were detected, the same GO terms were found in the two years (
Supplementary Figures S2–S7 and
Table 4). Identical GO terms were reported to be associated with ShB resistance in previous studies, such as ATP binding [
53,
54], the nucleus [
55,
56], and the plasma membrane [
57]. Moreover, four KEGG pathways were identified in the present study, and the TCA cycle [
58], glyoxylate and dicarboxylate metabolism, and lysine biosynthesis [
59,
60,
61,
62,
63] were reported to be related to disease resistance in plants in previous studies. The key genes regulating citrate synthase in the TCA cycle and glyoxylate and dicarboxylate pathway, relating to diaminopimelate decarboxylase in the lysine biosynthesis pathway, as well as controlling pre-mRNA-splicing factor 18 in the spliceosome pathway, should be further studied in the future to facilitate the breeding of rice with greater ShB resistance.


 and
and 



{kind=link}
{kind=link}
{kind=link}
{kind=link}