Abstract
Understanding the genetic background of elite cultivated tomato germplasm resources in crossbreeding and revealing the genetic basis of complex traits are vital for better-targeted germplasm expansion and the creation of strong hybrids. Here, we obtained approximately 21 million single-nucleotide polymorphisms (SNPs) based on the sequencing of 212 cultivated tomato accessions and the population structure of which was revealed. More importantly, we found that target genes distributed on chromosomes 1, 5, 9, and 11 may be actively selected in breeding. In particular, the significant signals related to soluble sugar content (chr1_94170222, chr1_96273188, chr9_4167512, and chr11_55592768), fruit firmness (chr5_4384919 and chr5_5111452) and gray leaf spot resistance (chr11_8935252 and chr11_9707993) were also detected on the corresponding chromosomes, respectively. Overall, we reported 28 significant association signals for nine agronomic traits based on a mixed linear model (MLM), including 114 genes. Among these signals, 21 contained potential novel genes for six fruit traits. These novel candidate genes located in genomic regions without previously known loci or on different chromosomes explained approximately 16% of the phenotypic variance on average in cultivated tomatoes. These findings could accelerate the identification and validation of novel and known candidate genes and QTLs, improving the understanding of the genetic structure of complex quantitative traits. These results also provide a basis for tomato breeding and improvement.
1. Introduction
Tomatoes (Solanum lycopersicum L.) are a staple vegetable crop with a global distribution. According to statistics from the Food and Agriculture Organization of the United Nations (FAO), the global production of tomatoes was more than 189.1 million tons from 5.17 million ha in 2021, which was approximately 4.3 million tons more than in the previous year (https://www.fao.org/faostat (accessed on 2 February 2023)). Although production is increasing, crop production is also facing major challenges, including improving the total quantity of production, optimizing the quality, enhancing resistance (particularly disease resistance), and coping with instability factors (global climate change)] [1]. It is critical to breed tomato varieties with a higher yield, more stable production, high nutritional value, resistance to diseases and insect pests, and greater adaptability to the environment to satisfy the divergent consumption requirements [2]. However, breeding elite varieties with multiple favorable characteristics and improved adaptability is a major challenge for scientists and breeders [3].
The elite commercial tomato cultivars, now widely planted worldwide, are generally hybrids, because crossbreeding, the most widely used and effective means of genetic improvement to select new varieties, can quickly integrate the genes required by multiple varieties to obtain new varieties with better traits [4,5]. Research has shown that heterosis mainly depends on the genetic nature, diversity, and heterogeneity of the parents [6]. Therefore, as a rich source of natural allelic variants, germplasm is essential for genetic analysis and subsequent breeding applications. In the process from wild type to large-fruit cultivated type, the genetic diversity of modern cultivated tomatoes has relatively progressively narrowed [7,8,9]. The continuous occurrence of artificial hybridization and recombination events, fuzzy division of dominant populations, lack of excellent core germplasm, and hybridization guidance theory limit the ability of traditional crossbreeding methods to develop new varieties with aggregated elite traits based primarily on complementary phenotypic crosses. Breeding practice shows that the location and cloning of numerous dominant related loci or genes have important guiding significance for the aggregation of excellent alleles, further improvement of modern elite cultivars, and better breeding of new varieties. A genome-wide association study (GWAS) combines the analysis of phenotypic trait data with genome-wide information to identify genes or genetic loci associated with traits [10]. It was initially applied to maize in plants to reveal the characteristics of candidate genes [11]. In the last decade, next-generation sequencing technologies (NGS) technologies have provided a powerful tool to elucidate the breeding history of many crops, such as grape [12], maize [13], cucumber [14], tomato [8], soybean [15], rice [16], Brassica rapa and Brassica oleracea [17], and peach [18]. With the development and intense application of NGS, GWAS has become a recognized strategy for decoding genotype–phenotype associations in various species [19,20]. Based on single-nucleotide polymorphism (SNP) marker sites, GWAS has successfully identified the genes and pathways underlying many economically valuable agronomic traits (particularly yield- and yield-related traits) of crops, including sorghum [21], rice [22], maize [23], and barley [24]. Currently, this approach is also widely used in tomatoes to investigate the potential regulatory mechanisms of traits related to yield [25,26] and quality [27,28,29,30,31,32]. Flowers and other plant characteristics have also been examined [3]. The results have shown associations with different traits, proving the potential of GWAS to reveal the genetic architecture of complex traits [11,33,34]. Although several loci associated with different traits have been identified in previous studies, these loci are responsible for only some of the genetic variation in each examined trait in tomatoes [26]. Additionally, previous authors have noted that only a limited portion of the phenotypic variation of a given trait can be explained in any given GWAS [35]. Hence, more work is required to reveal the genetic basis of complex traits to better assist molecular breeding.
Here, the population structure relationships in a collection of 212 tomato accessions representing different genetic backgrounds were revealed. This work reduces random chance in selecting parents for crosses based solely on phenotype and provides a basis for the classification and arrangement of germplasm resources and germplasm amplification. Furthermore, on the basis of the phenotypic identification of nine agronomic traits, GWAS was performed to identify and mine significant associated loci and candidate genes. These findings could accelerate the validation of novel and known candidate genes and QTLs, improving the understanding of the genetic structure of complex quantitative traits. These results also provide the basis for tomato breeding and improvement.
2. Materials and Methods
2.1. Materials
A set of 212 cultivated tomato accessions were collected and used for association analysis (Supplementary Materials Table S1). Traits of the set comprised diverse growth types, fruit colors, fruit sizes, fruit shapes, etc. The seeds of all accessions were selfed varieties above the 6th generation and were obtained from the Tomato Research Institute, Northeast Agricultural University, Harbin, Heilongjiang Province, China, which acts as a national key tomato genetics and breeding unit.
2.2. Field Experiment and Data Collection
Seeds of all accessions were sown at the Horticulture Experimental Station of Northeast Agricultural University (E 125°42′~130°10′, N 44°04′~46°40′) on 27 March 2018. The seedlings were transplanted to Xiang-yang Farm on 28 April 2018 and field-planted in plastic greenhouses at the farm on 28 May 2018. To reduce the impact of multiple factors, all seedlings were planted in the same greenhouses. Each accession was randomly numbered. Accessions were planted in sequence from L1 to L212. The mode, including ridge farming, black plastic film mulching, and double-row side-by-side planting, was used. Ridge width was 80 cm. Each accession was grown in a plot with one row, and each row consisted of 12 plants, with a plant spacing of 40 cm and a row spacing of 30 cm. To reduce deviation and improve precision in phenotyping, guidelines were fixed on the ground at both ends of each row, and the seedlings were planted carefully along the lines at the designated density. During the experiment, normal agronomic field management measures for tomato production under a shed were applied. The survey and statistics of related traits were conducted from June to August.
Nine important agronomic traits were comprehensively evaluated and phenotyped to study the genetic basis of yield, quality, and other related agronomic traits. These traits and their measuring standards are shown in Table 1.

Table 1.
List of 9 agro-morphological traits and their corresponding methods of determination.
2.3. Statistical Analyses
Statistical analysis and processing of raw data, such as phenotypes, were performed using Excel 2016. Cluster analysis of 212 accessions based on phenotypic data was performed by the software TBtools (Toolkit for Biologists integrating various big-data handling tools, http://cj-chen.github.io/tbtools/ (accessed on 5 September 2022)). Pearson’s correlation was performed for five fruit traits, including FW, LN, SSC, FF, and FS, by the Origin 2019 software (OriginLab company, Northampton, MA, USA). Histograms and heatmaps were mainly generated with Origin 2019 software. p-values were analyzed for significance by one-way ANOVA and the LSD test in Origin 2019.
2.4. Whole Genome Resequencing, Sequence Alignment, and Genotype Calling
Total DNA extraction was performed using the leaf tissues of approximately 4.5-month-old plants from each of the accessions. First, 848 paired-end sequencing libraries with an insert size of approximately 300 bp were constructed for the project. After sample QC, the qualified DNA samples were randomly fragmented by Covaris, and the fragments were collected using magnetic beads. Adenine was added to end-repaired DNA fragments before adaptor ligation. The fragments were then bridge amplified, and clusters were generated. Sequencing of these DNA libraries was performed using the Illumina HiSeq 4000 sequencing platform.
First, the reads with a high proportion of adaptors and unknown or low-quality reads in the raw data were removed. Here, we used the BGI in-house filter SOAPnuke to eliminate unwanted reads and bases. Clean data were yielded after three-step raw data filtration. Next, BWA was used to align filtered reads to the reference sequence (SL3.0) [36,37]. SNP and indel detection processes were performed with GATK (version 3.36) [38]. We used an analysis tool developed by BGI to perform SNP annotation and classification. The relevant steps and parameters were as follows:
- 1.
- Three-step raw data filtration:
- (1)
- Adaptor trimming. Any sequencing read with adaptor mapping rate higher than 50% is removed.
- (2)
- Low-quality reads trimming. Any sequencing read which consists of more than 50% of low-quality bases (Q20 < 50%) is removed.
- (3)
- Contiguous N bases trimming. Any sequencing read with over 2% of N base read is removed.
- 2.
- The steps of SNP and indel calling:
- (1)
- MarkDuplicates (Picard) was used for duplication trimming;
- (2)
- GATK was used for indel realignment to avoid calling errors caused by indels;
- (3)
- GATK was used for base recalibration;
- (4)
- GATK was used for variant, SNP, and indel calling;
- (5)
- The data were corrected. The filtration parameters for SNP calling: “QD < 2.0, FS > 60.0, MQ < 40.0, MQRankSum < −12.5, ReadPosRankSum < −8.0”. The filtration parameters for indel calling: “QD < 2.0, FS > 200.0, ReadPosRankSum < −20.0”.
2.5. Population Structure Analysis and Linkage Disequilibrium
The population structure was analyzed using the method of maximum likelihood method in Admixture 3.0 software [39]. Before using Admixture, we used Plink to obtain the required data files. The input parameter K varied from 2 to 10, which represented the assumed groups of simulated populations in ancient times. The source of each sample can be judged by the Bayesian clustering method. For each value of K, we set the burn-in to 1,000,000 and ran 20 repeats using different random seeds, and reported the lowest cross-validation score for each repeat. The cross-validation errors of each result were compared; finally, the appropriate K was selected as the optimal group stratification value for the GWAS. A neighbor-joining (NJ) tree and a principal component analysis (PCA) plot were used to infer the population structure. The phylogenetic tree was constructed using TreeBest software [40,41]. The genetic relatedness between individuals was constructed, and the PCA was plotted using the GAPIT tool [42].
For linkage disequilibrium analysis, Haploview software [43] (version 4.2) was used to calculate the correlation coefficients (r2) of different alleles. We calculated the mean r2 value in each length range and plotted the relationship between r2 values and paired SNP distances using the R language for the whole population.
2.6. Population SNP Filtering and Genotype Filling
Based on the results of data alignment to the reference genome (SL3.0), GATK software was used to identify the various sample genotypes. Next, the genotype differences between the samples were combined and integrated to produce a population SNP. Beagle software was used to predict and fill the site of genotype deletion in each sample, and the results were used for subsequent analysis. The filtering conditions to identify population SNPs were as follows:
- (1)
- The genotype of each sample had a quality ≥ 20;
- (2)
- The number of unique reads supported by each genotype was greater than 2;
- (3)
- The genotype copy number of each sample was >1.5;
- (4)
- Each population SNP site was biallelic (including only two alleles). The missing rate of each population SNP site was >0.4.
2.7. Genome-Wide Association Analysis
Based on the genotype dataset generated after the imputation of the missing genotypes, association analysis was performed using the general linear model (GLM) and mixed linear model (MLM) algorithms in Tassel 5.0 software [44]. The Manhattan and Q-Q plots were plotted in R language.
In the GLM analysis, the equation used was as follows:
Y = Xα + e
In the MLM analysis, the equation used was as follows:
where y is the phenotype, X is the genotype, P is the Q matrix of the results from the population structure analysis, and K represents the matrix of the relative kinship. Xα and Pβ are fixed effects, and Kμ and e are random effects. The P matrix was built via the top five principal components to correct the population structure. The K matrix was built via the simple matching coefficients matrix and then was compressed.
y = Xα + Pβ + Kμ + e.
3. Results
3.1. Phenotypic Variation and Correlation Analysis
The agricultural morphological characterization of germplasm provides essential information for crop breeding. In this study, the phenotypic values of all traits except for green shoulders and gray leaf spot resistance were approximately normally distributed (Supplementary Materials Figure S1). Phenotypic variation of the traits among accessions was characterized by the mean, standard deviation, range, and coefficient of variation (Table 2). The smallest and largest phenotypic variations were recorded in the first ripening stage (4.72%) and fruit weight (35.04%), respectively. According to the cluster analysis of the phenotypic data, the composition of the 212 accessions could be clearly indicated: cherry type with high soluble sugar content and small fruit weight, Roman type with large fruit shape, yellowish large-fruited tomato with large first ripening stage and fruit firmness, and non-yellowish large-fruited tomato (Figure 1A). The interrelationship among fruit-related traits was complex (Figure 1B). Especially for some antagonistic traits (such as quality and yield), balancing these relationships is critical to obtain varieties with comprehensive trait improvements. In summary, these data revealed extensive variation in the traits, suggesting the suitability of the genotypic panel for association analysis.
Table 2.
Basic statistics of the phenotypic variations observed for 7 quantitative traits.
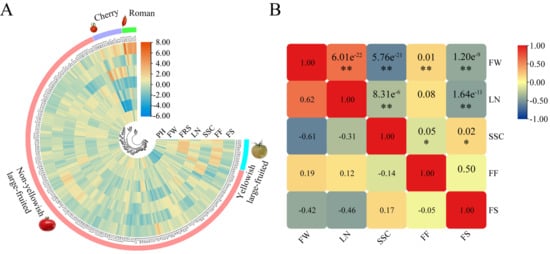
Figure 1.
Cluster analysis of 212 accessions based on phenotypic data and correlation analysis among fruit-related traits. (A) Cluster analysis. To reduce the range of fluctuation of the phenotypic value of each trait between different accessions, the phenotypic value was Log2 (Xi/X) normalized, where Xi represents the phenotypic value of each trait, and X represents the mean phenotypic value of the corresponding trait. The phenotypic levels were color-coded according to the normalized color scale. The following related parameters were used in clustering: Dist method—Euclidean; Cluster method—Average; Branch form—Equal. (B) Correlation analysis among fruit-related traits. The numbers in the top right box represent the p values and significance, and the numbers in the bottom left box represent the correlation coefficients. The correlation levels were color-coded according to the color scale in the top right corner. The symbols * and ** indicate significance at the 0.05 and 0.01 probability levels, respectively.
3.2. Resequencing and SNP Marker Statistics
To explore the genetic basis of phenotypic variation, the 212 accessions were resequenced at an average depth of 5×. The average coverage of the reference genome was above 84%, and the mapping rate of the samples varied from 97.69% to 99.85% (Supplementary Materials Table S2). In total, we obtained 2021.59 Gbp of clean data (Q20 ≥ 95.06%) after a three-step raw data filtration (Supplementary Materials Table S3).
After filtering out low-quality reads, 24,428,210 SNPs and indels (including 21,821,893 SNPs and 2,628,210 indels) were selected (Figure 2A). Among these variants, the fewest SNPs (518,799) were located on chromosome 2, and the largest number of SNPs were located on chromosome 9 (3,347,461). Among the identified SNPs, 2,701,235 SNPs (12.38%) were located in the genic region. Of them, a total of 408,807 SNPs (1.91%) were located in the CDS, including 237,015 non-synonymous and 179,861 synonymous mutations (Figure 2A). The most and fewest non-synonymous/synonymous mutations were found on chromosomes 1 and 11, respectively (Figure 2B). In the genome, the ratio (non-synonymous/synonymous) was 1.32 on average. Higher ratios were obtained for chromosomes 11, 5, and 9: 1.54, 1.52, and 1.51, respectively (Figure 2B). This suggests that amino acids were significantly altered. The distribution of so-called large-effect SNPs (SNPs representing potentially disabling gene functions) was further analyzed (Figure 2A). A total of 7720 SNPs were involved in the premature termination of codons, 3951 SNPs disrupted a splice donor or acceptor site in the genome, 8085 SNPs were related to a change in the initial methionine residue, and 1276 SNPs replaced the terminator with certain amino acid residues, resulting in a longer ORF.
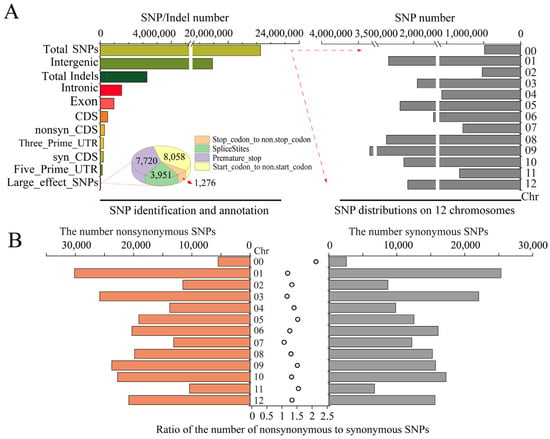
Figure 2.
SNP identification and distribution across the genome. (A) SNP identification and annotation across the genome. (B) The statistics of the non-synonymous and synonymous mutations on each chromosome.
3.3. Analyses of Population Structure and Linkage Disequilibrium
To truly reflect the genetic heterogeneity and evolutionary relationships among accessions, we ran Admixture v3.0 software with K values from 2 to 10 based on the previous population SNP dataset obtained by population sequencing. We clearly observed that the population could be divided into three different genetic groups when K = 3 (Figure 3B,C). Each group comprised cherry tomatoes, Roman tomatoes, and non-yellowish and yellowish large-fruited tomatoes, while cherry, Roman, and large-fruited tomatoes that formed clusters were not identified (Figure 3A). These results were consistent with those of PCA (Figure 3D). The results of the kinship analysis also indicated that genetic materials derived from germplasms of different genetic backgrounds infiltrated each other in the breeding process, resulting in the narrowing of the genetics of the offspring and the complexity of kinship (Figure 3E). Among the 212 accessions, 112 were clustered in group Ⅰ, 82 were scattered in group Ⅱ, and the remainder were located within group Ⅲ. Except for L115, the largest group Ⅰ could also be divided into two subgroups, I-1 (57 accessions) and I-2 (54 accessions). Therein, it contains a large number of yellowish late-ripening tomatoes: L38/L153/L155/L157/L158/L165/L166 (I-1) and L201/L205 (I-2); L186/L81/L80 (I-1) and L175/L177/L76 (I-2) are cherry tomatoes; and L179/L50 (I-1) and L174/L181/L6 (I-2) are Roman tomatoes. In group Ⅱ, L83, L185, L203, and L188 are cherry tomatoes; L172 and L51 are Roman tomatoes; and L116, L154, and L204 are yellowish large-fruited tomatoes. In the smallest group Ⅲ, L170 are cherry tomatoes; L169 and L173 are Roman tomatoes; and L156 are yellowish large-fruited tomatoes. The most yellowish late-maturing cultivars existed in group I-1, which may be related to the excessive selection of late-maturing-related genes for yield and fruit firmness. The measured LD level is the chromosome distance when the LD coefficient is reduced to half the maximum value. Within 10 kb, a decay distance of 6.2 kb for the LD coefficient was observed in the population (Figure 3F). This further indicated that the LD decay distance of the population was relatively larger, likely because of the decline in the genetic diversity of the population caused by the continuous selection of some preferred traits in the artificial crossing process.
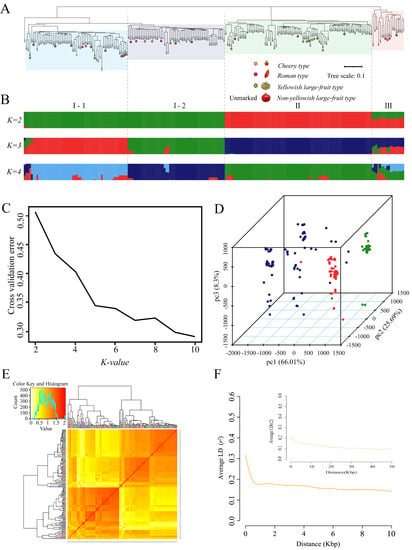
Figure 3.
Inferred population structure and analyses of linkage disequilibrium in the 212 tomato accessions. (A) Neighbor-joining tree. (B) Population structure based on K = 2–4 using Admixture. In the panel, each individual is indicated with a vertical bar partitioned into different colored segments, whose respective lengths represent the proportion of the individual’s genome in a given group. (C) Estimated cross-validation error of possible clusters (K) from 2 to 10. (D) PCA of the 212 tomato accessions. One dot represents each individual. (E) Heatmap of the pairwise kinship matrix of 212 genotypes. (F) Analyses of linkage disequilibrium within the 10 Kbp and 50 Kbp for 212 cultivated tomato accessions using the SNP data.
3.4. Genome-Wide Association Studies of Nine Agronomic Traits
After obtaining population analysis, the phenotypic data for nine agronomic traits were called. We performed GWAS via the GLM and MLM algorithms using the high-confidence SNP dataset obtained from the sequencing-based genotype dataset to uncover the most significant marker–trait associations. Next, the Q-Q and Manhattan plots were evaluated for evidence of p value inflation. The MLM approach, which considered genome-wide patterns of genetic relatedness, substantially reduced false positives, as shown in the Q-Q plots (Figure 4 and Figures S2–S8). After multiple tests using standard methods, we selected a high threshold of −log10 (P) > 8 as a parameter to avoid excessive false positives and false negatives, and obtain more significant associated loci. A total of 28 significant signals related to nine agronomic traits of interest were trapped via the MLM based on a significance threshold of −log10 (P) > 8 significant thresholds (Table 3). Although we also identified many associated signals via the GLM using the same threshold criteria, all were discarded because they were complex and likely had too many false positives. Finally, we focused on 28 significant association signals of the nine agronomic traits from the MLM. These signal regions contained a total of 114 genes, explaining approximately 16% (from 3% to 66% for different traits) of the observed phenotypic variance on average (Supplementary Materials Table S4).
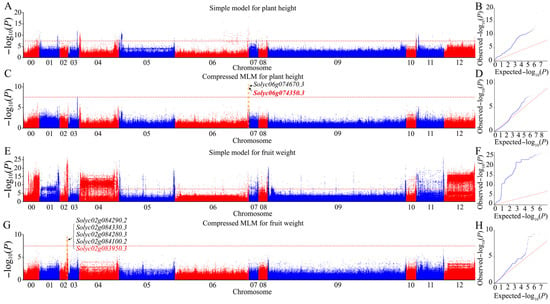
Figure 4.
Genome-wide association studies of plant height and fruit weight. (A,B) Manhattan and Q-Q plots of the simple model for plant height. (C,D) Manhattan and Q-Q plots of the compressed MLM for plant height. (E,F) Manhattan and Q-Q plots of the simple model for fruit weight. (G,H) Manhattan and Q-Q plots of the compressed MLM for fruit weight. Chr 00 represents unanchored scaffolds. The red horizontal dashed line indicates the genome-wide significance threshold. Gene numbers in red represent the known genes in the peak region, while gene numbers in black represent the top four unknown genes with a large phenotypic interpretation rate detected in the region. The yellow dotted line indicates the peak region where the labeled gene was located.
Table 3.
Genome-wide significant association signals of 9 agronomic traits from the compressed mixed linear model (MLM).
Plant height revealed associations with two genes located on chromosome 6, explaining 23.5–24.1% of the phenotypic variance (Figure 4C and Supplementary Materials Table S4). A novel candidate gene, solyc06g074670.3, encoding UDP-apiose/xylose synthase, was detected in the region of the known gene solyc06g074350.3 (self-pruning/SP) [45]. Fruit weight showed 15 associated genes on chromosome 2, explaining approximately 7.9% (5.5–13%) of the phenotypic variance on average (Figure 4G and Supplementary Materials Table S4). Within this region, in addition to the known gene solyc02g083950.3 (wuschel) [46], the protein kinase superfamily protein-encoding gene solyc02g084290.2 may also play an important role in phenotypic variance in this study. For the green shoulder, the 16 genes originating from two regions on chromosome 10 explained, on average, 18.9% (8.9–25.2%) of the phenotypic variance (Supplementary Materials Figure S2C and Table S4). Outside the region of the known gene solyc10g008160.3 (U) [47], a novel candidate gene solyc10g007158.1, encoding the transcription factor GTE4, was examined.
The 11 associated genes related to locule number on chromosome 2 explained 4.5–12.7% (average of 7.7%) of phenotypic variation (Supplementary Materials Figure S3C and Table S4). We found that these genes and the regions in which they were located were consistent with the fruit weight. It also indicates that the locule number was a major factor causing the change in fruit weight. Regarding the soluble sugar content, a total of 22 genes were identified on chromosomes 1, 9, and 11, explaining approximately 11.2% (7.8–16.9%) of the phenotypic variance on average (Supplementary Materials Figure S4C and Table S4). The 14 genes originating from two regions of chromosome 1 explained, on average, 11.8% of the phenotypic variance. Outside the region of the known gene solyc01g109790.3 (AgpL1) [48], a novel candidate gene, solyc10g007158.1, putatively encoding an HXXXD-type acyl-transferase family protein, was examined. The two genes on chromosome 9 explained, on average, 11% of the phenotypic variance. The six genes on chromosome 11 explained, on average, 10.7% of the phenotypic variance. Interestingly, the gene solyc11g071810.2 (fasciated), which is associated with increased fruit size due to an increase in locules [49], may also have played an important role in the phenotypic variance in this study. Four associated genes related to fruit firmness located in two regions of chromosome 5 explained 6.4–11.8% of the phenotypic variation, with an average of 8.6% (Supplementary Materials Figure S5C and Table S4). Of these genes, solyc05g011830.3 may also have played an important role in the phenotypic variance in this study.
Regarding fruit shape, 16 association genes on chromosomes 1, 2, 3, 9, and 11 explained approximately 15.7% of the phenotypic variance on average (Supplementary Materials Figure S6C and Table S4). Two genes on chromosome 1 explained, on average, 12.8% of the phenotypic variance. Five genes on chromosome 2 explained, on average, 10.7% of the phenotypic variance. Of them, solyc02g083950.3 (wuschel) [46] plays an important role in phenotypic variance. Five genes on chromosome 3 explained, on average, 28% of the phenotypic variance. A novel candidate gene solyc01g107080.3, encoding a Myb transcription factor, may also have played an important role in phenotypic variance in this study. Three genes on chromosome 9 explained, on average, 12.1% of the phenotypic variance. A novel candidate gene solyc11g032160.1 on chromosome 11, putatively encoding Gamma-irradiation and mitomycin c induced 1, may also have played an important role in phenotypic variance. The first ripening stage revealed associations with 18 genes located on chromosomes 1, 3, 5, and 12, explaining approximately 36.3% (5–66.2%) of the phenotypic variance on average (Supplementary Materials Figure S7C and Table S4). A novel candidate gene solyc01g094550.3 on chromosome 1, putatively encoding acyl-CoA thioesterase, may also have played an important role in phenotypic variance. Two genes on chromosome 3 explained, on average, 5.1% of the phenotypic variance. A gene on chromosome 5, encoding an MYB transcription factor, explained, on average, 7.1% of the phenotypic variance. Fourteen genes in seven regions of chromosome 12 explained 8.8–66.2% of the phenotypic variation, with an average of 40.7%. The genes solyc12g049100.2, solyc12g049160.1, and solyc12g049300.2 may also have played an important role in the phenotypic variance. Regarding gray leaf spot resistance, the 10 genes originating from two regions on chromosome 11 explained approximately 11.2% (4.3–17%) of the phenotypic variance on average (Supplementary Materials Figure S8C and Table S4). Two genes encoded disease resistance proteins, solyc11g020080.2 and solyc11g020100.2. The gene solyc11g018660.2, putatively encoding NAC domain-containing protein, may have played an important role in the phenotypic variance in this study. Here, we revalidated known locus and identified novel candidate locus, the latter of which will be attractive candidates for follow-up studies to advance our understanding of the genetic architecture of these traits.
4. Discussion
As shown in this study, there are obvious differences and complex correlations among phenotypes, and even the group division of phenotypes does not truly reflect the interindividual heterogeneity in the population structure. Although heterosis depends on the genetic nature, diversity, and heterogeneity of the parent [6], phenotypic heterogeneity complementarity is the principle applied in traditional hybrid breeding. This may be one of the factors contributing to the low efficiency and high random chance of traditional crossbreeding in production practice [50]. The ambiguity of the genetic background limit better breed improvement. Studies in maize have indicated that the magnitude of heterosis is correlated with the genetic distance among the parental inbred lines [51], and that intergroup hybrids from different heterotic groups are more vigorous [1]. For maize, the genetically different heterosis groups can be classified by genotype. The division of heterotic groups has significantly improved the breeding efficiencies in maize, rice, and other crops [52,53]. Although a few studies are being attempted in this field [54,55], representational heterotic groups characterized at the genetic and phenotypic levels have not been established in tomatoes. This topic will be the focus of future research emphasis to accelerate the breeding progress. Overall, the work reduces the blindness reduced in selecting cross-parents based solely on phenotype, and provides a basis for the classification and arrangement of germplasm resources, construction of dominant populations, and germplasm amplification and improvement.
Analyses of population structure and the genetic nature of traits help to accelerate the breeding process. So far, GWAS has become a recognized strategy to decode genotype–phenotype associations in species [19,20]. The main prerequisites for its success are the population size, differences in sample abundance, and marker density [10,29,30]. Hence, the establishment and adoption of large-scale heterotic groups may be more conducive to the discovery of significant loci regulating important agronomic traits. Compared with previous studies [56,57,58], a collection of 212 tomato accessions was used in this study, and our relatively large sample collection comprised only modern accessions, excluding types such as heirlooms and wild accessions. Although these conditions reduce the genetic diversity of the set, given the population size and wide variability among sample traits (Table 2), we believe that this collection was adequate for GWAS. A total of 28 significant signals related to nine agronomic traits of interest were recorded (Table 3). Of these, seven likely represent previously known loci for seven traits other than fruit firmness and the first ripening stage, respectively. These results demonstrate the feasibility and effectiveness of employing this collection to perform GWAS. The specific loci identified may also be related to the fact that large-effect QTLs regulating the corresponding traits are commonly stably selected in breeding and are easily detected in the population of modern cultivated tomatoes. We found that these known loci were not the most prominent in the association peak, which is consistent with previous findings that the peak signals of association loci often appeared near known genes [59]. For a known peak signal region of a related functional gene that has not been cloned, the true functional gene may be located near the peak. For instance, the previously reported gene solyc11g018743.1 plays an important role in the resistance to gray leaf spots [60], but we did not clone the resistance gene in the corresponding position, and in subsequent verification, the gene solyc11g020100.2 encoding the resistance protein was found and cloned in the region near the peak signal of this gene [61]. This also suggests that GWAS is a preliminary and reliable approach for identifying the locations of QTLs, and this phenomenon may provide insight helping us to verify and clone target functional genes.
Differences in the genetic background of the collection, year, environment and method of phenotyping and the analysis model used may influence the intensity of associated peak signals and the extent to which the signal peak deviates from target genes and allow the identification of new QTLs for the traits under study [26]. In contrast to previous studies, 21 significant association signals were identified in genomic regions without previously known loci for six traits (except plant height, fruit weight, and locule number), suggesting the discovery of novel potential QTLs. For five of these signals, candidate genes related to fruit quality (soluble sugar content and fruit firmness) and resistance were found on chromosomes 1, 5, 9, and 11. There were higher rates of non-synonymous and synonymous mutations in coding regions on chromosomes 5, 9, and 11 during SNP detection (Figure 3), indicating that target genes distributed on these chromosomes may be retained during breeding due to the active selection of related traits. Previous breeding goals have usually focused on traits such as resistance, yield, and fruit firmness, which also significantly affect tomato flavor [3]. Hence, the detection of a large number of QTLs related to fruit quality (soluble sugar content and fruit firmness) and resistance on these chromosomes may be a result of such positive selection. For instance, the previously known fruit firmness-related genes were solyc10g080210 [62], solyc06g051800 [63], and solyc03g111690 [64]. Currently, we detected novel association signals on chromosome 5. Additionally, previous studies have shown that the gene AGPL1 (95773188-96773188) can increase the starch content of immature fruits and the soluble solid content of mature fruits [48]. In genomic regions without previously known loci on chromosome 1 (93670222-94670222), we found a novel gene solyc01g107080.3, which was predicted to encode an HXXXD-type acyl-transferase family protein. Studies have shown that genes encoding this type of protein play an important role in the corresponding various chilling effects [65]. We speculated that the gene may function in regulating the accumulation of soluble sugar contents. These candidate loci explained >10% of the phenotypic variation in the two traits (Table 3). Thus, these genes may play an important role in regulating the corresponding traits.
Although fruit weight is one of the main factors affecting yield, the fruit weights and soluble sugar contents were significantly negatively correlated (Figure 2B). Improvements in yield frequently result in quality penalties, and there is thus considered to be a trade-off between quality and yield. Therefore, the coordination of these traits is critical for the integration of cultivars with overall excellent traits [66]. Currently, very little is known about the intrinsic regulatory mechanism of this coordinated relationship. Although many main effect sites that regulate fruit weight have been identified, such as fw1.1, fw1.2, fw2.1, fw2.2, fw2.3, fw3.1, fw3.2, fw4.1, fw9.1, fw11.3, lc, fas, FAB, and FIN, only four of these sites have been cloned: fw2.2, fw3.2, lc, and fas [67]. In this study, we only detected the lc gene, likely because of the selected population composition and differences in how these genes regulate fruit development. The gene lc showed associations with fruit weight, fruit shape, and locule number. Because the present and previous studies have reported phenotypic correlations among tomato fruit traits [58,68], the identification of QTLs with pleiotropic effects is expected. Interestingly, we found that fas, regulating the locule number, could explain 10.3% of the phenotypic variation in soluble sugar contents. Given the phenotypic relationship between the two traits, this gene may be used as a negative regulator of soluble sugar content. However, determining how it functions will certainly require follow-up research. In addition, although the traits were correlated, we found no other multipotent QTLs among fruit weight, first ripening stage, soluble sugar content, and fruit firmness, possibly due to the small number of association signals for these traits in our study. This result may be due to a small number of association signals for these traits in our study. Overall, we reported 28 significant association signals for nine traits. Of these, 21 represent potential novel QTLs for six fruit traits. Although further validation evidence is lacking, the mining of numerous yield- and quality-related genes provides useful information and insight for high-yield and high-quality tomato breeding.
5. Conclusions
Collectively, this work provides a molecular theoretical basis for the targeted expansion and improvement of excellent cultivated tomato germplasm resources used to breed strong hybrids. This work also highlighted the mining of yield- and quality-related genes. The identification of numerous novel major loci controlling fruit firmness, shape, weight, soluble sugar content, and maturity provides useful information and insight for breeding and reveals further opportunities for in-depth studies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/agronomy13051191/s1, Figure S1: Frequency distribution of variation of nice traits in 212 cultivated tomato accessions.; Figure S2: Genome wide association studies for green shoulder. (A) Manhattan plots of the simple model. Chr00 represents unanchored scaffolds. Red horizontal dashed line indicates the genome-wide significance threshold. (B) Q-Q plot of the simple model. (C) Manhattan plots of compressed MLM, as in A. (D) Q-Q plot of compressed MLM. Gene number in red represent known gene in the peak region, while gene number in black represent top four unknown genes with a large phenotypic interpretation rate detected in the region. The yellow dotted line indicates the peak region where the labeled gene was located.; Figure S3: Genome wide association studies for locule number. (A) Manhattan plots of the simple model. Chr00 represents unanchored scaffolds. Red horizontal dashed line indicates the genome-wide significance threshold. (B) Q-Q plot of the simple model. (C) Manhattan plots of compressed MLM, as in A. (D) Q-Q plot of compressed MLM. Gene number in red represent known genes in the peak region, while gene number in black represent top four unknown genes with a large phenotypic interpretation rate detected in the region. The yellow dotted line indicates the peak region where the labeled gene was located.; Figure S4: Genome wide association studies for soluble sugar content. (A) Manhattan plots of the simple model. Chr00 represents unanchored scaffolds. Red horizontal dashed line indicates the genome-wide significance threshold. (B) Q-Q plot of the simple model. (C) Manhattan plots of compressed MLM, as in A. (D) Q-Q plot of compressed MLM. Gene numbers in red represent known genes in the peak region, while gene number in black represent top four unknown gene with a large phenotypic interpretation rate detected in the region. Gene number in blue font indicate gene known to have other functions. The yellow dotted line indicates the peak region where the labeled gene was located.; Figure S5: Genome wide association studies for fruit firmness. (A) Manhattan plots of the simple model. Chr00 represents unanchored scaffolds. Red horizontal dashed line indicates the genome-wide significance threshold. (B) Q-Q plot of the simple model. (C) Manhattan plots of compressed MLM, as in A. (D) Q-Q plot of compressed MLM. Gene number in red represent known gene in the peak region, while gene number in black represent top four unknown genes with a large phenotypic interpretation rate detected in the region. The yellow dotted line indicates the peak region where the labeled gene was located.; Figure S6: Genome wide association studies for fruit shape. (A) Manhattan plots of the simple model. Chr00 represents unanchored scaffolds. Red horizontal dashed line indicates the genome-wide significance threshold. (B) Q-Q plot of the simple model. (C) Manhattan plots of compressed MLM, as in A. (D) Q-Q plot of compressed MLM. Gene number in red represent known genes in the peak region, while gene number in black represent top four unknown genes with a large phenotypic interpretation rate detected in the region. The yellow dotted line indicates the peak region where the labeled gene was located.; Figure S7: Genome wide association studies for first ripening stage. (A) Manhattan plots of the simple model. Chr00 represents unanchored scaffolds. Red horizontal dashed line indicates the genome-wide significance threshold. (B) Q-Q plot of the simple model. (C) Manhattan plots of compressed MLM, as in A. (D) Q-Q plot of compressed MLM. Gene number in red represent known gene in the peak region, while gene number in black represent top four unknown genes with a large phenotypic interpretation rate detected in the region. The yellow dotted line indicates the peak region where the labeled gene was located.; Figure S8: Genome wide association studies for gray leaf spot. (A) Manhattan plots of the simple model. Chr00 represents unanchored scaffolds. Red horizontal dashed line indicates the genome-wide significance threshold. (B) Q-Q plot of the simple model. (C) Manhattan plots of compressed MLM, as in A. (D) Q-Q plot of compressed MLM. Gene number in red represent known gene in the peak region, while gene number in black represent top four unknown genes with a large phenotypic interpretation rate detected in the region. The yellow dotted line indicates the peak region where the labeled gene was located.; Table S1: List of tomato samples with diverse traits; Table S2: The resequencing depth and mapping rate of sample; Table S3: Data quality of clean data; Table S4: Associated loci and candidate genes according to gene annotation.
Author Contributions
Conceptualization, J.L. and J.J.; methodology, Z.L.; software, Z.L.; validation, Z.L. and J.J.; formal analysis, Z.L.; investigation, Z.L. and J.J.; resources, J.J.; writing—original draft preparation, Z.L.; writing—review and editing, Z.L.; visualization, Z.L.; supervision, J.J.; project administration, J.J.; All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (32072589) and the Heilongjiang Province key research and development plan (2022ZX02B07), and by grants from the National Natural Science Foundation of China (U22A20496), the China Agriculture Research System (CARS-23-A11), the National Natural Science Foundation of China (32002059), the Heilongjiang Natural Science Foundation of China (LH2020C10).
Data Availability Statement
The raw sequencing data of this article are stored in the NCBI Sequence Read Archive under ac-cession number SUB11874557.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hochholdinger, F.; Baldauf, J.A. Heterosis in plants. Curr. Biol. 2018, 28, 1089–1092. [Google Scholar] [CrossRef]
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Mata-Nicolás, E.; Montero-Pau, J.; Gimeno-Paez, E.; Garcia-Carpintero, V.; Ziarsolo, P.; Menda, N.; Mueller, L.A.; Blanca, J.; Cañizares, J.; van der Knaap, E.; et al. Exploiting the diversity of tomato: The development of a phenotypically and genetically detailed germplasm collection. Hortic. Res. 2020, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, S.; Gong, J.; Zhao, Q.; Feng, Q.; Zhan, Q.; Zhao, Y.; Li, W.; Cheng, B.; Xia, J.; et al. Genomic architecture of heterosis for yield traits in rice. Nature 2016, 537, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Gao, S.; Chen, X.; Lei, T.; Li, W.; Huang, Y.; Li, Y.; Jiang, M.; Hu, D.; Duan, Y.; et al. Genetic analysis of main flower characteristics in the F1 generation derived from intraspecific hybridization between Plumbago auriculata and Plumbago auriculata f alba. Sci. Hortic. 2020, 274, 109652. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, J.; Ren, A.; Xu, X.; Zhang, H.; Zhao, T.; Jiang, X.; Sun, Y.; Li, J.; Yang, H. Heterosis and combining ability analysis of fruit yield, early maturity, and quality in tomato. Agronomy 2021, 11, 807. [Google Scholar] [CrossRef]
- García-Martínez, S.; Andreani, L.; Garcia-Gusano, M.; Geuna, F.; Ruiz, J.J. Evaluation of amplified fragment length polymorphism and simple sequence repeats for tomato germplasm fingerprinting: Utility for grouping closely related traditional cultivars. Genome 2006, 49, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.; Yu, Q.; Zheng, Z.; Zhang, Z.; Lun, Y.; Li, S.; Wang, X.; et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 2014, 46, 1220–1226. [Google Scholar] [CrossRef]
- Blanca, J.; Montero-Pau, J.; Sauvage, C.; Bauchet, G.; Illa, E.; Díez, M.J.; Francis, D.; Causse, M.; van der Knaap, E.; Cañizares, J. Genomic variation in tomato, from wild ancestors to contemporary breeding accessions. BMC Genom. 2015, 16, 257. [Google Scholar] [CrossRef]
- Huang, X.; Han, B. Natural Variations and Genome-Wide Association Studies in Crop Plants. Annu. Rev. Plant Biol. 2014, 65, 531–551. [Google Scholar] [CrossRef]
- Thornsberry, J.M.; Goodman, M.M.; Doebley, J.; Kresovich, S.; Nielsen, D.; Buckler, E.S. Dwarf 8 polymorphisms associate with variation in flowering time. Nat. Genet. 2001, 28, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Myles, S.; Boyko, A.R.; Owens, C.L.; Brown, P.J.; Grassi, F.; Aradhya, M.K.; Prins, B.; Reynolds, A.; Chia, J.-M.; Ware, D.; et al. Genetic structure and domestication history of the grape. Proc. Natl. Acad. Sci. USA 2011, 108, 3530–3535. [Google Scholar] [CrossRef]
- Hufford, M.B.; Xu, X.; van Heerwaarden, J.; Pyhäjärvi, T.; Chia, J.-M.; Cartwright, R.A.; Elshire, R.J.; Glaubitz, J.C.; Guill, K.E.; Kaeppler, S.M.; et al. Comparative population genomics of maize domestication and improvement. Nat. Genet. 2012, 44, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Liu, X.; Shen, D.; Miao, H.; Xie, B.; Li, X.; Zeng, P.; Wang, S.; Shang, Y.; Gu, X.; et al. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat. Genet. 2013, 45, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef]
- Meyer, R.S.; Choi, J.Y.; Sanches, M.; Plessis, A.; Flowers, J.M.; Amas, J.; Dorph, K.; Barretto, A.; Gross, B.; Fuller, D.Q.; et al. Domestication history and geographical adaptation inferred from a SNP map of African rice. Nat. Genet. 2016, 48, 1083–1088. [Google Scholar] [CrossRef]
- Cheng, F.; Sun, R.; Hou, X.; Zheng, H.; Zhang, F.; Zhang, Y.; Liu, B.; Liang, J.; Zhuang, M.; Liu, Y.; et al. Subgenome parallel selection is associated with morphotype diversification and convergent crop domestication in Brassica rapa and Brassica oleracea. Nat. Genet. 2016, 48, 1218–1224. [Google Scholar] [CrossRef]
- Li, Y.; Cao, K.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Zhao, P.; Guo, J.; Ding, T.; Guan, L.; et al. Genomic analyses of an extensive collection of wild and cultivated accessions provide new insights into peach breeding history. Genome Biol. 2019, 20, 36. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, C.; Liu, X.; Guo, Z.; Xu, X.; Wang, S.; Xie, C.; Li, W.-X.; Zou, C.; Xu, Y. Development of a multiple-hybrid population for genome-wide association studies: Theoretical consideration and genetic mapping of flowering traits in maize. Sci. Rep. 2017, 7, 40239. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Mol. Plant 2017, 10, 359–374. [Google Scholar] [CrossRef]
- Upadhyaya, H.D.; Wang, Y.-H.; Sastry, D.V.; Dwivedi, S.L.; Prasad, P.V.; Burrell, A.M.; Klein, R.R.; Morris, G.P.; Klein, P.E. Association mapping of germinability and seedling vigor in sorghum under controlled low-temperature conditions. Genome 2015, 59, 137–145. [Google Scholar] [CrossRef]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.-C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.; Bradbury, P.J.; Liu, X.; Lu, F.; Romay, C.M.; Glaubitz, J.C.; Wu, X.; Peng, B.; Shi, Y.; et al. Identification of genetic variants associated with maize flowering time using an extremely large multi-genetic background population. Plant J. 2016, 86, 391–402. [Google Scholar] [CrossRef]
- Maurer, A.; Draba, V.; Pillen, K. Genomic dissection of plant development and its impact on thousand grain weight in barley through nested association mapping. J. Exp. Bot. 2016, 67, 2507–2518. [Google Scholar] [CrossRef]
- Ye, J.; Wang, X.; Wang, W.; Yu, H.; Ai, G.; Li, C.; Sun, P.; Wang, X.; Li, H.; Ouyang, B.; et al. Genome-wide association study reveals the genetic architecture of 27 agronomic traits in tomato. Plant Physiol. 2021, 186, 2078–2092. [Google Scholar] [CrossRef]
- Kim, M.; Nguyen, T.T.P.; Ahn, J.-H.; Kim, G.-J.; Sim, S.-C. Genome-wide association study identifies QTL for eight fruit traits in cultivated tomato (Solanum lycopersicum L.). Hortic. Res. 2021, 8, 203. [Google Scholar] [CrossRef]
- Ranc, N.; Muños, S.; Xu, J.; Le Paslier, M.-C.; Chauveau, A.; Bounon, R.; Rolland, S.; Bouchet, J.-P.; Brunel, D.; Causse, M. Genome-wide association mapping in tomato (Solanum lycopersicum) is possible using genome admixture of Solanum lycopersicum var. cerasiforme. G3 Genes|Genomes|Genetics 2012, 2, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ranc, N.; Muños, S.; Rolland, S.; Bouchet, J.-P.; Desplat, N.; Le Paslier, M.-C.; Liang, Y.; Brunel, D.; Causse, M. Phenotypic diversity and association mapping for fruit quality traits in cultivated tomato and related species. Theor. Appl. Genet. 2013, 126, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, V.; Francese, G.; Sacco, A.; D’Alessandro, A.; Rigano, M.M.; Parisi, M.; Milone, M.; Cardi, T.; Mennella, G.; Barone, A. An association mapping approach to identify favourable alleles for tomato fruit quality breeding. BMC Plant Biol. 2014, 14, 337. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, C.; Segura, V.; Bauchet, G.; Stevens, R.; Do, P.T.; Nikoloski, Z.; Fernie, A.R.; Causse, M. Genome-wide association in tomato reveals 44 candidate loci for fruit metabolic traits. Plant Physiol. 2014, 165, 1120–1132. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, J.; Xu, Y.; Liang, J.; Chang, P.; Yan, F.; Li, M.; Liang, Y.; Zou, Z. Genome-wide association mapping for tomato volatiles positively contributing to tomato flavor. Front. Plant Sci. 2015, 6, 1042. [Google Scholar] [CrossRef]
- Burgos, E.; De Luca, M.B.; Diouf, I.; Haro, L.A.; Albert, E.; Sauvage, C.; Tao, Z.J.; Bermudez, L.; Asís, R.; Nesi, A.N.; et al. Validated MAGIC and GWAS population mapping reveals the link between vitamin E content and natural variation in chorismate metabolism in tomato. Plant J. 2020, 105, 15077. [Google Scholar] [CrossRef]
- Beló, A.; Zheng, P.; Luck, S.; Shen, B.; Meyer, D.J.; Li, B.; Tingey, S.; Rafalski, A. Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize. Mol. Genet. Genom. 2008, 279, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Brown, M.A.; McCarthy, M.I.; Yang, J. Five Years of GWAS Discovery. Am. J. Hum. Genet. 2012, 90, 7–24. [Google Scholar] [CrossRef]
- Maher, B. Personal genomes: The case of the missing heritability. Nature 2008, 456, 18–21. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Han, B.; Huang, X. Sequencing-based genome-wide association study in rice. Curr. Opin. Plant Biol. 2013, 16, 133–138. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Pnueli, L.; Carmel-Goren, L.; Hareven, D.; Gutfinger, T.; Alvarez, J.; Ganal, M.; Zamir, D.; Lifschitz, E. The SELF-PRUNING gene of tomato regulates vegetative to reproductive switching of sympodial meristems and is the ortholog of CEN and TFL. Development 1998, 125, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Muños, S.; Ranc, N.; Botton, E.; Bérard, A.; Rolland, S.; Duffé, P.; Carretero, Y.; Le Paslier, M.-C.; Delalande, C.; Bouzayen, M.; et al. Increase in tomato locule number is controlled by two single-nucleotide polymorphisms located near WUSCHEL. Plant Physiol. 2011, 156, 2244–2254. [Google Scholar] [CrossRef]
- Powell, A.L.T.; Nguyen, C.V.; Hill, T.; Cheng, K.L.; Figueroa-Balderas, R.; Aktas, H.; Ashrafi, H.; Pons, C.; Fernández-Muñoz, R.; Vicente, A.; et al. Uniform ripening encodes a Golden 2-like transcription factor regulating tomato fruit chloroplast development. Science 2012, 336, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Petreikov, M.; Shen, S.; Yeselson, Y.; Levin, I.; Bar, M.; Schaffer, A.A. Temporally extended gene expression of the ADP-Glc pyrophosphorylase large subunit (AgpL1) leads to increased enzyme activity in developing tomato fruit. Planta 2006, 224, 1465–1479. [Google Scholar] [CrossRef]
- Cong, B.; Barrero, L.S.; Tanksley, S.D. Regulatory change in YABBY-like transcription factor led to evolution of extreme fruit size during tomato domestication. Nat. Genet. 2008, 40, 800–804. [Google Scholar] [CrossRef]
- Li, J.F. (Ed.) Chinese Tomato Breeding; China Agriculture Publishing House: Beijing, China, 2011; Volume 140, pp. 125–127. [Google Scholar]
- Moll, R.H.; Lonnquist, J.H.; Fortuno, J.V.; Johnson, E.C. The relationship of heterosis and genetic divergence in maize. Genet. 1965, 52, 139–144. [Google Scholar] [CrossRef]
- Xie, F.; He, Z.; Esguerra, M.Q.; Qiu, F.; Ramanathan, V. Determination of heterotic groups for tropical Indica hybrid rice germplasm. Theor. Appl. Genet. 2013, 127, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Suwarno, W.B.; Pixley, K.V.; Palacios-Rojas, N.; Kaeppler, S.M.; Babu, R. Formation of Heterotic Groups and Understanding Genetic Effects in a Provitamin A Biofortified Maize Breeding Program. Crop Sci. 2014, 54, 14–24. [Google Scholar] [CrossRef]
- He, L.S. Study on division of processing tomato geterosis group and utilization of heterosis models. North. Hortic. 2012, 18, 25–27. [Google Scholar]
- Jin, L.; Zhao, L.P.; Wang, Y.L.; Xu, L.P.; Zhou, R.; Song, L.X. Combining ability and division of heterosis group in tomato. Jiangsu J. Agric. Sci. 2019, 35, 667–675. [Google Scholar]
- Ranc, N.; Muños, S.; Santoni, S.; Causse, M. A clarified position for solanum lycopersicum var. cerasiformein the evolutionary history of tomatoes (solanaceae). BMC Plant Biol. 2008, 8, 130. [Google Scholar] [CrossRef]
- Albert, E.; Segura, V.; Gricourt, J.; Bonnefoi, J.; Derivot, L.; Causse, M. Association mapping reveals the genetic architecture of tomato response to water deficit: Focus on major fruit quality traits. J. Exp. Bot. 2016, 22, 6413–6430. [Google Scholar] [CrossRef]
- Phan, M.A.T.; Bucknall, M.P.; Arcot, J. Co-ingestion of red cabbage with cherry tomato enhances digestive bioaccessibility of anthocyanins but decreases carotenoid bioaccessibility after simulated in vitro gastro-intestinal digestion. Food Chem. 2019, 289, 125040. [Google Scholar] [CrossRef]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Su, X.; Zhu, G.; Huang, Z.; Wang, X.; Guo, Y.; Li, B.; Du, Y.; Yang, W.; Gao, J. Fine mapping and molecular marker development of the Sm gene conferring resistance to gray leaf spot (Stemphylium spp.) in tomato. Theor. Appl. Genet. 2018, 132, 871–882. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Jiang, J.; Liu, M.; Liu, Z.; Tan, Y.; Zhao, T.; Zhang, H.; Chen, X.; Li, J.; et al. The Sm gene conferring resistance to gray leaf spot disease encodes an NBS-LRR (nucleotide-binding site-leucine-rich repeat) plant resistance protein in tomato. Theor. Appl. Genet. 2022, 135, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Bird, C.R.; Smith, C.J.S.; Ray, J.A.; Moureau, P.; Bevan, M.W.; Bird, A.S.; Hughes, S.; Morris, P.; Grierson, D.; Schuch, W. The tomato polygalacturonase gene and ripening-specific expression in transgenic plants. Plant Mol. Biol. 1988, 11, 651–662. [Google Scholar] [CrossRef]
- Brummell, D.A.; Harpster, M.H.; Civello, P.M.; Palys, J.M.; Bennett, A.B.; Dunsmuir, P. Modification of expansin protein abundance in tomato fruit alters softening and cell wall polymer metabolism during ripening. Plant Cell 1999, 11, 2203–2216. [Google Scholar] [CrossRef] [PubMed]
- Uluisik, S.; Chapman, N.H.; Smith, R.; Poole, M.; Adams, G.; Gillis, R.B.; Besong, T.M.D.; Sheldon, J.; Stiegelmeyer, S.; Perez, L.; et al. Erratum: Corrigendum: Genetic improvement of tomato by targeted control of fruit softening. Nat. Biotechnol. 2016, 34, 950–952. [Google Scholar] [CrossRef]
- Hao, X.; Wang, B.; Wang, L.; Zeng, J.; Yang, Y.; Wang, X. Comprehensive transcriptome analysis reveals common and specific genes and pathways involved in cold acclimation and cold stress in tea plant leaves. Sci. Hortic. 2018, 240, 354–368. [Google Scholar] [CrossRef]
- He, G.M.; Deng, X.W. On the molecular basis of heterosis in plant: Opportunity and challenge. China Basic Sci. 2016, 18, 28–34, 64. [Google Scholar]
- Du, M.M.; Zhou, M.; Deng, L.; Li, C.Y.; Li, C.B. Current status and prospects on tomato molecular breeding—From gene cloning to cultivar improvement. Acta Hortic. Sin. 2017, 44, 581–600. [Google Scholar] [CrossRef]
- Hernández-Bautista, A.; Lobato-Ortiz, R.; Cruz-Izquierdo, S.; García-Zavala, J.J.; Chávez-Servia, J.L.; Hernández-Leal, E.; Bonilla-Barrientos, O. Fruit size QTLs affect in a major proportion the yield in tomato. Chil. J. Agric. Res. 2015, 75, 402–409. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).