
Grape Endophytic Microbial Community Structures and Berry Volatile Components Response to the Variation of Vineyard Sites

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Description and Sample Collection
2.2. Analysis of the Physicochemical Parameters of Berries
2.3. Volatile Compound Analysis Using HS-SPME/GC-MS
2.4. Sample Preparation for Microbial Community Analysis
2.5. DNA Extraction, Microbial Amplicon Sequence Analysis
2.6. Statistical Analyses
3. Results and Discussion
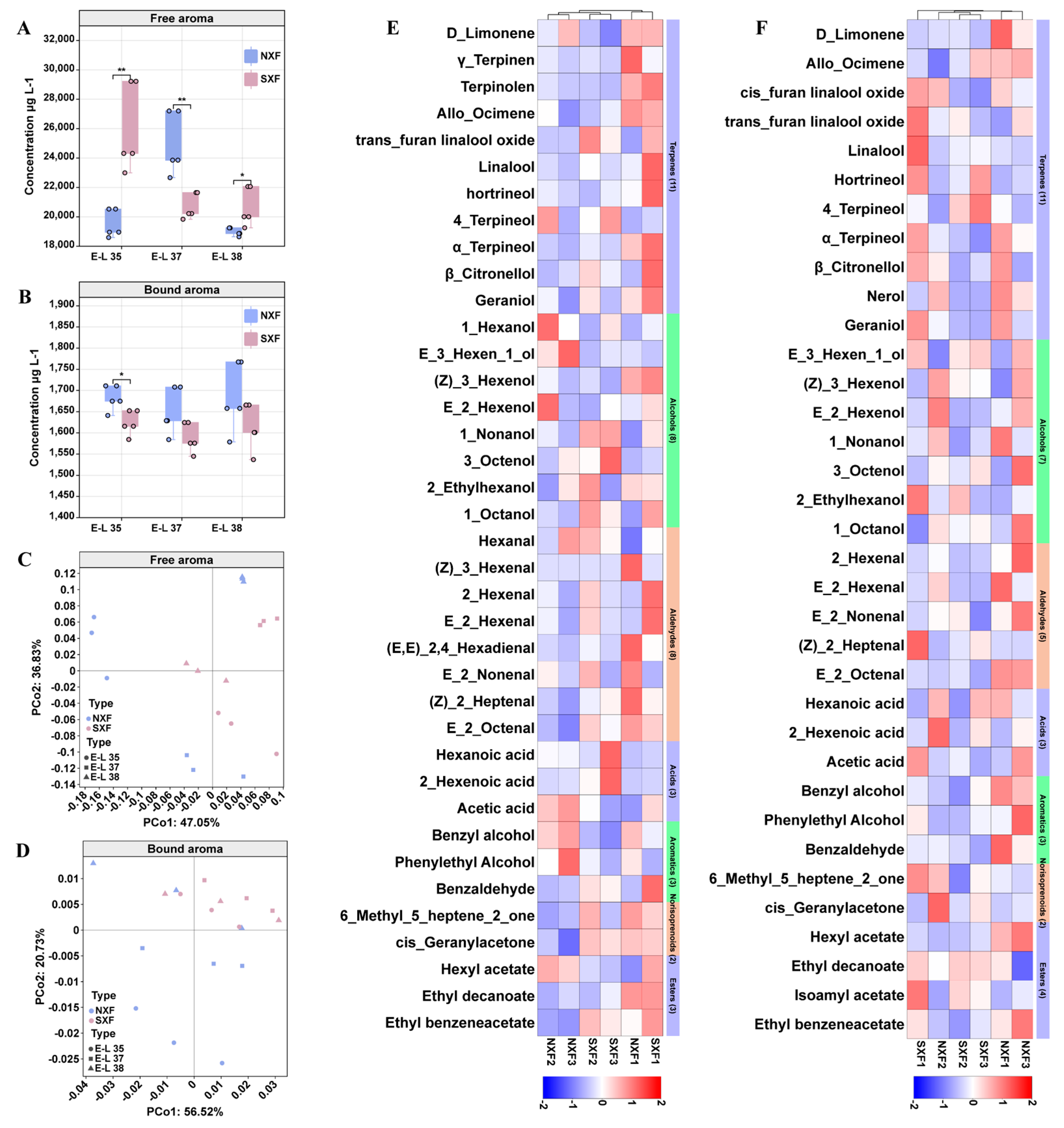
3.1. Comparison of Volatile Compounds during Grape Ripening
3.2. Comparison of Microbial Diversity during Grape Ripening
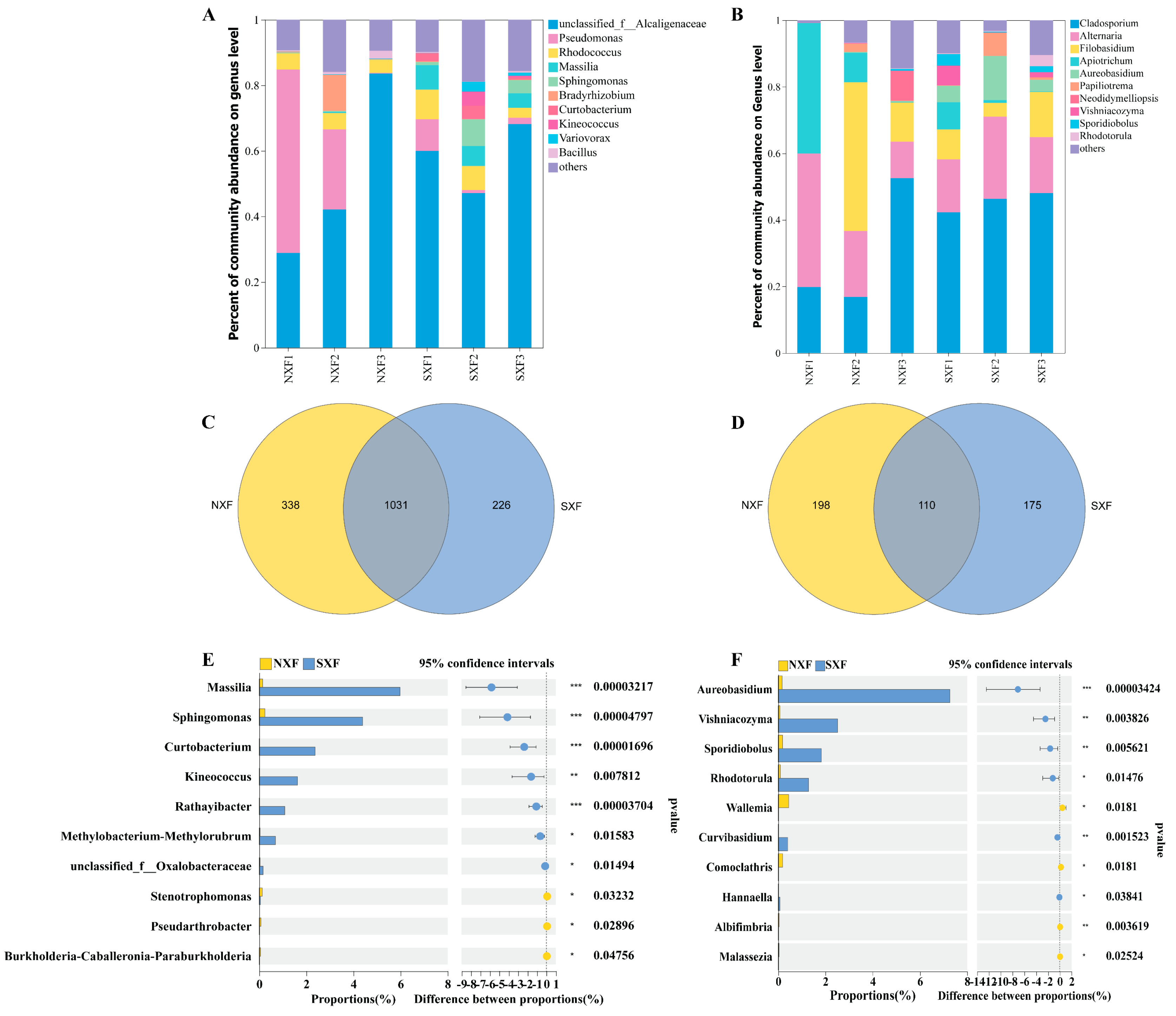
3.3. Comparison of Microbial Community Structure during Grape Ripening
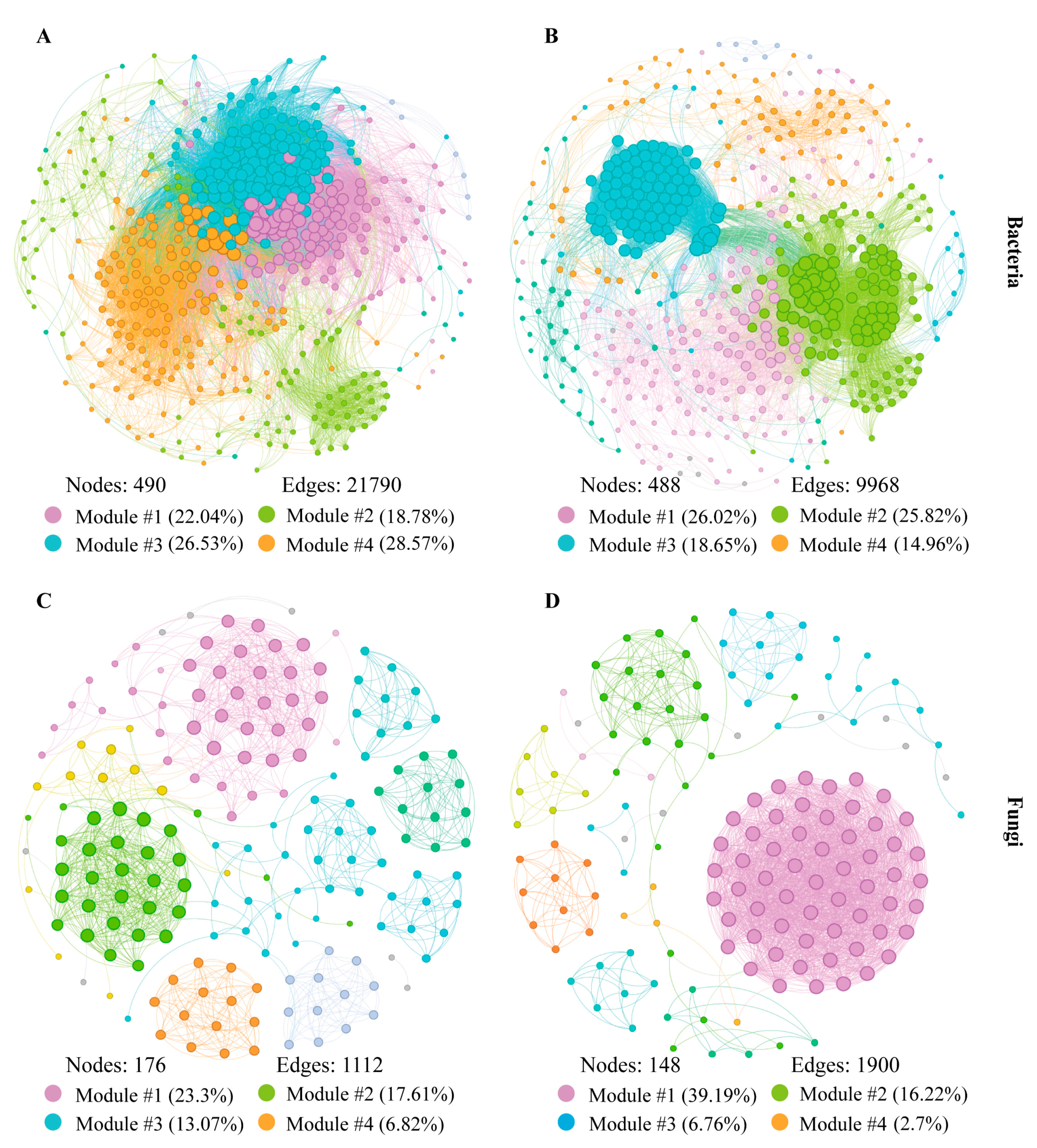
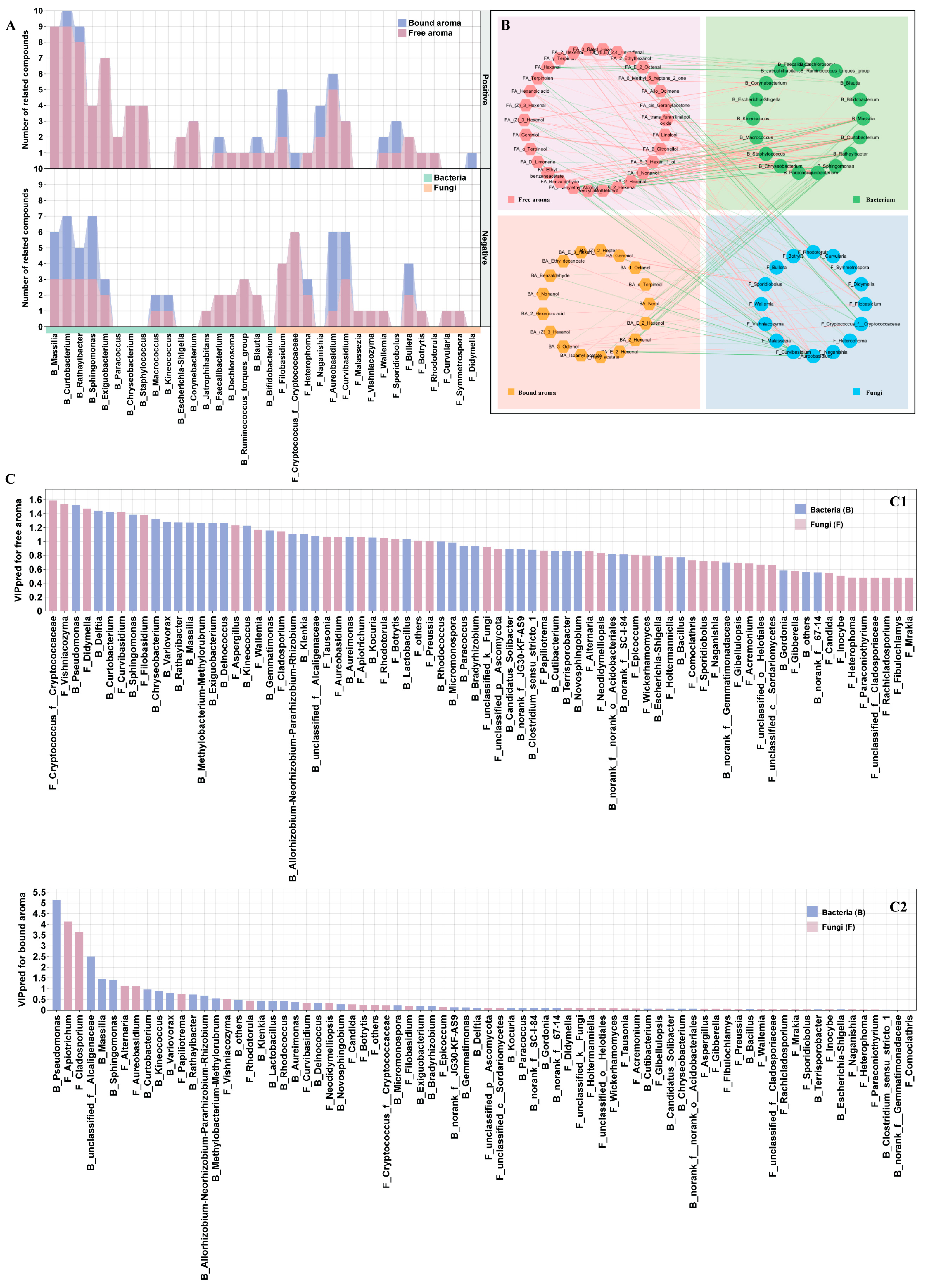
3.4. Distinct Endophytic Bacteria and Fungi Co-Occurrence Networks in Grape Berries
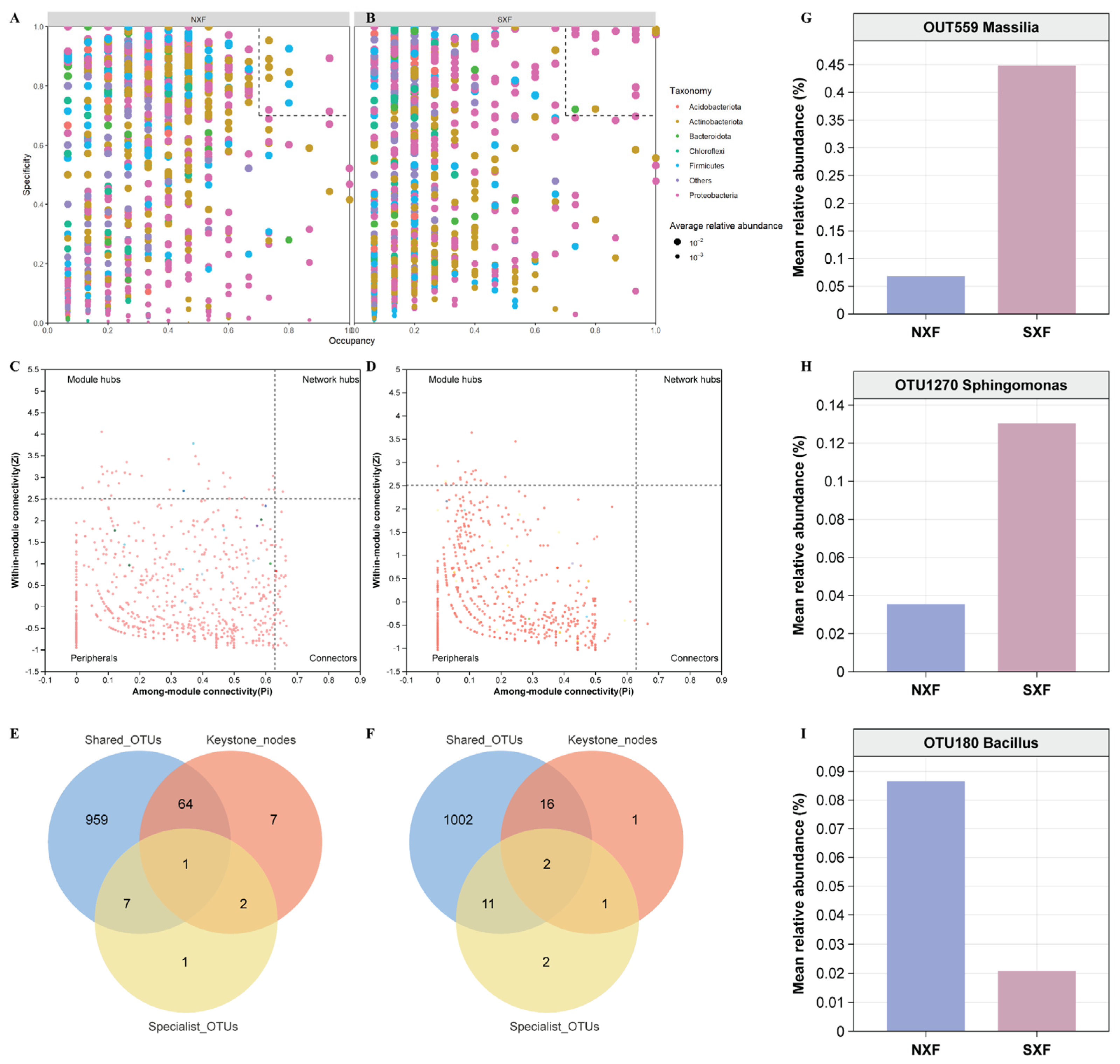
3.5. Identifying Core Taxa and Their Effects on Berry Volatiles
3.6. Correlations between Microbial Community and Berry Volatile Profiles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lund, S.; Bohlmann, J. The molecular basis for wine grape quality-a volatile subject. Science 2006, 311, 804–805. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; De Luca, V. The biosynthesis and regulation of biosynthesis of Concord grape fruit esters, including ‘Foxy’ methylanthranilate. Plant J. 2005, 44, 606–619. [Google Scholar] [CrossRef]
- Bokulich, N.; Thorngate, J.; Richardson, P.; Mills, D. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. USA 2013, 111, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Kustos, M.; Gambetta, J.; Jeffery, D.; Heymann, H.; Goodman, S.; Bastian, S. A matter of place: Sensory and chemical characterisation of fine Australian Chardonnay and Shiraz wines of provenance. Food Res. Int. 2020, 130, 108903. [Google Scholar] [CrossRef]
- Priori, S.; Pellegrini, S.; Perria, R.; Puccioni, S.; Storchi, P.; Valboa, G.; Costantini, E. Scale effect of terroir under three contrasting vintages in the Chianti Classico Area (Tuscany, Italy). Geoderma 2019, 334, 99–112. [Google Scholar] [CrossRef]
- Lola, D.; Miliordos, D.; Goulioti, E.; Kontoudakis, N.; Myrtsi, E.; Haroutounian, S.; Kotseridis, Y. Assessment of the volatile and non-volatile profile of Savatiano PGI wines as affected by various terroirs in Attica, Greece. Food Res. Int. 2023, 174, 113649. [Google Scholar] [CrossRef] [PubMed]
- Olimi, E.; Kusstatscher, P.; Wicaksono, W.; Abdelfattah, A.; Cernava, T.; Berg, G. Insights into the microbiome assembly during different growth stages and storage of strawberry plants. Environ. Microbiome 2022, 17, 21. [Google Scholar] [CrossRef]
- Gao, Y.; Li, D.; Liu, Y. Microbiome research outlook: Past, present, and future. Protein Cell 2023, 14, 709–712. [Google Scholar] [CrossRef]
- Li, Z.; Wen, W.; Qin, M.; He, Y.; Xu, D.; Li, L. Biosynthetic mechanisms of secondary metabolites promoted by the interaction between endophytes and plant hosts. Front. Microbiol. 2022, 13, 928967. [Google Scholar] [CrossRef]
- Sangiorgio, D.; Cellini, A.; Spinelli, F.; Pastore, C.; Farneti, B.; Savioli, S.; Rodriguez-Estrada, M.; Donati, I. Contribution of fruit microbiome to raspberry volatile organic compounds emission. Postharvest Biol. Technol. 2022, 183, 111742. [Google Scholar] [CrossRef]
- Nasopoulou, C.; Pohjanen, J.; Koskimäki, J.; Pirttilä, A. Localization of strawberry (Fragaria x Ananassa) and methylobacterium extorquens genes of strawberry flavor biosynthesis in strawberry tissue by in situ hybridization. J. Plant Physiol. 2014, 171, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Escobar Rodríguez, C.; Novak, J.; Buchholz, F.; Uetz, P.; Bragagna, L.; Gumze, M.; Antonielli, L.; Mitter, B. The bacterial microbiome of the tomato fruit is highly dependent on the cultivation approach and correlates with flavor chemistry. Front. Plant Sci. 2021, 12, 775722. [Google Scholar] [CrossRef]
- Sangiorgio, D.; Cellini, A.; Spinelli, F.; Farneti, B.; Khomenko, I.; Muzzi, E.; Savioli, S.; Pastore, C.; María, T.; Rodriguez-Estrada, M. Does organic farming increase raspberry quality, aroma and beneficial bacterial biodiversity? Microorganisms 2021, 9, 1617. [Google Scholar] [CrossRef] [PubMed]
- Semwal, P.; Misra, S.; Misra, A.; Kar, S.; Majhi, B.; Mishra, S.; Srivastava, S.; Chauhan, P. Endophytic Bacillus strains enhance biomass and bioactive metabolites of Gloriosa Superba. Ind. Crops Prod. 2023, 204, 117296. [Google Scholar] [CrossRef]
- Tripathi, A.; Awasthi, A.; Singh, S.; Sah, K.; Maji, D.; Patel, V.K.; Verma, R.; Kalra, A. Enhancing artemisinin yields through an ecologically functional community of endophytes in Artemisia annua. Ind. Crop. Prod. 2020, 150, 112375. [Google Scholar] [CrossRef]
- Mezzasalma, V.; Sandionigi, A.; Guzzetti, L.; Galimberti, A.; Grando, M.; Tardaguila, J.; Labra, M. Geographical and cultivar features differentiate grape microbiota in Northern Italy and Spain vineyards. Front. Microbiol. 2018, 9, 946. [Google Scholar] [CrossRef]
- Yue, X.; Liu, S.; Wei, S.; Fang, Y.; Zhang, Z.; Ju, Y. Transcriptomic and metabolic analyses provide new insights into the effects of exogenous sucrose on monoterpene synthesis in “Muscat Hamburg” grapes. J. Agric. Food Chem. 2021, 69, 4164–4176. [Google Scholar] [CrossRef]
- Tian, M.; Ma, W.; Xia, N.; Peng, J.; Hu, R.; Duan, C.; He, F. Soil variables and reflected light revealed the plasticity of grape and wine composition: Regulation of the flavoromics under inner row gravel covering. Food Chem. 2023, 414, 135659. [Google Scholar] [CrossRef]
- Yue, X.; Ju, Y.; Cui, Y.; Wei, S.; Xu, H.; Zhang, Z. Evolution of green leaf volatile profile and aroma potential during the berry development in five Vitis vinifera L. Cultivars. Food Chem. X 2023, 18, 100676. [Google Scholar] [CrossRef]
- Ren, J.; Dong, G.; Yan, dh. Organs, cultivars, soil, and fruit properties affect structure of endophytic mycobiota of Pinggu peach trees. Microorganisms 2019, 7, 322. [Google Scholar] [CrossRef]
- Edgar, R.; Haas, B.; Clemente, J.; Quince, C.; Knight, R. Uchime improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, L.; Ma, T. Easy Amplicon: An easy-to-use, open-source, reproducible, and community-based pipeline for amplicon data analysis in microbiome research. iMeta 2023, 2, e83. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, Y.; Huang, L. ImageGP: An easy-to-use data visualization web server for scientific researchers. iMeta 2022, 1, e5. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Wang, T.; Huang, Q.; Hanyue, G.; Zhang, H.; Xu, Q.; Shen, Q.; Ling, N. Core species impact plant health by enhancing soil microbial cooperation and network complexity during community coalescence. Soil Biol. Biochem. 2024, 188, 109231. [Google Scholar] [CrossRef]
- Yue, X.; Wang, S.; Dong, Y.; Chen, W.; Wang, Y.; Xu, H.; Zhang, Z.; Fang, Y.; Ju, Y. Targeted metabolomics analysis based on HS-SPME-GC-MS to discriminate geographical origin of ‘Muscat Hamburg’ grape and wine. Food Res. Int. 2024, 181, 114120. [Google Scholar] [CrossRef]
- Xie, S.; Lei, Y.; Wang, Y.; Wang, X.; Ren, R.; Zhang, Z. Influence of continental climates on the volatile profile of Cabernet Sauvignon grapes from five Chinese viticulture regions. Plant Growth Regul. 2019, 87, 83–92. [Google Scholar] [CrossRef]
- Aleynova, O.; Nityagovsky, N.; Ananev, A.; Suprun, A.; Ogneva, Z.; Dneprovskaya, A.; Beresh, A.; Dubrovina, A.; Chebukin, P.; Kiselev, K. Bacterial and fungal endophytes of grapevine cultivars growing in Primorsky Krai of Russia. Horticulturae 2023, 9, 1257. [Google Scholar] [CrossRef]
- Wijekoon, C.; Quill, Z. Fungal endophyte diversity in table grapes. Can. J. Microbiol. 2020, 67, 29–36. [Google Scholar] [CrossRef]
- Compant, S.; Clément, C.; Sessitsch, A. Plant growth-promoting bacteria in the rhizo- and endosphere of plants: Their role, colonization, mechanisms involved and prospects for utilization. Soil Biol. Biochem. 2010, 42, 669–678. [Google Scholar] [CrossRef]
- Liu, D.; Howell, K. Community succession of the grapevine fungal microbiome in the annual growth cycle. Environ. Microbiol. 2020, 23, 1842–1857. [Google Scholar] [CrossRef]
- Li, Y.; Jin, J.; Li, P.; Wang, Q.; Xu, L.; Wei, G. Regional variations and plant compartments shape the community structures of the endophytic microbiome and secondary metabolites of Astragalus mongholicus. Ind. Crops Prod. 2023, 192, 116037. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.; Tringe, S.; Sa, T.; Singh, B. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, A.; Acedo, A.; Imam, N.; Santini, R.; Ortiz Álvarez, R.; Ellegaard-Jensen, L.; Belda, I.; Hansen, L. A global microbiome survey of vineyard soils highlights the microbial dimension of viticultural terroirs. Commun. Biol. 2022, 5, 241. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Lehosmaa, K.; Toth, K.; Koskimäki, J.; Häggman, H.; Pirttilä, A. Weather in two climatic regions shapes the diversity and drives the structure of fungal endophytic community of bilberry (Vaccinium myrtillus L.) fruit. Environ. Microbiome 2024, 19, 7. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Abdelfattah, A.; Wasserman, B.; Wisniewski, M.; Droby, S.; Fazio, G.; Mazzola, M.; Wu, X. Contrasting effects of genotype and root size on the fungal and bacterial communities associated with apple rootstocks. Hortic. Res. 2022, 9, 1842–1857. [Google Scholar] [CrossRef]
- Liu, Y.; Chu, G.; Stirling, E.; Zhang, H.; Chen, S.; Xu, C.; Zhang, X.; Ge, T.; Wang, D. Nitrogen fertilization modulates rice seed endophytic microbiomes and grain quality. Sci. Total Environ. 2023, 857, 159181. [Google Scholar] [CrossRef]
- Coleman-Derr, D.; Desgarennes, D.; Fonseca, C.; Gross, S.; Clingenpeel, S.; Woyke, T.; North, G.; Visel, A.; Partida Martinez, L.; Tringe, S. Plant compartment and biogeography affect microbiome composition in cultivated and native agave species. New Phytol. 2015, 209, 798–811. [Google Scholar] [CrossRef]
- Qin, D.; You, C.; Lan, W.; Wang, Y.; Yu, B.; Peng, Y.; Xu, J.; Dong, J. Microbial assemblages of schisandraceae plants and the correlations between endophytic species and the accumulation of secondary metabolites. Plant Soil 2023, 483, 85–107. [Google Scholar] [CrossRef]
- Wang, H.; Guo, D.; Zhang, M.; Wu, G.; Shi, Y.; Zhou, J.; Ding, N.; Chen, X.; Li, X. Correlation study on microbial communities and volatile flavor compounds in Cigar Tobacco leaves of diverse origins. Appl. Microbiol. Biotechnol. 2024, 108, 236. [Google Scholar] [CrossRef]
- Miao, S.; Xia, Y.; Cui, J.; Wang, J.; Wang, M. Correlation analysis between differential metabolites and bacterial endophytes of Ephedra Sinica in different years. Ind. Crops Prod. 2022, 175, 114250. [Google Scholar] [CrossRef]
- He, X.; Wang, D.; Jiang, Y.; Li, M.; Delgado-Baquerizo, M.; McLaughlin, C.; Marcon, C.; Guo, L.; Baer, M.; Moya, Y. Heritable microbiome variation is correlated with source environment in locally adapted maize varieties. Nat. Plants 2024, 10, 598–617. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, R.; Zeng, M.; Liu, Y.; Shi, J.; Wan, Z.; Wang, M.; Zhang, S.; Zhang, Z.; Zeng, Q. Grape Endophytic Microbial Community Structures and Berry Volatile Components Response to the Variation of Vineyard Sites. Agronomy 2024, 14, 2186. https://doi.org/10.3390/agronomy14102186
Ren R, Zeng M, Liu Y, Shi J, Wan Z, Wang M, Zhang S, Zhang Z, Zeng Q. Grape Endophytic Microbial Community Structures and Berry Volatile Components Response to the Variation of Vineyard Sites. Agronomy. 2024; 14(10):2186. https://doi.org/10.3390/agronomy14102186
Chicago/Turabian StyleRen, Ruihua, Maoyu Zeng, Yunqi Liu, Jingjing Shi, Zhuowu Wan, Miaomiao Wang, Shibo Zhang, Zhenwen Zhang, and Qingqing Zeng. 2024. "Grape Endophytic Microbial Community Structures and Berry Volatile Components Response to the Variation of Vineyard Sites" Agronomy 14, no. 10: 2186. https://doi.org/10.3390/agronomy14102186