Abstract
The grain filling rate (GFR) plays a crucial role in determining grain yield. However, the regulatory and molecular mechanisms of the grain filling rate (GFR) in foxtail millet remains unclear. In this study, we found that the GFR of ′Changnong No.47′ (CN47) was significantly higher at 14 DAF (days after flowering) and 21 DAF in comparison to ‘Changsheng 13’ (CS13). Furthermore, CN47 also exhibited higher a thousand-grain weight and yield than CS13. Therefore, RNA-seq and UHPLC-MS/MS were used to conduct transcriptome and metabolome analyses during two stages of grain filling in both cultivars. Conjoint analysis of transcriptomics and metabolomics was adopted in order to analyze the biological processes and functional genes associated with GFR. The results identified a total of 765 differentially expressed genes (DEGs) and 246 differentially accumulated metabolites (DAMs) at the 14 DAF stage, while at the 21 DAF stage, a total of 908 DEGs and 268 DAMs were identified. The integrated analysis of co-mapped DAMs and DEGs revealed enriched pathways, including flavonoid biosynthesis, plant hormone signal transduction, tyrosine metabolism, ATP-binding cassette (ABC) transporters, and beta-Alanine metabolism, as well as stilbenoid, diarylheptanoid, and gingerol biosynthesis. In order to elucidate their potential functions in the context of GFR, we developed a gene–metabolite regulatory network for these metabolic pathways. Notably, we found that some genes associated with ABC transporters and the plant hormone signal transduction pathway were implicated in auxin transport and signal transduction, highlighting the crucial role of auxin during grain filling. These findings provide initial insights into the regulatory and molecular mechanisms underlying GFR in foxtail millet, as well as offering valuable genetic resources for further elucidation of GFR in future studies. The findings have also established a theoretical basis for improving the efficiency of yield breeding in foxtail millet.
1. Introduction
Foxtail millet (Setaria italica L.) is an ancient crop originating in China, renowned for its high drought tolerance [1]. It has emerged as a prominent cereal in regions with limited water resources, including China, India, and certain African countries [2]. Foxtail millet grains are abundant in essential nutrients, such as organic acids [3], vitamin E, various amino acids, and high-quality protein [4]. Additionally, it serves as a valuable source of trace elements like zinc and iron [5]. Its bran also contains substantial amounts of linoleic and linolenic acids [6,7], making it an excellent crude fiber source that aids intestinal digestion and promotes digestive health [8]. These nutritional attributes have garnered increasing global attention towards this grain. However, its yield per unit area is significantly lagging behind staple crops like maize, wheat, and rice, and the planting areas and total yield are showing a downward trend [9]. Therefore, enhancing the yield potential of foxtail millet becomes imperative.
Cereal crops’ grain yield is determined by three component traits—the number of panicles, the number of grains per panicle, and thousand kernel weight (TKW) [10]. Kernel weight, the most crucial factor determining yield, is largely influenced by the grain filling duration (GFD) and the grain filling rate (GFR) [11]. Studies show that GFD is greatly affected by temperature [12,13] and nitrogenous fertilizer [14], especially when the grain filling period occurs under water stress [15]. However, the stability of GFR and its limited susceptibility to environmental factors [16,17] suggest a potential genetic determination for this parameter [18]. More importantly, the GFR plays a crucial role in enhancing grain productivity by facilitating endosperm development and nutrient accumulation [19]. The performance of cultivars with high filling rates is generally superior under adverse conditions such as abiotic and biotic stresses, as they produce seeds that are fuller and exhibit mitigated effects compared to cultivars with low filling rates [20]. Thus, the attainment of crops with high GFR can be accomplished through genetic breeding, and the strategic selection and breeding of genotypes exhibiting high GFR is very likely to be a successful approach in ensuring optimal grain yield. Furthermore, by exploring and identifying regulatory genes and key loci associated with GFR, as well as analyzing and defining the regulatory pathways and molecular mechanisms governing GFR, we can facilitate the precise breeding of rapid filling characteristics, thereby enhancing both grain yield and quality.
The identification of several pivotal genes involved in GFR, along with the elucidation of their molecular pathways in major crops, has significantly contributed to our understanding of this intricate process. In rice, the enzymes associated with its assimilate synthesis and transport—including those involved in starch synthesis and sucrose metabolism—exert a significant impact on GFR. Grain incomplete filling 2 (GIF2), encoding an ADP-Glc pyrophosphorylase large subunit, is involved in starch biosynthesis during seed development. The mutants of GIF2 show lower GFR and yield [21]. The sucrose transport-related genes Sucrose Transporter1 (OsSUT1), OsSUT2, OsSUT3, and OsSUT4 have a significant influence on grain filling due to their involvement in the transportation of sucrose. OsNF-YB1 acts as an upstream regulator of OsSUT3 and OsSUT4 expression, specifically expressed in the aleurone layer [22,23]. Moreover, the SWEET genes, such as ZmSWEET4c in maize and OsSWEET4 in rice, depend on transepithelial hexose transport to regulate seed filling [24]. In maize, Opaque2 activates the expression of the sucrose synthase genes SUS1 and SUS2 and promotes grain filling [25]. Furthermore, several findings have indicated that the members of the cell wall invertase (CIN) gene family impact the GFR. The cell wall convertase genes OsGIF1/OsCIN2 have been shown to domesticate selective genes involved in carbon allocation during the early growth stage. These genes regulate the transport and unloading of sucrose and affect GFR [26]. The level of plant hormone also strongly affects GFR by regulating the expression levels of key genes. Ethylene (ETH) and abscisic acid (ABA) influence the expression levels of starch synthesis genes, thereby further inhibiting carbohydrate biosynthesis in spikelets, reducing GFR, and ultimately affecting yield [27,28]. And low concentrations of auxin (IAA) and cytokinin (CK) in rice lead to a decrease in GFR, resulting in poor grain filling, ultimately leading to reduced grain weight and yield [29,30]. DEP1/qPE9-1 is the rice G protein γ subunit, which regulates GFR by increasing the auxin and CK contents of grains [31]. In addition, microRNAs (miRNAs) play a crucial role during grain filling, and the suppression of miR1432’s expression enhances GFR, leading to a significantly increased grain yield in rice [19]. GRAIN-FILLING RATE1 (GFR1) encodes a membrane-localized protein in rice. It can enhance the expression levels of Rubisco genes involved in the Calvin cycle, thereby promoting the synthesis of sucrose and increasing the GFR and the yield [32]. And QGfr.sicau-7D.1 is a newly discovered QTL related to GFR in wheat [33].
However, research on the molecular mechanisms and regulatory genes related to GFR in foxtail millet is rarely reported compared to that of major cereal crops. The whole genome sequencing of foxtail millet has facilitated further research in the field of molecular biology and functional genomics for this crop [34,35]. High-throughput sequencing technology and multi-omics analysis are among the means used to look for genes affecting grain filling. By comparing the differences in transcriptome- [9,36,37], metabolome- [38], and genome-wide DNA methylation in foxtail millet [39] at different grain filling stages, numerous potential regulators of grain filling and the accumulation of grain metabolites have been identified [35,39]. Furthermore, one paper has provided evidence of differences in the gene expressions and molecular mechanisms related to seed filling among foxtail millet cultivars with different panicle types in the North China summer sowing region [40]. However, that research focused solely on elucidating the mechanism underlying the foxtail millet grain filling process, without delving into the mechanisms of differences in the grain filling processes of two genotypes (high GFR and low GFR). In particular, the molecular pathways and associated genes governing the GFR of foxtail millet have not been identified.
In this study, we conducted a systematic analysis of agronomic traits, as well as the variations in thousand millets weight (de-husked grains) and GFR across six different developmental stages, utilizing the grains of Changnong No.47 (CN47, high GFR) and Changsheng 13 (CS13, low GFR) foxtail millet cultivars. Then, the stages with the most significant disparities in GFR between the two cultivars were chosen for transcriptome and metabolome investigation using the technology of RNA-seq and UHPLC-MS/MS. Our comprehensive transcriptomic and metabolomic analyses revealed potentially crucial metabolites, transcriptional regulators, and several genes related to GFR in various metabolic pathways, shedding light on the molecular regulatory mechanism underlying GFR. In addition, the reliability of the transcriptome analysis was validated by measuring the relative expression levels of several selected genes using qRT-PCR. The aims of this study are to investigate the changes in thousand millets weight (de-husked grains) and GFR at different stages of grain filling in two foxtail millet cultivars, as well as determining the key transcriptional regulators, metabolic pathways, and differentially expressed genes (DEGs) of GFR. Furthermore, the results would preliminarily reveal the molecular mechanism of GFR, providing a theoretical foundation for achieving high-yield and high-quality molecular design breeding of foxtail millet.
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
The foxtail millet cultivars (Setaria italica L.) utilized in this study were Changsheng 13 (CS13, low GFR) and Changnong No. 47 (CN47, high GFR), which were provided by the Millet Research Institute of Shanxi Agricultural University (Changzhi, Shanxi, China). The two cultivars were cultivated at the experimental station of the Millet Research Institute of Shanxi Agricultural University in Changzhi, Shanxi, China (36°21′ N, 113°14′ E) from May to October in 2023. The experimental field featured a temperate semi-humid continental monsoon climate, characterized by an average annual temperature ranging from 8.6 °C to 10.5 °C; an average annual rainfall exceeding 600 mm; and a frost-free period lasting between 155 and 184 days. A randomized block design was employed for the field experiments, with each plot measuring 5 m × 2.4 m, and three biological replicates were stablished. The soil consisted of brown loam with nutrient contents as follows: organic matter (33.51 g/kg), available nitrogen (46.14 µg/g), available phosphorus (20.14 µg/g), and available potassium (168.76 µg/g). The field was subjected to the implementation of routine agronomic practices.
2.2. Investigation of Grain Development and GFR
The experimental plots were labeled with sixty plants exhibiting spikelets of the same age and size in the central sections, all flowering synchronously. On each plant, the glume surface of 200 flowers was carefully annotated with a black pen. After 7 days of labeling, we selected fresh grains from five random plants. The grains were harvested and then subjected to oven drying at a temperature of 105 °C for 30 min. They were subsequently dried in another oven at 80 °C until the weight remained constant. Subsequently, these dried grains were manually dehulled and weighed using a precision balance with an accuracy of up to 0.001 g. Two hundred foxtail millet grains, chosen randomly per plant, were weighed and the average weight was calculated. The fresh grains collected at the 7-, 14-, 21-, 28-, 35-, and 42-day labeling points later underwent meticulous procedures including harvesting, drying, hulling, and weighing. The method of calculating the grain filling rate was adapted from Rice′s technique [32]. The GFR was then calculated for each stage using the following formula:
GFRi = (GWi − GWi−1)/7.
Among these, GFRi refers to the grain filling rate (mg grain−1 d−1) at stage i and GWi represents dried grain weight (mg grain−1) at stage i (i = 1, 2, 3, 4, 5, 6).
A stereo microscope (SZX10, Olympus, Tokyo, Japan) was employed to document the changes in developing grains at different stages including 7, 14, 21, 28, 35, and 42 DAF (days after flowering).
In addition, grains of the same age and size from the central sections of marked plants were sampled at 14 and 21 DAF. Three biological replicates were selected for each developmental stage, with the samples collected from three distinct field plots. The grains from five panicles within the same plot were combined as a single biological replicate. The samples were promptly flash-frozen in liquid nitrogen and subsequently stored at −80 °C for further experimentation.
2.3. RNA Sequencing and Transcriptomic Profiling Analysis
The grains of CS13 and CN47 at the 14 DAF stage and the 21 DAF stage were collected for RNA extraction and sequencing. RNA-seq was performed by Novogene Co., Ltd. (Beijing, China). Each treatment group consisted of three biological replicates. The concentration, integrity, and quality of RNA was assessed using the Bioanalyzer 5400 system (Agilent Technologies) and agarose gel electrophoresis (Figure S1). An RNA integrity index (RIN) was used to evaluate RNA integrity, and those with scores greater than 6 were used for sequencing. The cDNA libraries were constructed, and sequencing was conducted with the Illumina (HiSeq/MiSeq) system by Genepioneer. Subsequently, clean reads were obtained by filtering out unqualified raw reads, based on Q20, Q30, and GC content criteria. The clean reads were trimmed with Trimmomatic (version 0.36) [41] while HISAT2 (version v2.0.5) [42] mapped them to the reference genome of foxtail millet [39]. Differential expression analysis of genes was performed using the DESeq package with BaseMean value estimation [43]. Differentially expressed genes (DEGs) were identified based on |log2(fold change)| ≥ 1 and adjusted p-value < 0.05 as screening criteria. Gene Ontology (GO) [44] and Kyoto Encyclopedia of Genes and Genomes (KEGG) [45] pathway enrichment analyses were conducted using cloud analysis tools provided by Novogene (https://magic.novogene.com/, accessed on 16 April 2024).
2.4. Metabolome Profiling Analysis
The metabolome analysis was performed using material consistent with that used in Section 2.3., but with six biological replicates for each group. Similarly, Novogene Co., Ltd. in Beijing, China, conducted the metabolite extraction and LC-MS analysis. UHPLC-MS/MS analyses were performed with a Vanquish UHPLC system (ThermoFisher), coupled with an Orbitrap Q ExactiveTM HF mass spectrometer or Orbitrap Q ExactiveTMHF-X mass spectrometer (Thermo Fisher, Germany) [46]. The acquired raw data files from UHPLC-MS/MS analysis were processed by Compound Discoverer 3.3 (CD3.3, ThermoFisher) software for tasks such as peak alignment, selection, and metabolite quantification. Principal component analysis (PCA) and partial least squares discriminant analysis (PLS-DA) were performed using metaX (a flexible and comprehensive software for processing metabolomics data) for metabolites [47]. The VIP value was calculated for each metabolite. For the univariate analysis, t-tests were used to determine the statistical significance (p-value) and fold change (FC-value) of metabolites between the two groups. The metabolites with VIP > 1 and p-value < 0.05, along with |log2 (fold change)| > 0.5, were considered as differential metabolites (DAMs). These DAMs were annotated using the KEGG database (https://www.genome.jp/kegg/pathway.html, accessed on 16 April 2024) and mapped onto KEGG metabolic pathways for pathway analysis and enrichment.
2.5. Conjoint Analysis of Transcriptomes and Metabolomes
By simultaneously mapping DEGs and DAMs to the KEGG pathway database, we obtained their shared pathway information. Correlation analysis was performed using quantitative values of genes and metabolites in all samples. Spearman’s correlation coefficient (Cor) and p-value were calculated using the Cor function of the R language. Finally, a network map of DEGs–DAMs based on Spearman’s correlation coefficient (with |Cor| > 0.8 and p ≤ 0.05) was constructed using Cytoscape.
2.6. Quantitative qRT-PCR
Total RNA was extracted from all samples with the TaKaRa MiniBEST Plant Total RNA Extraction Kit. The SynScript®Ⅲ RT SuperMix for qPCR was used to reverse transcribe RNA. The synthesized first-strand cDNA was diluted 5-fold before being used as a template for qRT-PCR. qPCR was conducted using the ArtiCanCEO SYBR qPCR Mix and Bio-Rad CFX96 (BIORAD). The reaction conditions were as follows: pre-denaturation at 95 °C for 5 min, 40 cycles of denaturation at 95 °C for 15 s, annealing, extension at 60 °C for 20 s, and extension at 72 °C for 20 s. The relative expression levels of genes were determined using the 2−∆∆Ct method. Each pair of primers was set up with three biological replicates and three technical replicates. The gene expression was standardized by normalizing it to the housekeeping gene Actin 1. The primers are listed in Table S1.
2.7. Statistical Analysis
The statistical analyses of phenotype values and the relative expression levels of genes by qRT-PCR were performed using SPSS 27.0 software. The t-test was considered to have a statistically significant difference when the p-value was ≤ 0.05. The figures were generated using Origin 2021 software.
3. Results
3.1. Characterization of Grain Filling Rate-Related Phenotypes in Foxtail Millet
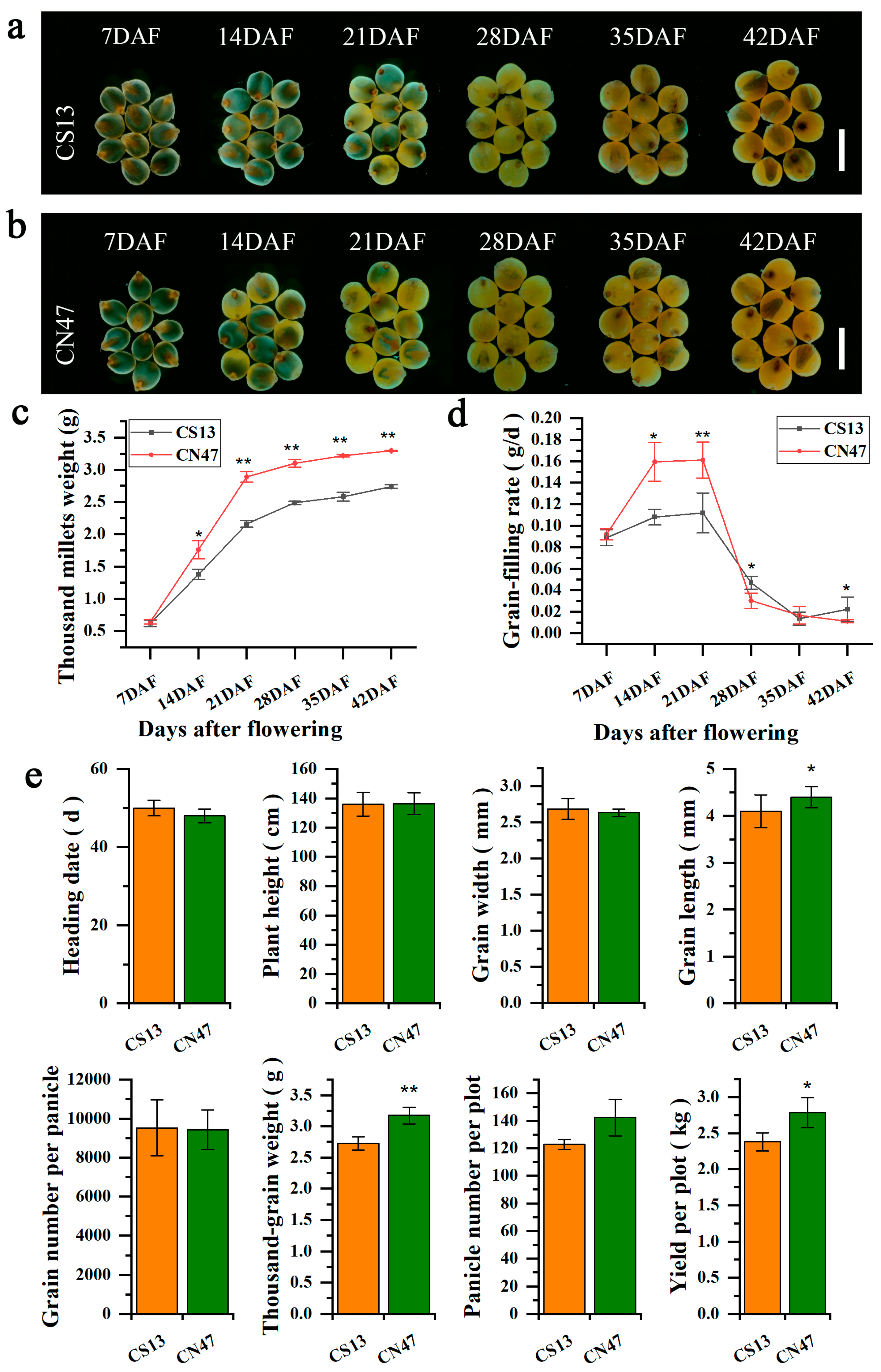
To investigate dynamic changes in foxtail millet grain development after flowering, spikelets were collected from two cultivars (CS13 and CN47) at 7, 14, 21, 28, 35, and 42 days after flowering (DAF) during the grain filling period. The growth morphology changes in de-husked grains of CS13 and CN47 were observed across six stages using a stereo microscope (Figure 1a,b). Additionally, the grain filling rate and the thousand millets weight (de-husked grains) were measured for both cultivars at each time point mentioned (Figure 1c,d). There were distinct variations in the GFR and thousand millets weight between CS13 and CN47, with CN47 consistently exhibiting a higher thousand millets weight from 14 DAF to 42 DAF. The highest grain filling rates were observed at 21 DAF for both cultivars, with CN47 exhibiting higher rates at 14 DAF and 21 DAF compared to CS13. No significant differences were found among the agronomic traits analyzed, including heading date (HD), plant height (PH), grain width (GW), grain number per panicle, and panicle number per plot. However, significant differences were observed in grain length (GL), thousand-grain weight (TGW), and yield per plot (YPP) between CS13 and CN47 (Figure 1e). Analysis of the agronomic traits indicated that the relatively high values of GL, TGW, and YPP in CN47 could be attributed to its elevated grain filling rate.
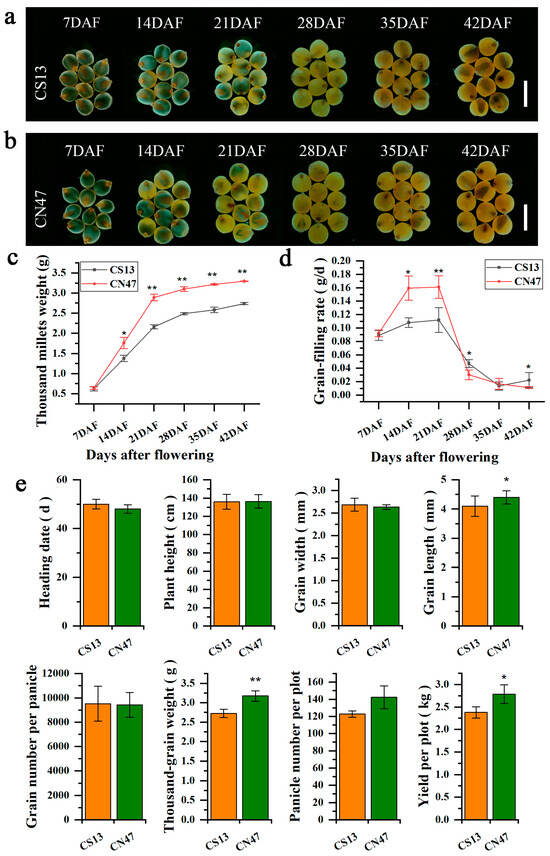
Figure 1.
Phenotypic changes related to grain filling rate in foxtail millets (a,b) Morphological changes in foxtail millet grains of CS13 (a) and CN47 (b) at six stages. Bars = 500 μm. (c) Changes in thousand millets weights (de-husked grains) of CS13 and CN47 at six stages. (d) GFR changes of CS13 and CN47 at six stages. (e) Comparison of the agronomic traits between CS13 and CN47. Values are presented as means ± SD (n = 3). Significance was assessed using the t-test (*, p ≤ 0.05 and **, p ≤ 0.01).
3.2. Transcriptomic Differences in Grains during Grain Filling Stages between Foxtail Millet Cultivars
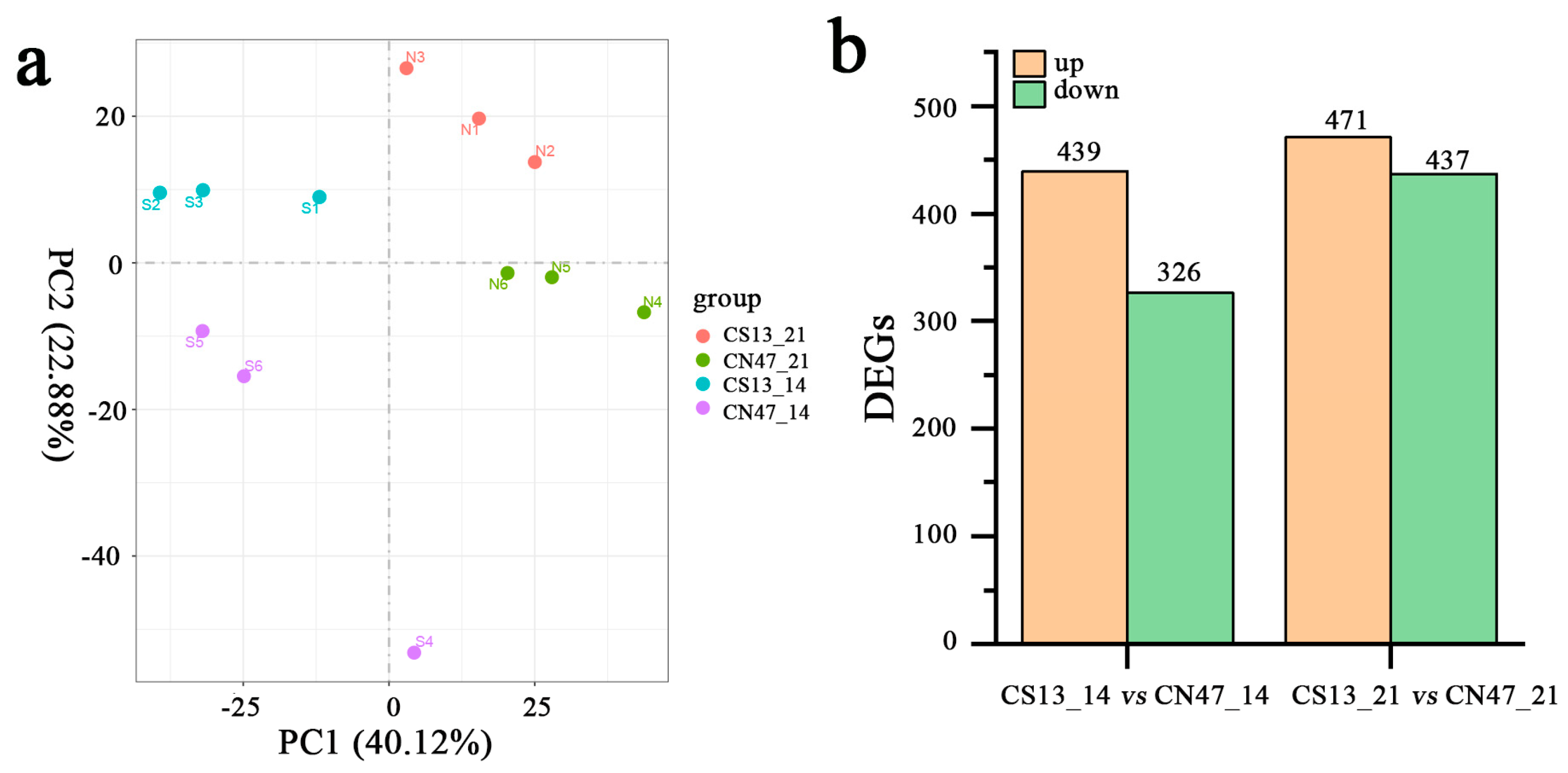
The transcription profiles of CS13 and CN47 were examined at the 14 DAF and 21 DAF stages, with three biological replicates used for each stage. Twelve cDNA libraries were sequenced separately by Illumina sequencing. An average of 6.7 Gb clean bases per library were generated after removing the adapter sequences and the low quality or contaminated reads, with a mean Q30 level of 94.8%. This indicated that the library contained high-quality raw reads (Table S2). The read mapping ratio to the reference genome per sample ranged from 92% to 94% (Table S3). Gene expression levels were quantified using the fragments mapped (FPKM). Principal component analysis (PCA) revealed distinct clustering of the samples, with tight clusters formed by the three biological replicates within each group (Figure 2a). Differential expression analysis was performed to compare CS13_14 DAF vs. CN47_14 DAF and CS13_21 DAF vs. CN47_21 DAF, with a fold change threshold of >1 and false discovery rate of FDR < 0.05. The CS13_14 DAF vs. CN47_14 DAF comparison revealed a total of 765 differentially expressed genes (DEGs), with 439 genes up-regulated and 326 genes down-regulated. Similarly, 908 DEGs were identified in the CS13_21 DAF vs. CN47_21 DAF comparison, with 471 genes up-regulated and 439 genes down-regulated (Figure 2b).
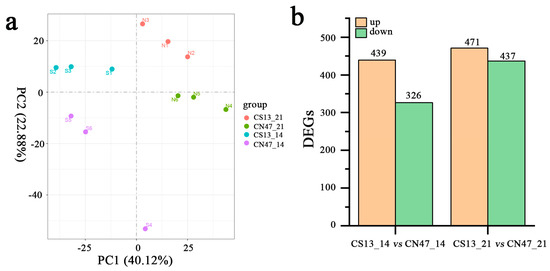
Figure 2.
The transcriptome analysis and the total number of differentially expressed genes of the two foxtail millets at two stages during grain filling. (a) The principal component analysis (PCA) plot analysis of transcriptomes; the x-axis shows principal component 1 (PC1) and the Y-axis shows principal component 2 (PC2). S1–S3, S4–S6, N1–N3, and N4–N6 represent the three biological replicates, corresponding to 14 DAF of CS13, 21 DAF of CS13, 14 DAF of CN47, and 21 DAF of CN47. (b) The statistics of expressed genes (DEGs) in the CS13_14 vs. CN47_14 and CS13_21 vs. CN47_21 comparison.
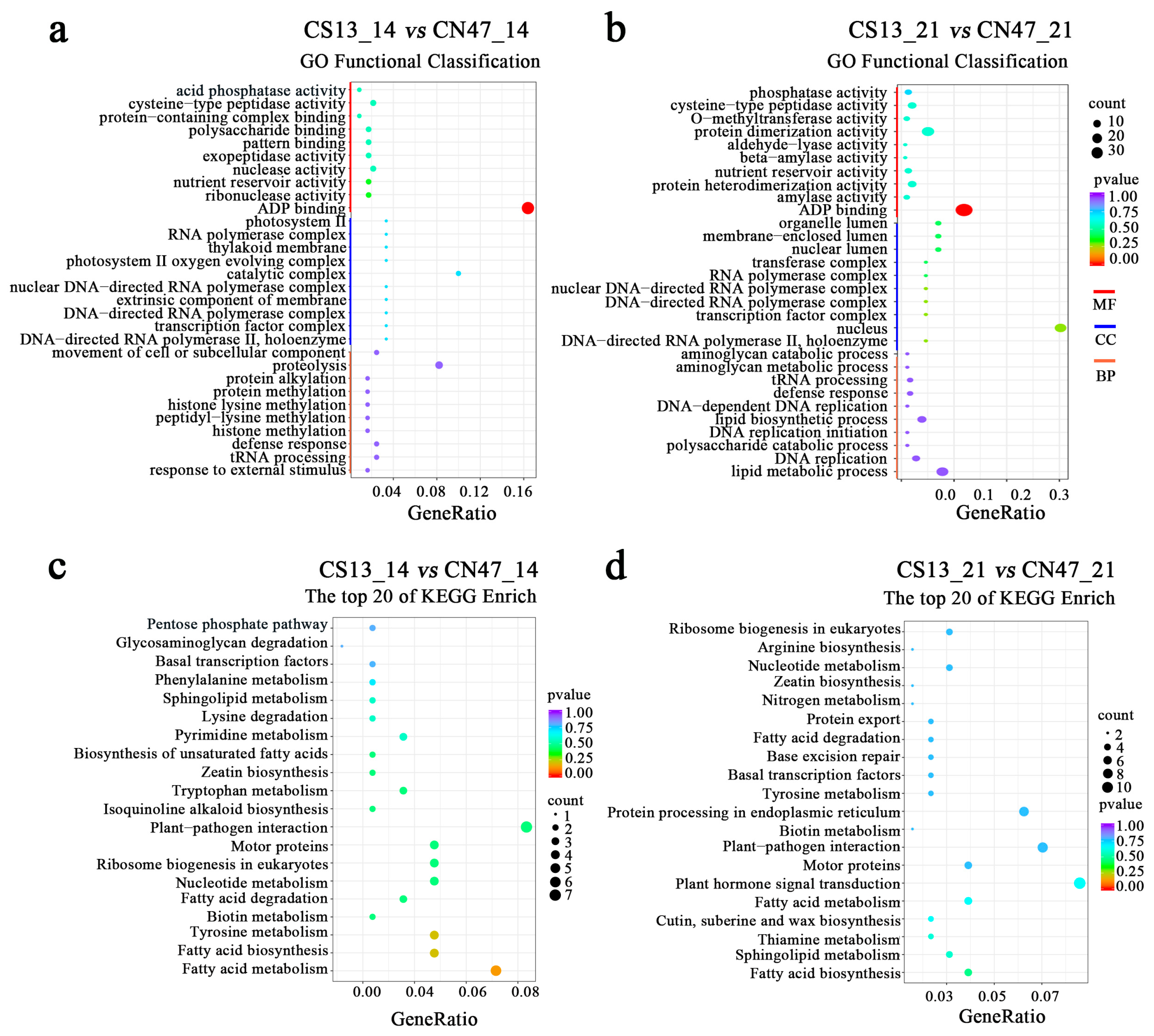
Gene Ontology (GO) enrichment analyses were performed to gain further insights into the functions and associated biological processes in which the DEGs participated (Figure 3). The results showed that the DEGs were classified into cellular components (CC), molecular functions (MF), and biological processes (BP). We further analyzed the top 10 categories for CC, MF, and BP in the GO annotation (Figure 3a,b). In terms of molecular functions, we observed an enrichment of shared GO terms such as ADP binding, nutrient reservoir activity, and cysteine-type peptidase activity. Regarding cellular components, both comparison groups showed enrichment of GO terms related to the DNA-directed RNA polymerase II, holoenzyme, transcription factor complex; the DNA-directed RNA polymerase complex; the nuclear DNA-directed RNA polymerase complex; and the RNA polymerase complex. Different GO terms were enriched in the two comparison groups for biological processes, with only two shared terms identified. These findings indicate that various factors influence the differences in grain filling rates between CS13 and CN47.
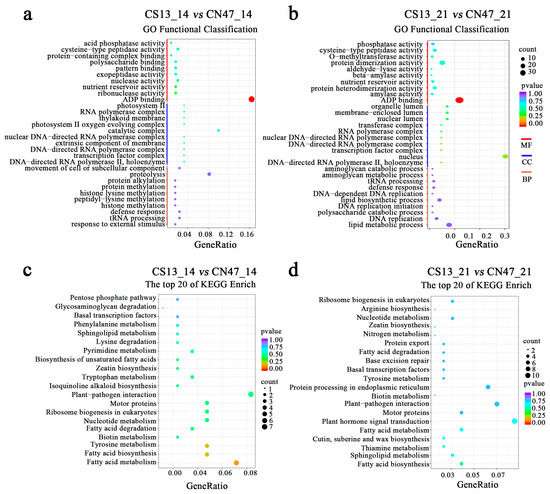
Figure 3.
(a,b)—The top 25 GO classifications in each category of DEGs in the CS13_14 vs. CN47_14 and CS13_21 vs. CN47_21 comparisons of foxtail millet. (c,d)—The top 20 KEGG classifications in each category of DEGs in the CS13_14 vs. CN47_14 and CS13_21 vs. CN47_21 comparisons of foxtail millet. |log2(fold change)| ≥ 1 and p-value < 0.05 were used as screening criteria of DEGs.
To investigate the biological pathways related to GFR, a pathway enrichment analysis of the DEGs was also performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG), with the results depicted in Figure 3c,d. The KEGG enrichment analysis revealed that 12 out of the top 20 terms were shared between the two stages, indicating a high consistency in the pathways involved in grain filling. We observed significant enrichment in pathways, including basal transcription factor, sphingolipid metabolism, zeatin biosynthesis, plant–pathogen interaction, motor proteins, ribosome biogenesis in eukaryotes, nucleotide metabolism, fatty acid degradation, biotin metabolism, tyrosine metabolism, fatty acid biosynthesis, and fatty acid metabolism. Moreover, the pathways of plant hormone signal transduction and nitrogen metabolism were identified as pivotal for the process of grain filling. These findings provide further evidence that the regulation of the grain filling rate involves multiple pathways.
3.3. Metabolite Differences in Grains during Grain Filling Stages between Foxtail Millet Cultivars
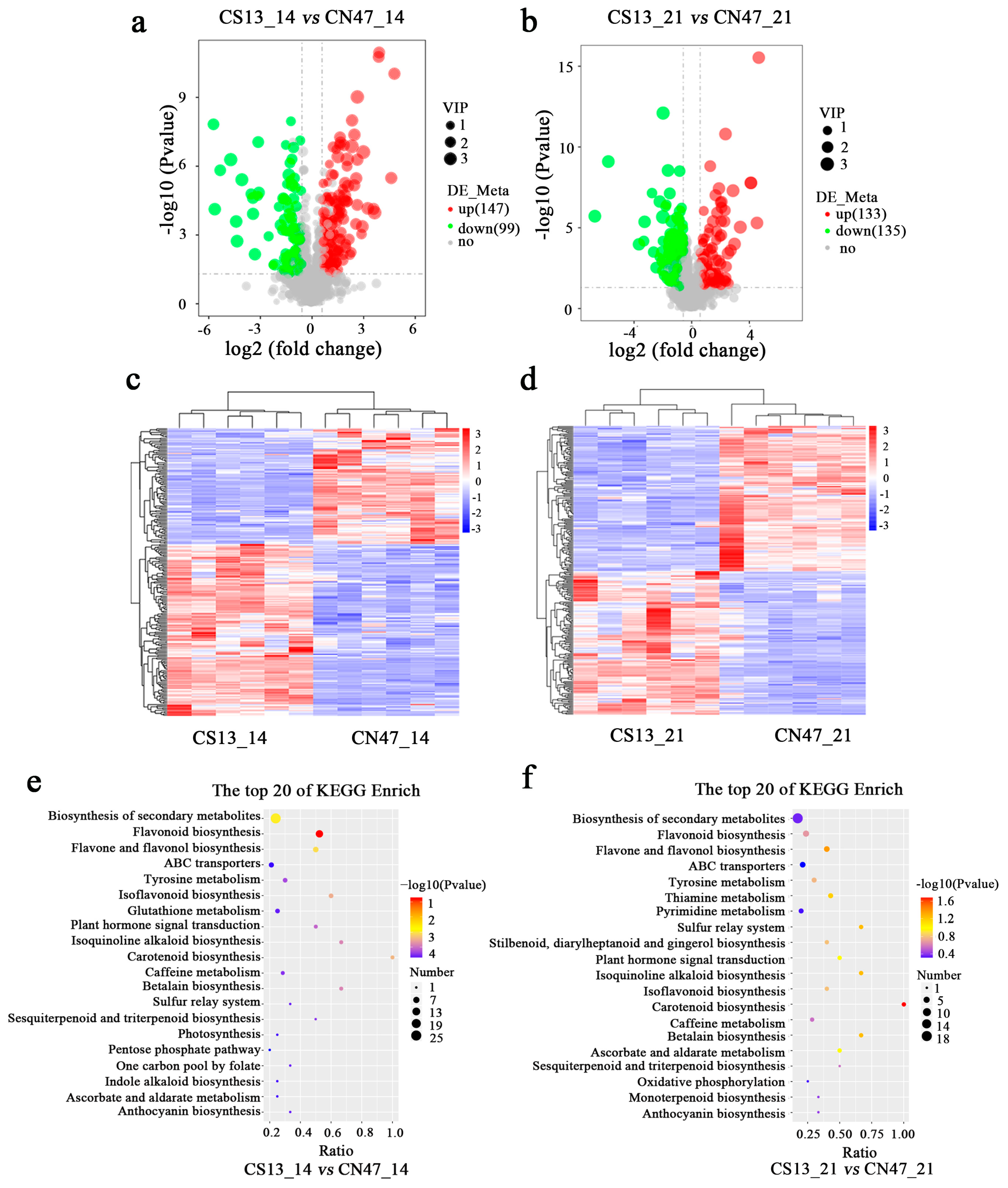
In order to identify differential metabolites between the two cultivars at the 14 DAF and 21 DAF stages, a non-targeted metabolic analysis was conducted, which revealed distinct patterns of metabolite accumulation. Metabolites were primarily categorized into two modes: the positive ion mode (POS) and the negative ion mode (NEG). Metabolites exhibiting |log2 (fold change)| > 0.5 and VIP ≥ 1 were chosen as differentially accumulated metabolites (DAMs). In the POS and NEG modes, a total of 246 DAMs (146 up-regulated, 100 down-regulated) were identified between CS13_14 vs. CSN47_14 DAF, while 268 DAMs (133 up-regulated, 135 down-regulated) were found between CS13_21 DAF vs. CSN47_21 DAF (Table S4; Figure 4a,b). The gene clustering heatmap analysis demonstrated excellent biological repeatability, with clear distinctions among the samples (Figure 4c,d). At the 14 DAF stage, 44 DAMs were annotated to the KEGG pathway and mapped to 24 pathways (Table S5). At the 21 DAF stage, 40 DAMs were annotated to the KEGG pathway and mapped to 23 pathways (Table S6). The top 20 pathways are depicted in Figure 4e,f. A large number of identical pathways were discovered at the 14 DAF and 21 DAF stages. These included flavonoid biosynthesis; biosynthesis of secondary metabolites; flavone and flavonol biosynthesis; carotenoid biosynthesis; isoflavonoid biosynthesis; isoquinoline alkaloid biosynthesis; betalain biosynthesis; plant hormone signal transduction; tyrosine metabolism; sesquiterpenoid and triterpenoid biosynthesis; caffeine metabolism; anthocyanin biosynthesis; sulfur relay system; glutathione metabolism; ascorbate and aldarate metabolism; ATP-binding cassette (ABC) transporters; beta-Alanine metabolism; and stilbenoid, diarylheptanoid, and gingerol biosynthesis. These results suggest that these metabolic pathways are exhibited similarly between the two stages during grain filling.
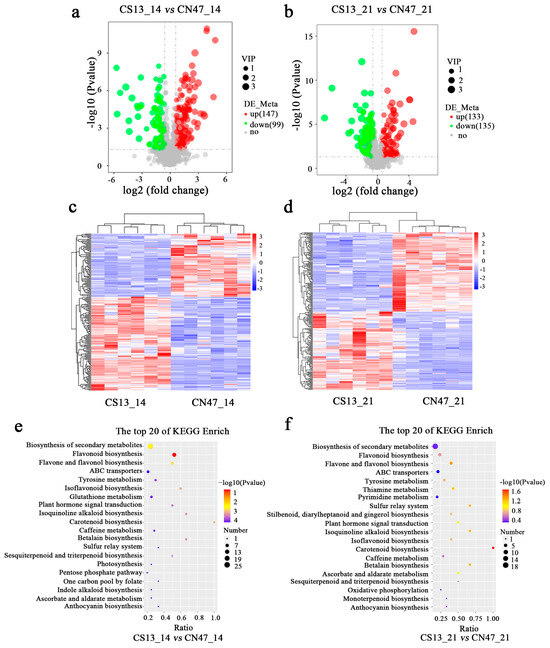
Figure 4.
The metabolomic profiling analysis at two stages during grain filling of the two foxtail millets. (a,b)—A volcano plot of DAMs in the CS13_14 vs. CN47_14 and CS13_21 vs. CN47_21 comparisons, respectively. (c,d)—A heatmap analysis of DAMs in the CS13_14 vs. CN47_14 and CS13_21 vs. CN47_21 comparisons, respectively; red means high-level and blue means low-level. (e,f)—The top 20 KEGG pathway classifications of DAMs in the CS13_14 vs. CN47_14 and CS13_21 vs. CN47_21 comparisons, respectively.
3.4. Integrated Transcriptome and Metabolome Analysis
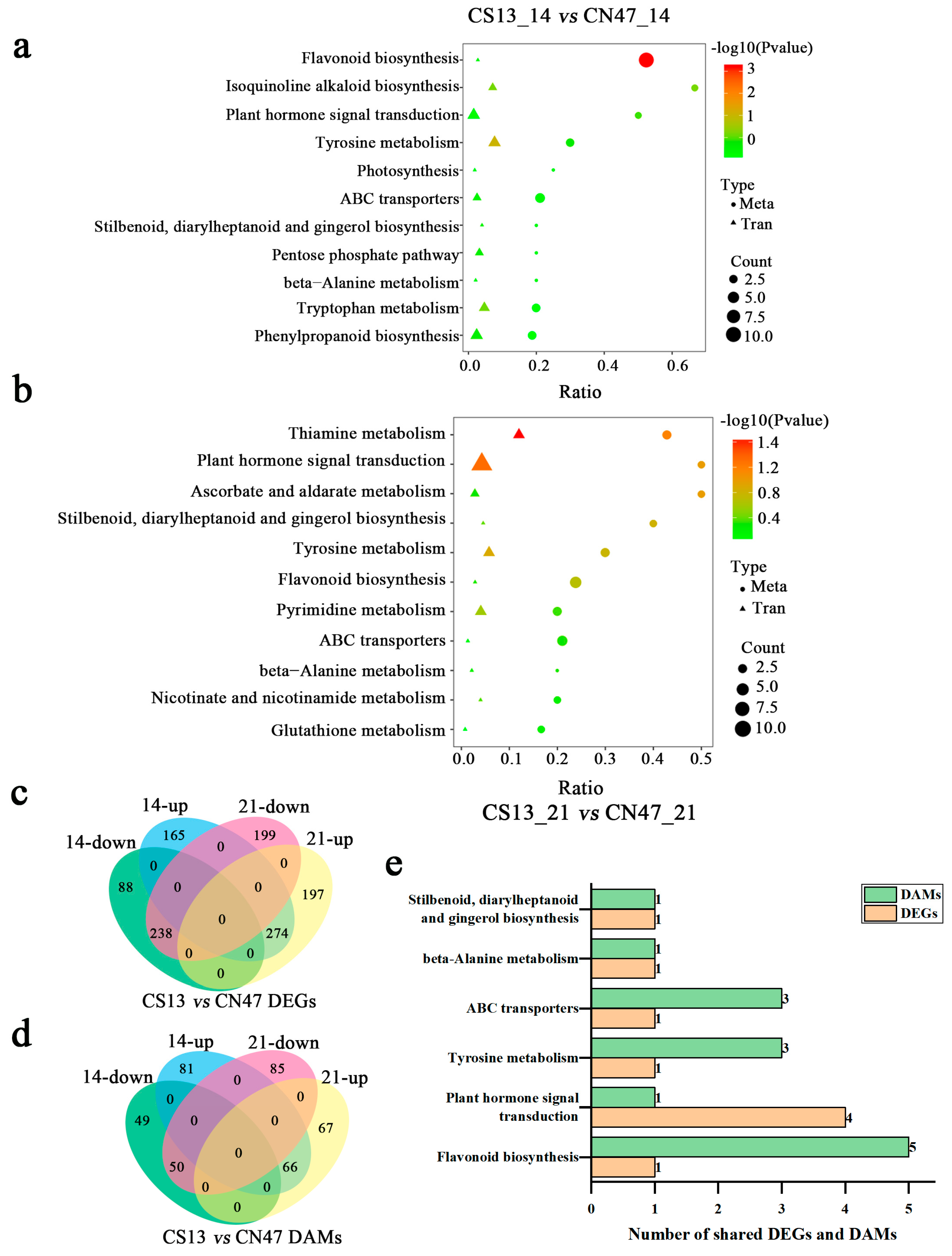
Based on the DAMs and DEGs results, a joint analysis of the metabolome and transcriptome was then conducted in order to unveil their relationship and regulatory mechanisms. Figure 5 displays the KEGG pathways that were co-enriched in both DAMs and DEGs during the 14 DAF and 21 DAF stages. The results show that flavonoid biosynthesis, plant hormone signal transduction, tyrosine metabolism, ABC transporters, beta-Alanine metabolism, and stilbenoid, diarylheptanoid, and gingerol biosynthesis were identified as shared pathways in both comparisons of CS13_14 DAF vs. CSN47_14 DAF and CS13_21 DAF vs. CSN47_21 DAF (Figure 5a,b). However, the DAMs and DEGs mapped to shared pathways differed at different stages (Table S7).
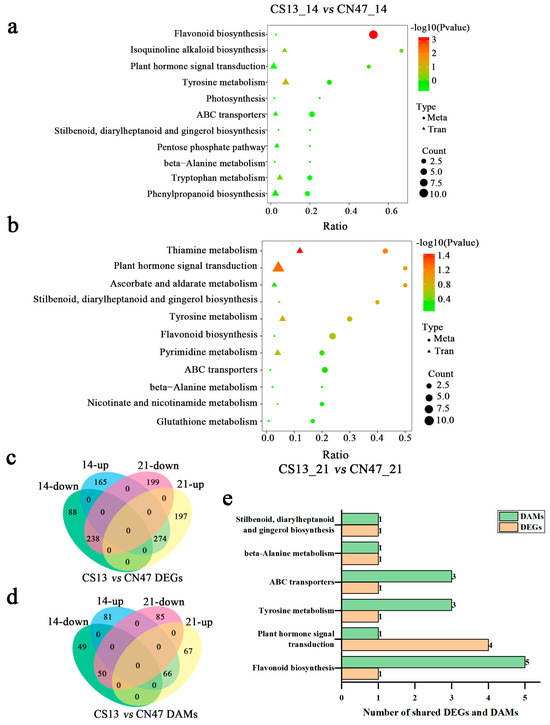
Figure 5.
Co-mapped pathways of DAMs and DEGs at two stages during grain filling of the two foxtail millets. (a,b)—The KEGG pathway analyses of the simultaneous annotations of DAMs and DEGs in the CS13_14 vs. CN47_14 and CS13_21 vs. CN47_21 comparisons, respectively. (c,d)—The Venn diagrams of DEGs and DAMs in the CS13_14 vs. CN47_14 and CS13_21 vs. CN47_21 comparisons, respectively. (e)—The number of shared DAMs and DEGs related to co-mapped pathways. |Cor| > 0.8 and p ≤ 0.05 were used as screening criteria on the co-enrichment pathway of DEGs-DAMs.
To identify key genes related to grain filling rate, a Venn diagram analysis was conducted to compare all DEGs and DAMs at the 14 DAF and 21 DAF stages of two foxtail millet cultivars. As shown in Figure 5c, there were 512 identical DEGs for the comparisons of CS13 and CSN47, with 274 genes up-regulated and 238 genes down-regulated, showing similar trends of change. At the same time, we found 116 identical metabolites, including 66 up-regulated metabolites and 55 down-regulated metabolites (Figure 5d). The change trend in the DAMs was also the same. Further analysis reveals that shared DEGs and DAMs were enriched the same pathway as mentioned above (Table S8; Figure 5e). It can be inferred that flavonoid biosynthesis, plant hormone signal transduction, tyrosine metabolism, ABC transporters, beta-Alanine metabolism, and stilbenoid, diarylheptanoid, and gingerol biosynthesis may be related to the high GFR of CN47.
3.5. IAA and CTK Were Key Regulators of the Grain Filling Rate
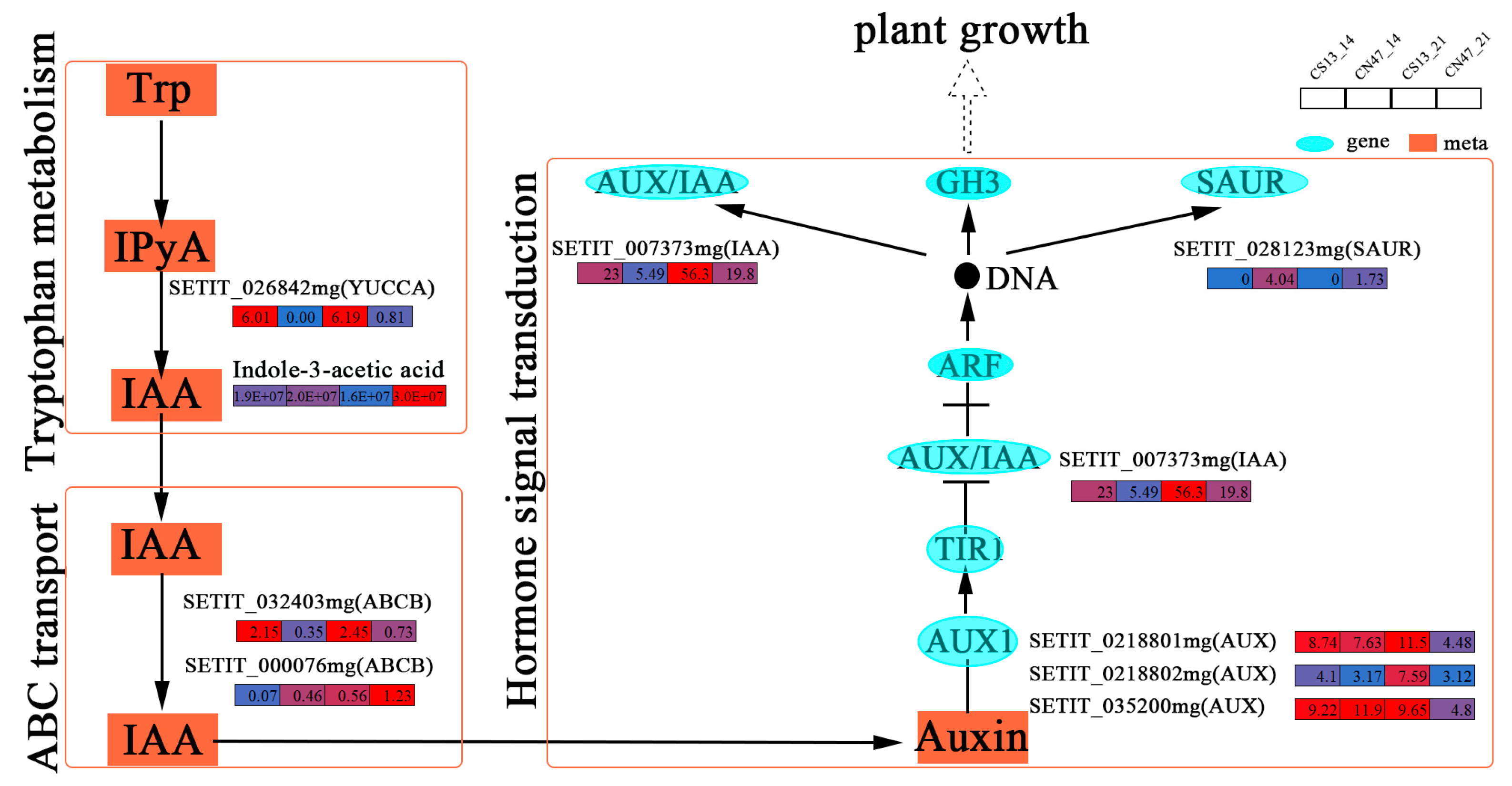
KEGG pathway classification analysis was performed on shared DEGs and DAMs, showing a significant enrichment of shared DEGs in hormone signaling. A total of 11 genes involved in phytohormone signaling were discovered, including auxin-, cytokinin (CTK)-, and ethylene (ETH)-related genes. And the expression patterns of phytohormone-related genes were displayed. Of the enriched phytohormones, auxin played a vital role in the grain filling stages [48]. We found several key genes enriched in the auxin biosynthesis (tryptophan metabolism), transport (ABC transport), and signal transduction pathways at the 14 DAF and 21 DAF stages. And a network was established to display the relationship between auxin and plant growth and development (Figure 6). The tryptophan-dependent pathway is the primary pathway responsible for the production of indole-3-acetic acid (IAA) [49]. SETIT_026842mg was identified, which encodes a YUCCA protein associated with tryptophan metabolism that acts as a rate-limiting enzyme for auxin biosynthesis. This gene was expressed at low levels in CN47 at 14 DAF and 21 DAF, and it may negatively regulate GFR since the IAA concentration of CN47 was higher than that of CS13. And two genes related to ABCB1 were identified, namely SETIT_032403mg and SETIT_000076mg. They are linked to ABC transport and code an ABCB protein that interacts with PIN protein to regulate polar auxin transport. In CN47, SETIT_032403mg showed low expression levels at the 14 DAF and 21 DAF stages of grain filling progress. Conversely, the expression patterns of SETIT_000076mg were different. Additionally, we identified several key genes involved in the auxin signal transduction pathway, including three AUX1; one AUX/IAA; and one SAUR, SETIT_0218801mg, SETIT_0218802mg, SETIT_035200mg, SETIT_007373mg, and SETIT_028123mg. Compared with the 14 DAF stage, the down-regulated genes SETIT_0218801mg, SETIT_0218802mg, SETIT_035200mg, and SETIT_007373mg showed high expression differences at 21 DAF in CN47 compared to CS13. The SAUR family is one of the most important families of auxin-responsive proteins, which regulate foxtail millet grain development [50]. Furthermore, SETIT_028123mg, which was highly expressed in CN47 at the 14 DAF and 21 DAF stages, has been identified as being related to SUAR. It may be positively correlated with the GFR of foxtail millet, and these results suggest its potential role in facilitating extracellular-to-intracellular auxin transport. Notably, SETIT_007373mg and SETIT_028123mg, as auxin early response genes, could regulate downstream gene expression. These data suggest that these genes related to auxin signaling are involved in grain filling progress and GFR. Therefore, the potential function of these key genes related to auxin biosynthesis, transport, and signal transduction underscores the significance of IAA during grain filling and GFR.
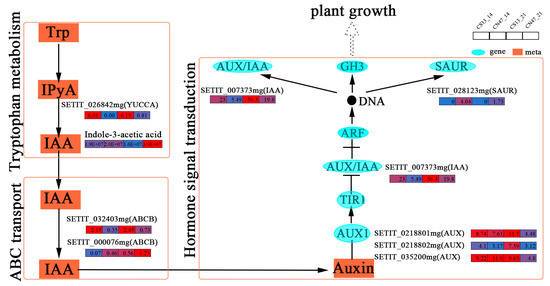
Figure 6.
The gene expressions of the auxin biosynthesis, transport, and signal transduction pathway. The rectangles’ colors represent the expression levels of genes—blue means low-level, red means high-level.
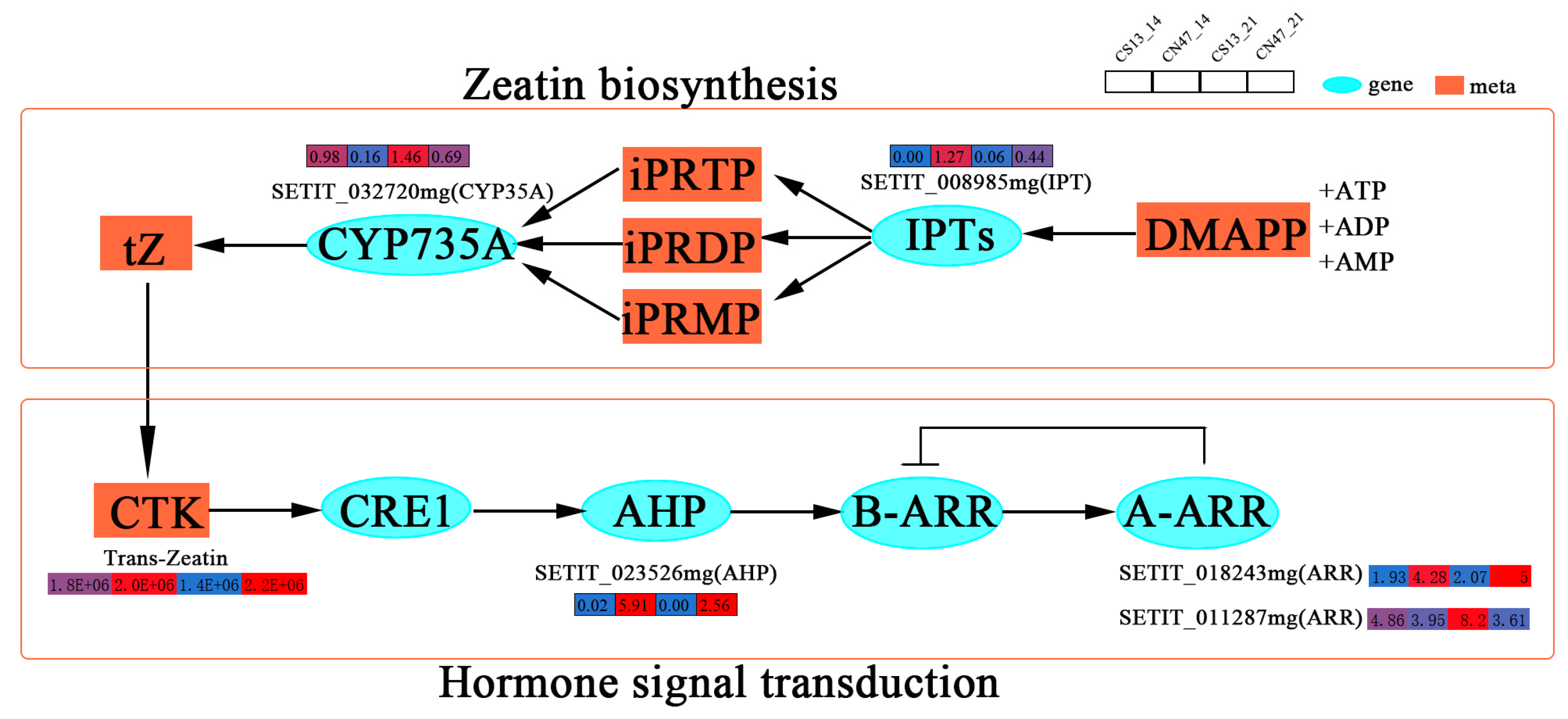
Cytokinin (CTK), an important phytohormone, plays a significant role in grain development [51]. The CTK biosynthesis (Zeatin metabolism) and signal transduction pathway exhibited enrichment of several genes during the grain filling stages at 14 DAF and 21 DAF, as depicted in Figure 7. Furthermore, a network was established to illustrate the correlation between CTK and plant growth and development. It is noteworthy that the Zeatin metabolism pathway plays an important role in the production of CTK. Its genes include a prenyltransferase (SETIT_008985mg) and a cytochrome monooxygenase (SETIT_032720mg), which, respectively, encode the IPTs protein and the CYP735A protein associated with zeatin metabolism. These enzymes act as rate-limiting enzymes for CTK biosynthesis. The expression level of SETIT_008985mg was higher at the 14 DAF and 21 DAF stages in CN47, while the CTK contents were also higher in CN47 compared to CN13 at these developmental stages. This suggests that SETIT_008985mg may be positively correlated with the GFR of foxtail millet. Additionally, we identified three key genes involved in the CTK signal transduction pathway at the 14 DAF and 21 DAF stages. These genes consisted of SETIT_023526mg, SETIT_018243mg, and SETIT_011287mg. Specifically, SETIT_023526mg was shown to encode the AHPs protein, which facilitates the transfer of phosphate groups to response regulators (SETIT_018243mg, SETIT_011287mg), thereby inducing gene expression and regulating plant growth and development. This gene exhibited high expression levels in CN47 at the 14 DAF and 21 DAF stages, suggesting a potential positive correlation with foxtail millet’s GFR. Additionally, SETIT_018243mg and SETIT_011287mg, as members of the type-A (ARR) family of response regulators, displayed distinct expression patterns between CN47 and CS13 during the 14 DAF and 21 DAF stages. These three genes collectively functioned as rate-limiting enzymes for CTK signal transduction. In addition, this study identified SETIT_029213mg and SETIT_009909mg, which are associated with the ethylene-insensitive 3 (EIN3)-like 1 (EIL1) protein and EIN3-like 5 (EIL5) protein, respectively (Figure S2). These genes exhibited high expression levels in CN47 during the 21 DAF stage, suggesting that they may have a positive correlation with GFR.
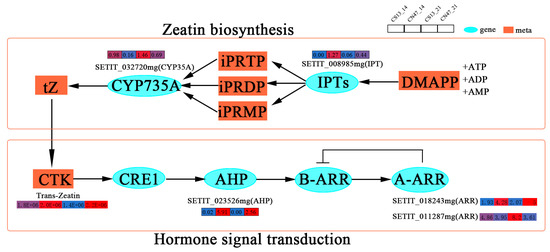
Figure 7.
The gene expressions of the cytokinin (CTK) biosynthesis, transport, and signal transduction pathway. The rectangles’ colors represent the expression levels of genes—blue means low-level, red means high-level.
3.6. qRT-PCR Verification of RNA-Seq Data
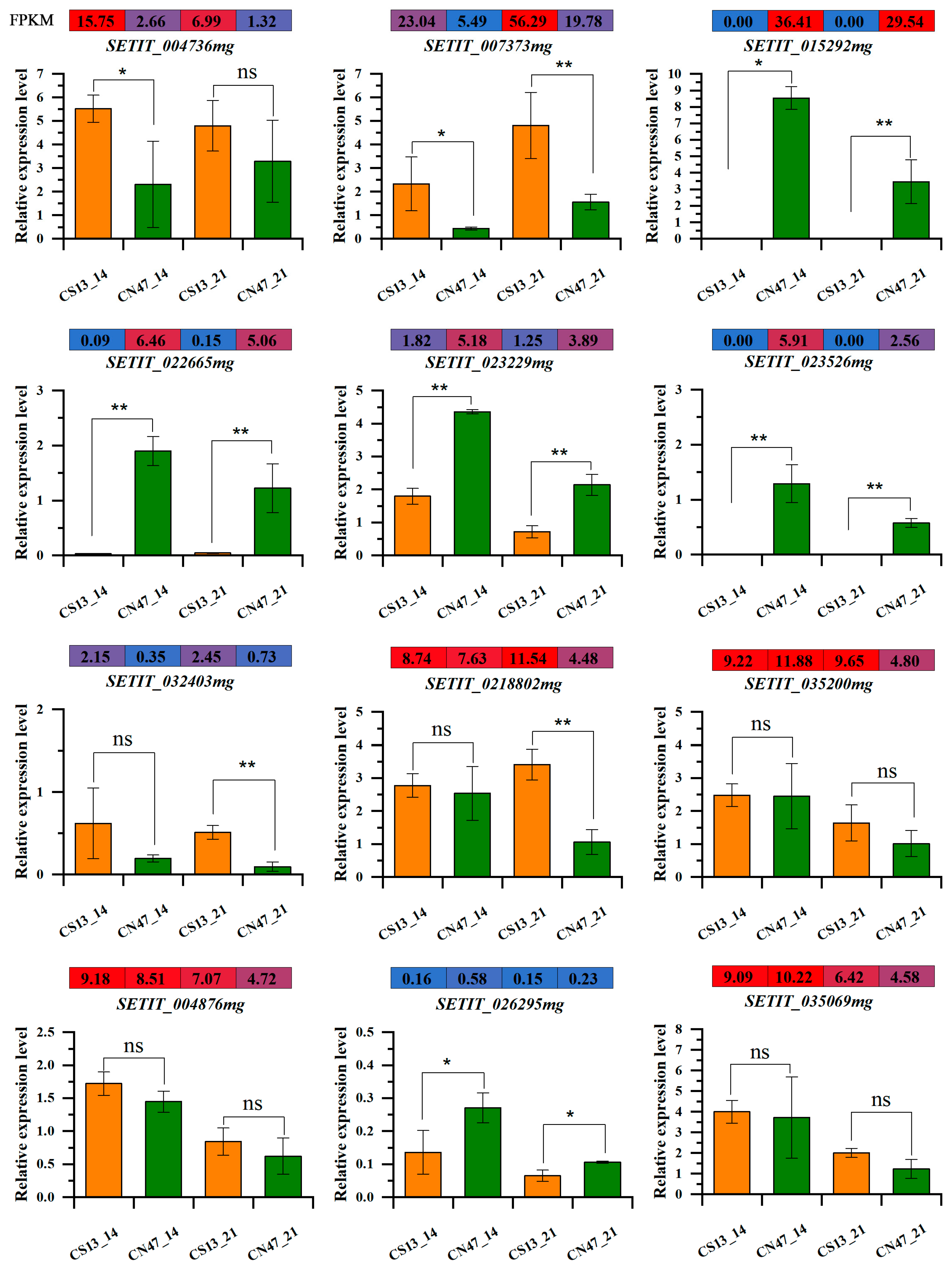
To ensure the accuracy of our RNA-seq data, 12 key genes were selected for qRT-PCR validation (Figure 8). These genes are involved in plant hormone signal transduction, flavonoid biosynthesis, ABC transporters, tyrosine metabolism, and beta-Alanine metabolism, as well as stilbenoid, diarylheptanoid, and gingerol biosynthesis. The results demonstrate that gene expression levels recorded by qRT-PCR align with those obtained from the RNA-seq data, which suggests the reliability of our analytical findings.
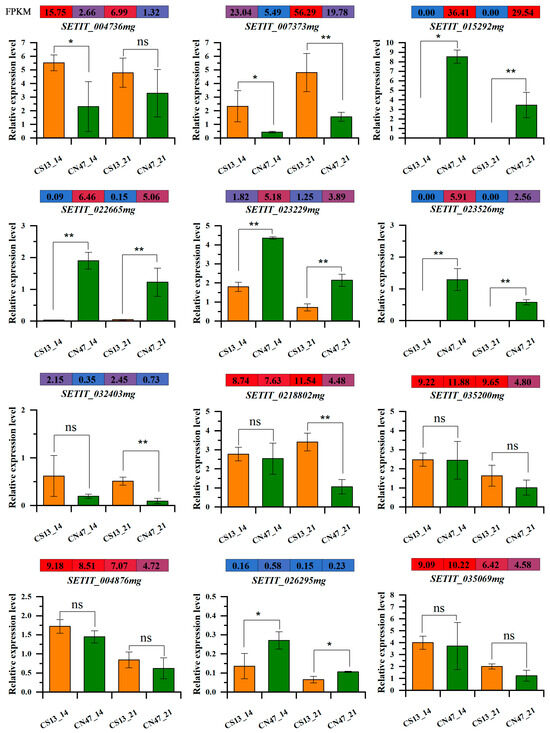
Figure 8.
Quantitative real-time PCR (qRT-PCR) validation and RNA-seq data of 12 selected DEGs. Significance was assessed using the t-test (*, p ≤ 0.05 and **, p ≤ 0.01, ns, p > 0.05).
4. Discussion
Foxtail millet grain filling progress is crucial for seed setting, grain weight, quality, and yield [39]. This process is highly sophisticated, involving the accumulation of carbohydrates and other nutrients by the developing endosperm to synthesize starch and enhance grain development [52]. The interaction of phytohormones, the activity of starch biosynthesis enzymes, the levels of polyamines, and the synthesis and translocation of assimilates all play essential roles in grain filling [24,53,54]. Various factors, such as photosynthetic products, transportation of stored substances, transport tissues, physiological activities of the grains, regulation of plant hormones, and environmental factors, can restrict this progress [55]. Enhancing GFR, particularly improving the rate at which grains are filled with nutrients and water, has been demonstrated as an effective strategy to enhance crop yield and quality [56]. The grain filling rate is an important agronomic trait primarily influenced by genetic factors, while research on the molecular mechanisms of grain development in cereal crops has only been carried out in recent years. Some genes and quantitative trait loci (QTLs) associated with grain size and GFR have been identified and cloned in rice, maize, and wheat [32,33,57]. However, the molecular mechanisms related to GFR in foxtail millet have not yet been reported. Therefore, this study conducted a preliminary investigation into the regulation of fox millet’s grain filling rate and its underlying molecular mechanism.
CS13 and CN47 are well-suited for cultivation in the mid–late mature spring foxtail millet region of northwest China, exhibiting a growth period of 122 days and a heading date ranging from 48 to 50 days [58,59]. The two cultivars have similar agronomic traits, except for the thousand-grain weight. Nevertheless, we found that CN47 has a significantly higher grain filling rate at the 14 DAF and 21 DAF stages compared to CS13. Therefore, CN47 exhibits a high grain filling rate and a great yield phenotype, while CS13 is the opposite. The significant disparity in grain filling rate between CN47 and CS13 makes them the optimal materials for investigating the grain filling rate of spring foxtail millet in the middle and late maturity regions of northwest China. So we sampled and sequenced the grains of foxtail millet in both periods. It can be seen that this study has provided experimental materials for exploring genes and pathways related to the regulation of grain filling rate.
The utilization of RNA-seq is indispensable in the investigation of plant development processes, facilitating our comprehension of gene expression levels across various stages and cultivars [60]. The metabolites, serving as the ultimate outcomes of gene expression, are intricately associated with the development of plant traits. The integrated analysis of transcriptomes and metabolomes is a common approach to identifying pivotal genes and elucidating the mechanisms underlying phenotypic variations. The transcriptome analysis conducted on foxtail millet at five distinct grain filling stages initially revealed the involvement of pathways related to starch biosynthesis, cell-wall invertases, hormone signal transduction, and polyamine metabolism in the process of grain filling [36]. Wang et al. found the intricate mechanisms underlying plant hormone signaling, starch and sugar metabolism, carotenoid metabolism, flavonoid biosynthesis, and folate metabolism pathways during grain development in foxtail millet [9]. Moreover, a comparative transcriptome analysis also revealed significant variations in the expression levels of genes related to the ABC transporter, photosynthesis, and the photosynthesis-antenna protein pathway. Besides, a large-scale metabolome study indicated dynamic changes in flavonoid, glutathione, linoleic acid, starch, sucrose, valine, leucine, and isoleucine during foxtail millet grain filling [38]. In this study, the transcriptome analysis conducted at the 14 DAF and 21 DAF stages revealed inconsistencies with previous reports in terms of significantly enriched pathways. A total of 765 DEGs and 246 DAMs were found at the 14 DAF stage, and 908 DEGs and 268 DAMs were found at the 21 DAF stage. However, RNA-seq data analysis indicated that a limited number of DEGs were implicated in pathways associated with the process of grain filling. Metabolome analysis revealed that pathways such as flavonoid biosynthesis, ABC transporters, photosynthesis, and plant hormone signal transduction were consistent with previous research findings. The findings indicated that a limited number of DEGs were implicated in pathways associated with the process of grain filling. The DEGs and DAMs identified between CS13 and CN47 at the 14 DAF and 21 DAF stages were found to be associated with pathways such as flavonoid biosynthesis, plant hormone signal transduction, tyrosine metabolism, ABC transporters, and beta-Alanine metabolism, as well as stilbenoid, diarylheptanoid, and gingerol biosynthesis. The results obtained above demonstrate that flavonoid biosynthesis, ABC transporters, and plant hormone signal transduction pathways play a crucial role in influencing the grain filling process of foxtail millet. Importantly, these pathways have also been identified as potentially significant factors contributing to the disparity in GFR between CS13 and CN47.
Flavonoids, as a natural oxidizer, are considered to confer nutritional and pharmacological benefits to plants [61]. They play essential roles in enhancing the overall quality of plants, encompassing aspects such as color and flavor [62]. This study revealed that foxtail millets are abundant in flavonoids, such as vitexin, naringenin, and naringenin chalcone. It is widely acknowledged that the flavonoid metabolic pathway significantly influences the development of a yellow hue in de-husked grains. In essence, the flavonoid metabolic pathway plays a crucial role in GFR. In this study, the integration of the DEGs and DAMs between CS13 and CN47 at the 14 DAF and 21 DAF stages identified co-enrichment of flavonoid biosynthesis. Specifically, the pathway only contained one gene, SETIT_015292mg, which encodes a methyltransferase involved in the post-modification of flavonoids. The gene clearly encodes flavonoid O-methyltransferase (FOMT), which serves as a key modifying enzyme in the flavonoid metabolic pathway by catalyzing the synthesis of O-methylated derivatives of flavonoids [63]. Furthermore, SETIT_015292mg also plays a crucial role in the stilbenoid, diarylheptanoid, and gingerol biosynthesis pathway (Table S8). Interestingly, the expression of SETIT_015292mg was observed exclusively in CN47, but not in CS13, which could potentially serve as a significant contributing factor to the high GFR in CN47 (Figure S3).
The ABC transporters are essential for normal plant development and are essential to diverse biological processes, encompassing auxin transport and seed development in plants [64]. In cereal crops, numerous genes related to ABC transporters have been found and identified as being linked with grain development and grain filling. TaABCC3 promotes grain formation in wheat, while its suppression results in a reduction in the number of grains [65]. And wheat ABCC13 is reported to be functionally essential for grain development [66]. Similarly, strong expression of OsABCI15 and OsABCI16 is exhibited in the seeds of rice, indicating their involvement in rice seed development. Overexpression of either gene significantly enhances the grain yield, while CRISPR/Cas9-mediated loss-of-function mutations in these genes result in incomplete filling of developing seeds [67]. The expression levels of several ABC transporter-related DEGs, including ABCB and ABCC genes, have been found to be significantly up-regulated during the advanced stages of grain development in foxtail millets [47]. In contrast, our study found that two ABCB1 genes in the ABC transporters pathway exerted influences on grain filling at different stages by regulating auxin transport. These genes, SETIT_032403mg and SETIT_000076mg, exhibited different expression patterns between CS13 and CN47 at the 14 DAF and 21 DAF stages, suggesting diverse roles of ABC transporters in GFR.
The process of grain filling is largely regulated by phytohormones. So far, numerous genes related to plant hormones have been identified. The development of rice panicles is regulated by auxin through the modulation of growth gene expression [68]. Furthermore, a study has reported that auxin-responsive factors potentially influence early grain filling in foxtail millet [47]. Additionally, some studies have shown that CTK and ETH are involved in regulating grain filling [69,70]. In the present study, many differentially expressed genes, including YUCCA, ABCB, AUXIN1, AUX/IAA, SAUR, IPTs, AHP, ARR, and EIN3, were found to be associated with the biosynthesis as well as the signaling pathways of plant hormones. Notably, Indole-3-acetic acid (IAA) was detected as a DAM. The majority of genes associated with auxin were found within the plant hormone signal transduction pathway. The identification of genes related to the auxin signaling pathway could provide useful materials for the molecular breeding of foxtail millet. Therefore, our primary focus lies on elucidating the mechanisms underlying auxin-mediated grain filling and exploring strategies to regulate the growth factor receptor.
Auxin, a phytohormone, is known to be crucial for endosperm development and grain filling [57]. YUCCA flavin monooxygenases are involved in auxin biosynthesis. OsYUC11, the auxin biosynthesis gene, is indispensable to the process behind grain plumpness in rice. Mutations of OsYUC11 have been shown to hinder grain filling and the accumulation of storage proteins [71,72]. Blocking auxin transport leads to abnormalities in seeds [73]. The intercellular transport of auxin, facilitated by PIN [74], ABCB [75], and AUXIN [76], is heavily dependent on auxin establishing and maintaining an optimal concentration. The involvement of ABCB proteins in auxin transport has been reported, with ABCB1, ABCB4, and ABCB19 being responsible for its directional transport in rice [77]. The auxin signal transduction pathway, encompassing TIR1/AFB co-receptors, Aux/IAA repressors, and ARF transcription factors, is recognized for its regulatory role in grain yield. In rice, the module of OsSK41-OsIAA10-OsARF is known to regulate grain yield [78]; meanwhile, research has shown that the functions of OsTIR1/AFB genes are redundant for yield in rice [79]. Nevertheless, the module’s involvement in the grain filling process remains ambiguous. The OsARF12-mediated auxin signal regulates grain filling and grain size by regulating cell division [80]. SAUR genes, which are early auxin-responsive genes, play a role in plant growth, particularly in cell elongation [81]. In this study, we identified several vital genes related to auxin biosynthesis, transport, and signal transduction. Contrary to findings in other plant species, the expression of a YUCCA gene was found to be higher in CS13 with low GFR compared to CN47 with high GFR at 14 DAF. Interestingly, the variation in auxin levels between the two cultivars was observed at 21 DAF instead of at 14 DAF. It is hypothesized that YUCCA may exert a negative regulatory effect on auxin synthesis, with its impact being delayed. Furthermore, several genes related to the auxin transport pathway have been identified, belonging to the ABCB gene family and the AUXIN gene family (Figure 6). In rice, the low expression of OsLAX1/OsAUX1 was found to increase root length [82]. Similarly, the low expression levels of four genes in CN47 may also lead to an increase in grain length by promoting the grain filling rate. The expression levels of SETIT_007373mg and SETIT_028123mg as auxin response genes exhibited contrasting trends. The expression level of the AUX/IAA gene SETIT_007373mg was found to be higher in CS13, whereas a decrease in expression was observed for the SAUR gene SETIT_028123mg. The expression profiles of different auxin response genes may vary in their responsiveness to auxin signaling pathways. Based on these findings, it is reasonable to suggest that these genes possibly exert diverse regulatory functions across various stages. The genes YUCCA, AUX/IAA, and SAUR in foxtail millet are major candidates and further investigation is required to elucidate their involvement in the grain filling process.
GFR, as a pivotal agronomic trait and a complex quantitative trait, necessitates the exploration and investigation of the regulatory genes and superior alleles associated with GFR in order to achieve precise breeding for rapid grain filling characteristics, thereby augmenting the yield and quality of foxtail millet. In recent decades, the breeding of hybrid crops, including hybrid foxtail millet, and their large-scale popularization have made a universally recognized contribution to improving yield per unit area. However, these hybrid crops often exhibit inadequate grain filling, suboptimal plumpness, and a failure to achieve the expected spikelet count in terms of yield. Therefore, it is important to improve grain filling, especially the grain filling rate, to increase hybrid grain yield [40]. Currently, research on the genetic analysis of GFR is still in its nascent stages, with limited comprehension of the molecular regulatory mechanism and network underlying GFR. The molecular genetic analysis of grain-related characteristics and traits, such as GFR, is poised to become a focal point that can yield pivotal breakthroughs in the field of crop research, including research on foxtail millet. The application of microCT and other phenomics-related technologies in grain filling research [83] is expected to enable high-throughput real-time measurement of GFR under different panicle positions and environmental conditions. This will facilitate the analysis of the molecular genetic mechanisms underlying the spatiotemporal complexity and environmental variability of GFR.
5. Conclusions
The present study adopted a joint analysis of transcriptomes and metabolomes at the 14 DAF and 21 DAF stages to investigate the molecular mechanism regulating GFR in CS13 (low GFR) and CN47 (high GFR). The results identified a total of 765 DEGs and 908 DEGs, as well as 246 DAMs and 268 DAMs, at the 14 DAF and 21 DAF stages in CS13 and CN47, respectively. The functional analysis of DEGs and DAMs revealed stage-specific properties in the GFR of foxtail millet. The study lays a foundation for understanding the regulatory network related to GFR and complex metabolic genes. Several important pathways—such as flavonoid biosynthesis, plant hormone signal transduction, tyrosine metabolism, ABC transporters, and beta-Alanine metabolism, as well as stilbenoid, diarylheptanoid, and gingerol biosynthesis—were enriched via a conjoint analysis of DEGs and DAMs. These results indicate a direction to follow in order to study the metabolic pathway genes related to GFR. Notably, the majority of shared DEGs were enriched within hormone signaling pathways, including auxin, cytokinin, and so on. In particular, several key genes related to auxin biosynthesis (YUCCA), transport (ABCB1), and signal transduction (AUX1, AUX/IAA, SAUR) pathways were identified in the ABC transporters and plant hormone signal transduction pathways, which provide valuable candidate genes for improving GFR and grain production. These findings highlight the significant role of auxin in regulating GFR and grain filling progress, and also provide valuable molecular insights for investigating differences in GFR among foxtail millet cultivars and theoretical mechanisms for high-yield breeding.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/agronomy14061114/s1, Figure S1: The peak and gel maps of 12 RNA samples; Figure S2: The gene expressions of the ethylene (ETH) transport and signal transduction pathways; Figure S3: The expression levels (FPKM) of SETIT_015292mg at 14 DAF and 21 DAF between CS13 and CN47; Table S1: Primers used for qRT-PCR; Table S2: Summary of sequencing data quality and transcriptome statistics; Table S3: Sample comparison regional statistics; Table S4: Statistical information of the identified metabolites; Table S5: KEGG pathway classification of DAMs at the 14 DAF stage; Table S6: KEGG pathway classification of DAMs at the 21 DAF stage; Table S7: DAMs and DEGs mapped to shared pathways at the 14 DAF and 21 DAF stages; Table S8: Shared DAMs and DEGs mapped to shared pathways.
Author Contributions
Y.H., P.Z. and A.Z., conceptualization; G.W., software; Y.H. and P.Z., validation; M.L., formal analysis; Y.H., P.Z., Y.Z. and E.G., investigation; P.Z., data curation; Y.H. and P.Z., writing—original draft; A.Z., writing—review and editing; A.Z., funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Key Research and Development Program of China (2023YFD1202704); the Construction Project of the National Modern Agricultural Industry Technology System (CARS-06-14.5-A21); and the Construction Project of the Modern Agricultural Industrial Technology System in Shanxi Province (2023CYJSTX04-04).
Data Availability Statement
Available upon request from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
Correction Statement
This article has been republished with a minor correction to resolve spelling and grammatical errors. This change does not affect the scientific content of the article.
References
- Lata, C.; Gupta, S.; Prasad, M. Foxtail millet: A model crop for genetic and genomic studies in bioenergy grasses. Crit. Rev. Biotechnol. 2013, 33, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Memariani, Z.; Abbas, S.; Hassan, S.; Ahmadi, A.; Chabra, A. Naringin and naringenin as anticancer agents and adjuvants in cancer combination therapy: Efficacy and molecular mechanisms of action, a comprehensive narrative review. Pharmacol. Res. 2021, 171, 105264. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Purewal, S.; Sandhu, K.; Kaur, M.; Salar, R. Millets: A cereal grain with potent antioxidants and health benefits. J. Food Meas. Charact. 2019, 13, 793–806. [Google Scholar] [CrossRef]
- Sachdev, N.; Goomer, S.; Singh, L. Foxtail millet: A potential crop to meet future demand scenario for alternative sustainable protein. J. Sci. Food Agric. 2021, 101, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Sushree, S.; Rana, S.; Suranjika, S.; Muthamilarasan, M.; Parida, A.; Prasad, M. Genetic determinants of micronutrient traits in graminaceous crops to combat hidden hunger. Theor. Appl. Genet. 2021, 134, 3147–3165. [Google Scholar] [CrossRef] [PubMed]
- Amadou, I.; Amza, T.; Shi, Y.; Le, G. Chemical analysis and antioxidant properties of foxtail millet bran extracts. Songklanakarin J. Sci. Technol. 2011, 33, 509–515. [Google Scholar]
- Liang, S.; Yang, G.; Ma, Y. Chemical characteristics and fatty acid profile of foxtail millet bran oil. J. Am. Oil Chem. Soc. 2010, 87, 63–67. [Google Scholar] [CrossRef]
- Sharma, N.; Niranjan, K. Foxtail millet: Properties, processing, health benefits, and uses. Food Rev. Int. 2018, 34, 329–363. [Google Scholar] [CrossRef]
- Wang, D.; Su, M.; Hao, J.; Li, Z.; Dong, S.; Yuan, X.; Li, X.; Gao, L.; Chu, X.; Yang, G.; et al. Dynamic transcriptome landscape of foxtail millet grain development. Seed Biol. 2023, 2, 19. [Google Scholar] [CrossRef]
- Xing, Y.; Zhang, Q. Genetic and molecular bases of rice yield. Annu. Rev. Plant Biol. 2010, 61, 421–442. [Google Scholar] [CrossRef]
- Li, X.; Pan, Z. A study on the grain filling characteristic of different weight wheat. Rev. China Agric. Sci. Technol. 2005, 7, 26–30. [Google Scholar] [CrossRef]
- Shouichi, Y.; Hara, T. Effects of air temperature and light on grain filling of an indica and a japonica rice (Oryza sativa L.) under controlled environmental conditions. J. Soil. Sci. Plant Nutr. 1977, 23, 15. [Google Scholar] [CrossRef]
- Wiegand, C.; Cuellar, J. Duration of grain filling and kernel weight of wheat as affected by temperature. Crop Sci. 1981, 21, 95–101. [Google Scholar] [CrossRef]
- Ma, J.; Ming, D.; Ma, W.; Xu, F. Effects of different nitrogen application periods on rice starch accumulation and starch synthesis Studies on the activity changes of related enzymes. Sci. Agric. Sin. 2005, 38, 290–296. [Google Scholar]
- Wang, Z.; Xu, Y.; Chen, T.; Zhang, H.; Yang, J.; Zhang, J. Abscisic acid and the key enzymes and genes in sucrose-to-starch conversion in rice spikelets in response to soil drying during grain filling. Planta 2015, 241, 1091–1107. [Google Scholar] [CrossRef]
- Sanford, D. Variation in kernel growth characters among soft red winter wheats. Crop Sci. 1985, 25, 626–630. [Google Scholar] [CrossRef]
- Mashiringwani, N.; Mashingaidze, K.; Kangai, J.; Olsen, K. Genetic basis of grain filling rate in wheat (Triticum aestivum L. emend. Thell.). Euphytica 1994, 76, 33–44. [Google Scholar] [CrossRef]
- Jones, D.; Peterson, M.; Geng, S. Association between grain filling rate and duration and yield components in rice. Crop Sci. 1979, 19, 641–644. [Google Scholar] [CrossRef]
- Zhao, Y.; Peng, T.; Sun, H.; Teotia, S.; Wen, H.; Du, Y.; Zhang, J.; Li, J.; Tang, G.; Xue, H.; et al. miR1432-OsACOT (acyl-CoA thioesterase) module determines grain yield via enhancing grain filling rate in rice. Plant Biotechnol. J. 2019, 17, 712–723. [Google Scholar] [CrossRef]
- Zhu, D.; Wang, H.; Liu, D.; Gao, D.; Lu, G.; Wang, J.; Gao, Z.; Lu, C. Characteristics of grain filling and dehydration in wheat. Sci. Agric. Sin. 2019, 52, 4251–4261. [Google Scholar] [CrossRef]
- Wei, X.; Jiao, G.; Lin, H.; Sheng, Z.; Shao, G.; Xie, L.; Tang, S.; Xu, Q.; Hu, P. GRAIN INCOMPLETE FILLING 2 regulates grain filling and starch synthesis during rice caryopsis development. J. Integr. Plant Biol. 2017, 59, 134–153. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Okamura, K.; Miyazaki, M.; Phan, T.; Yuasa, T.; Iwaya-Inoue, M. Expression of rice sucrose transporter gene OsSUT1 in sink and source organs shaded during grain filling may affect grain yield and quality. Environ. Exp. Bot. 2014, 97, 49–54. [Google Scholar] [CrossRef]
- Bai, A.; Lu, X.; Li, D.; Liu, J.; Liu, C. NF-YB1-regulated expression of sucrose transporters in aleurone facilitates sugar loading to rice endosperm. Cell Res. 2016, 26, 384–388. [Google Scholar] [CrossRef]
- Sosso, D.; Luo, D.; Li, Q.; Sasse, J.; Yang, J.; Gendrot, G.; Suzuki, M.; Koch, K.; McCarty, D.; Chourey, P.; et al. Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat. Genet. 2015, 47, 1489–1493. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, J.; Zhang, Z.; Wu, Y. Transactivation of Sus1 and Sus2 by Opaque2 is an essential supplement to sucrose synthase-mediated endosperm filling in maize. Plant Biotechnol. J. 2020, 18, 1897–1907. [Google Scholar] [CrossRef]
- Wang, E.; Wang, J.; Zhu, X.; Hao, W.; Wang, L.; Li, Q.; Zhang, L.; He, W.; Lu, B.; Lin, H.; et al. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 2008, 40, 1370–1374. [Google Scholar] [CrossRef]
- Kuanar, S.; Molla, K.; Chattopadhyay, K.; Sarkar, R.; Mohapatra, P. Introgression of Sub1 (SUB1) QTL in mega rice cultivars increases ethylene production to the detriment of grain-filling under stagnant flooding. Sci. Rep. 2019, 9, 18567. [Google Scholar] [CrossRef]
- Zhu, G.; Ye, N.; Yang, J.; Peng, X.; Zhang, J. Regulation of expression of starch synthesis genes by ethylene and ABA in relation to the development of rice inferior and superior spikelets. J. Exp. Bot. 2011, 62, 3907–3916. [Google Scholar] [CrossRef]
- Zhang, W.; Cao, Z.; Zhou, Q.; Chen, J.; Xu, G.; Gu, J.; Liu, L.; Wang, Z.; Yang, J.; Zhang, H. Grain filling characteristics and their relations with endogenous hormones in large- and small-grain mutants of rice. PLoS ONE 2016, 11, e0165321. [Google Scholar] [CrossRef] [PubMed]
- Panda, B.; Sekhar, S.; Dash, S.; Behera, L.; Shaw, B. Biochemical and molecular characterisation of exogenous cytokinin application on grain filling in rice. BMC Plant Biol. 2018, 18, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, M.; Zhou, Y.; Wang, Y.; Shen, J.; Chen, H.; Zhang, L.; Lü, B.; Liang, G.; Liang, J. The Rice G Protein gamma Subunit DEP1/qPE9-1 Positively Regulates Grain-Filling Process by Increasing Auxin and Cytokinin Content in Rice Grains. Rice 2019, 12, 91. [Google Scholar] [CrossRef]
- Liu, E.; Zeng, S.; Zhu, S.; Liu, Y.; Wu, G.; Zhao, K.; Liu, X.; Liu, Q.; Dong, Z.; Dang, X.; et al. Favorable Alleles of GRAIN-FILLING RATE1 increase the grain-filling rate and yield of rice. Plant Physiol. 2019, 181, 1207–1222. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, X.; Yang, T.; Yang, X.; Wang, Z.; Wu, F.; Liu, S.; Li, C.; Deng, M.; Ma, J.; et al. Identifcation and validation of stable quantitative trait loci for grain filling rate in common wheat (Triticum aestivum L.). Theor. Appl. Genet. 2020, 133, 2377–2385. [Google Scholar] [CrossRef]
- Bennetzen, J.; Schmutz, J.; Wang, H.; Percifield, R.; Hawkins, J.; Pontaroli, A.; Estep, M.; Feng, L.; Vaughn, J.; Grimwood, J.; et al. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 2012, 30, 555–561. [Google Scholar] [CrossRef]
- Jia, G.; Huang, X.; Zhi, H.; Zhao, Y.; Zhao, Q.; Li, W.; Chai, Y.; Yang, L.; Liu, K.; Lu, H.; et al. A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet (Setaria italica). Nat. Genet. 2013, 45, 957–961. [Google Scholar] [CrossRef]
- Wang, T.; Song, H.; Li, P.; Wei, Y.; Hu, N.; Chen, Z.; Wang, W.; Liu, J.; Zhang, B.; Peng, R. Transcriptome Analysis Provides Insights into Grain Filling in Foxtail Millet (Setaria italica L.). Int. J. Mol. Sci. 2020, 21, 5031. [Google Scholar] [CrossRef]
- Wang, T.; Wang, S.; Tong, Y.; Wei, Y.; Li, P.; Hu, N.; Liu, Y.; Zhao, Z.; Zhao, Y.; Chen, H.; et al. Spatiotemporal dynamics of the foxtail millet transcriptome during grain filling. Physiol. Plant 2024, 176, e14157. [Google Scholar] [CrossRef]
- Wang, T.; Xing, L.; Song, H.; Wei, Y.; Li, P.; Lu, Q.; Hu, N.; Liu, Y.; Zhao, Y.; Liu, J.; et al. Large-scale metabolome analysis reveals dynamic changes of metabolites during foxtail millet grain filling. Food Res. Int. 2023, 165, 112516. [Google Scholar] [CrossRef]
- Wang, T.; Lu, Q.; Song, H.; Hu, N.; Wei, Y.; Li, P.; Liu, Y.; Zhao, Z.; Liu, J.; Zhang, B.; et al. DNA methylation and RNA-sequencing analysis show epigenetic function during grain filling in foxtail millet (Setaria italica L.). Front. Plant Sci. 2021, 12, 741415. [Google Scholar] [CrossRef]
- Song, H.; Wang, T.; Li, L.; Xing, L.; Xie, H.; Feng, B.; Liu, J. Comparative transcriptome analysis provides insights into grain filling commonalities and differences between foxtail millet [Setaria italica (L.) P. Beauv.] varieties with different panicle types. Peer J. 2022, 10, e12968. [Google Scholar] [CrossRef]
- Bolger, A.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, 106. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.; Blake, J.; Botstein, D.; Butler, H.; Cherry, J.; Davis, A.; Dolinski, K.; Dwight, S.; Eppig, J.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef]
- Want, E.; Masson, P.; Michopoulos, F.; Wilson, I.; Theodoridis, G.; Plumb, R.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J. Global metabolic profiling of animal andhuman tissues via UPLC-MS. Nat. Protoc. 2012, 8, 17–32. [Google Scholar] [CrossRef]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef]
- Zhang, H.; Tan, G.; Yang, L.; Yang, J.; Zhang, J.; Zhao, B. Hormones in the grains and roots in relation to post-anthesis development of inferior and superior spikelets in japonica/indica hybrid rice. Plant Physiol. Biochem. 2009, 47, 195–204. [Google Scholar] [CrossRef]
- Zhao, Y. Auxin biosynthesis: A simple two-step pathway converts tryptophan to indole-3-acetic acid in plants. Mol. Plant 2012, 5, 334–338. [Google Scholar] [CrossRef]
- Ma, X.; Dai, S.; Qin, N.; Zhu, C.; Qin, J.; Li, J. Genome-wide identification and expression analysis of the SAUR gene family in foxtail millet (Setaria italica L.). BMC Plant Biol. 2023, 23, 31. [Google Scholar] [CrossRef]
- Mok, M. Cytokinins and plant development—An overview. In Cytokinins, 1st ed.; Mok, M.C., Ed.; CRC Press: Boca Raton, FL, USA, 2019; pp. 15–66. [Google Scholar] [CrossRef]
- Ishimaru, T.; Matsuda, T.; Ohsugi, R.; Yamagishi, T. Morphological development of rice caryopses located at the different positions in a panicle from early to middle stage of grain filling. Funct. Plant Biol. 2003, 30, 1139–1149. [Google Scholar] [CrossRef]
- Liu, D.; Zhao, H.; Xiao, Y.; Zhang, G.; Cao, S.; Yin, W.; Qian, Y.; Yin, Y.; Zhang, J.; Chen, S.; et al. A cryptic inhibitor of cytokinin phosphorelay controls rice grain size. Mol. Plant 2022, 15, 293–307. [Google Scholar] [CrossRef]
- Zhao, Y. Essential roles of local auxin biosynthesis in plant development and in adaptation to environmental changes. Annu. Rev. Plant Biol. 2018, 69, 417–435. [Google Scholar] [CrossRef]
- Slafer, G.; Foulkes, M.; Reynolds, M.; Murchie, E.; Carmo-Silva, E.; Flavell, R.; Gwyn, J.; Sawkins, M.; Griffiths, S. A ‘wiring diagram’ for sink strength traits impacting wheat yield potential. J. Exp. Bot. 2023, 74, 40–71. [Google Scholar] [CrossRef]
- Wang, G.; Kang, M.; Moreno, O. Genetic analyses of grain-filling rate and duration in maize. Field Crop Res. 1999, 61, 211–222. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Z.; Cui, Z.; Hu, Y.; Wang, B.; Tang, J. Genetic analysis of grain filling rate using conditional QTL mapping in maize. PLoS ONE 2013, 8, e56344. [Google Scholar] [CrossRef]
- Hao, X.; Wang, G.; Wang, X.; Yang, H.; Cheng, Q.; Qin, Y. Breeding and cultivation techniques of millet variety Changsheng 13 suitable for mechanized production. China Seed Ind. 2019, 10, 74–76. [Google Scholar] [CrossRef]
- Fan, H.; Zhao, P.; Zhang, A.; Li, Y.; Wang, L.; Wang, R.; Guo, E. Breeding of high-quality herbicide-resistant millet variety Changnong 47 and its high-yield cultivation techniques. J. Hebei Agric. Sci. 2022, 26, 1–5. [Google Scholar]
- Garg, R.; Jain, M. RNA-Seq for Transcriptome Analysis in Non-model Plants. Methods Mol. Biol. 2013, 1069, 43–58. [Google Scholar] [CrossRef]
- Li, X.; Gao, J.; Song, J.; Guo, K.; Hou, S.; Wang, X.; He, Q.; Zhang, Y.; Yang, Y.; Tang, J.; et al. Multi-omics analyses of 398 foxtail millet accessions reveal genomic regions associated with domestication, metabolite traits, and anti-inflammatory effects. Mol. Plant 2022, 15, 1367–1383. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, J.; Qie, Q.; Yang, Y.; Hou, S.; Wang, X.; Li, X.; Han, Y. Comparative Analysis of Flavonoid Metabolites in Foxtail Millet (Setaria italica) with Different Eating Quality. Life 2021, 11, 578. [Google Scholar] [CrossRef]
- Schubert, H.; Blumenthal, R.; Cheng, X. Many paths to methyltransfer: A chronicle of convergence. Trends Biochem. Sci. 2003, 28, 329–335. [Google Scholar] [CrossRef]
- Rea, P. Plant ATP-Binding cassette transporters. Annu. Rev. Plant Biol. 2007, 58, 347–375. [Google Scholar] [CrossRef]
- Walter, S.; Kahla, A.; Arunachalam, C.; Perochon, A.; Khan, M.; Scofield, S.; Doohan, F. A wheat ABC transporter contributes to both grain formation and mycotoxin tolerance. J. Exp. Bot. 2015, 66, 2583–2593. [Google Scholar] [CrossRef]
- Bhati, K.; Alok, A.; Kumar, A.; Kaur, J.; Tiwari, S.; Pandey, A. Silencing of ABCC13 transporter in wheat reveals its involvement in grain development, phytic acid accumulation and lateral root formation. J. Exp. Bot. 2016, 67, 4379–4389. [Google Scholar] [CrossRef]
- Ma, B.; Cao, X.; Li, X.; Bian, Z.; Zhang, Q.; Fang, Z.; Liu, J.; Li, Q.; Liu, Q.; Zhang, L.; et al. Two ABCI family transporters, OsABCI15 and OsABCI16, are involved in grain-filling in rice. J. Genet. Genom. 2023; in press. [Google Scholar] [CrossRef]
- Gao, F.; Wang, K.; Liu, Y.; Chen, Y.; Chen, P.; Shi, Z.; Luo, J.; Jiang, D.; Fan, F.; Zhu, Y.; et al. Blocking miR396 increases rice yield by shaping inflorescence architecture. Nat. Plants 2015, 2, 15196. [Google Scholar] [CrossRef]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef]
- Li, H.; Lu, Q.; Deng, J.; Huang, J.; Cai, F.; Liang, C.; Chen, Q.; Wang, Y.; Zhu, L.; Zhang, X.; et al. Transcriptome analysis reveals key seed-development genes in common buckwheat (Fagopyrum esculentum). Int. J. Mol. Sci. 2019, 20, 4303. [Google Scholar] [CrossRef]
- Xu, X.; E, Z.; Zhang, D.; Yun, Q.; Zhou, Y.; Niu, B.; Chen, C. OsYUC11-mediated auxin biosynthesis is essential for endosperm development of rice. Plant Physiol. 2021, 185, 934–950. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, M.; Liang, J. RGB1 Regulates Grain Development and Starch Accumulation Through Its Effect on OsYUC11-Mediated Auxin Biosynthesis in Rice Endosperm Cells. Front. Plant Sci. 2021, 12, 585174. [Google Scholar] [CrossRef] [PubMed]
- Forestan, C.; Meda, S.; Varotto, S. ZmPIN1-mediated auxin transport is related to cellular differentiation during maize embryogenesis and endosperm development. Plant Physiol. 2010, 152, 1373–1390. [Google Scholar] [CrossRef] [PubMed]
- Adamowski, M.; Friml, J. PIN-dependent auxin transport: Action, regulation, and evolution. Plant Cell 2015, 27, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Bailly, A.; Zwiewka, M.; Sovero, V.; Di, D.; Ge, P.; Oehri, J.; Aryal, B.; Hao, P.; Linnert, M.; et al. TWISTED DWARF1 mediates the action of auxin transport inhibitors on actin cytoskeleton dynamics. Plant Cell 2016, 28, 930–948. [Google Scholar] [CrossRef] [PubMed]
- Marchant, A.; Kargul, J.; May, S.; Delbarre, A.; Perrot-Rechenmann, C.; Bennett, M. AUX1 regulates root gravitropism in Arabidopsis by facilitating auxin uptake within root apical tissues. Embo J. 2014, 18, 2066–2073. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Lee, Z.; Cho, H. ATP-binding cassette B4, an auxin-efflux transporter, stably associates with the plasma membrane and shows distinctive intracellular trafficking from that of PIN-FORMED proteins. Plant Physiol. 2012, 159, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhang, F.; Zhu, Y.; Lan, D.; Yan, P.; Wang, Y.; Hu, Z.; Zhang, X.; Hu, J.; Niu, F.; et al. Auxin signaling module OsSK41- OsIAA10-OsARF regulates grain yield traits in rice. J. Integr. Plant Biol. 2023, 65, 1753–1766. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Huang, Y.; Qi, P.; Lian, G.; Hu, X.; Han, N.; Wang, J.; Zhu, M.; Qian, Q.; Bian, H. Functional analysis of auxin receptor OsTIR1/OsAFB family members in rice grain yield, tillering, plant height, root system, germination, and auxinic herbicide resistance. New Phytol. 2021, 229, 2676–2692. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, X.; Cheng, Y.; Du, X.; Teotia, S.; Miao, C.; Sun, H.; Fan, G.; Tang, G.; Xue, H.; et al. The miR167-OsARF12 module regulates grain filling and grain size downstream of miR159. Nat. Commun. 2023, 4, 100604. [Google Scholar] [CrossRef]
- Huang, X.; Lu, Z.; Zhai, L.; Li, N.; Yan, H. The Small Auxin-Up RNA SAUR10 Is Involved in the Promotion of Seedling Growth in Rice. Plants 2023, 12, 3880. [Google Scholar] [CrossRef]
- Yu, C.; Sun, C.; Shen, C.; Wang, S.; Liu, F.; Liu, Y.; Chen, Y.; Li, C.; Qian, Q.; Aryal, B.; et al. The auxin transporter, OsAUX1, is involved in primary root and root hair elongation and in Cd stress responses in rice (Oryza sativa L). Plant J. 2015, 83, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, C.; Jiang, Y.; Huang, C.; Liu, Q.; Xiong, L.; Yang, W.; Chen, F. Nondestructive 3D image analysis pipeline to extract rice grain traits using X-ray computed tomography. Plant Phenom. 2020, 2020, 3414926. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).