
Unveiling Microbial Dynamics and Gene Expression in Legume–Buffel Grass Coculture Systems for Sustainable Agriculture

 ,
,  ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
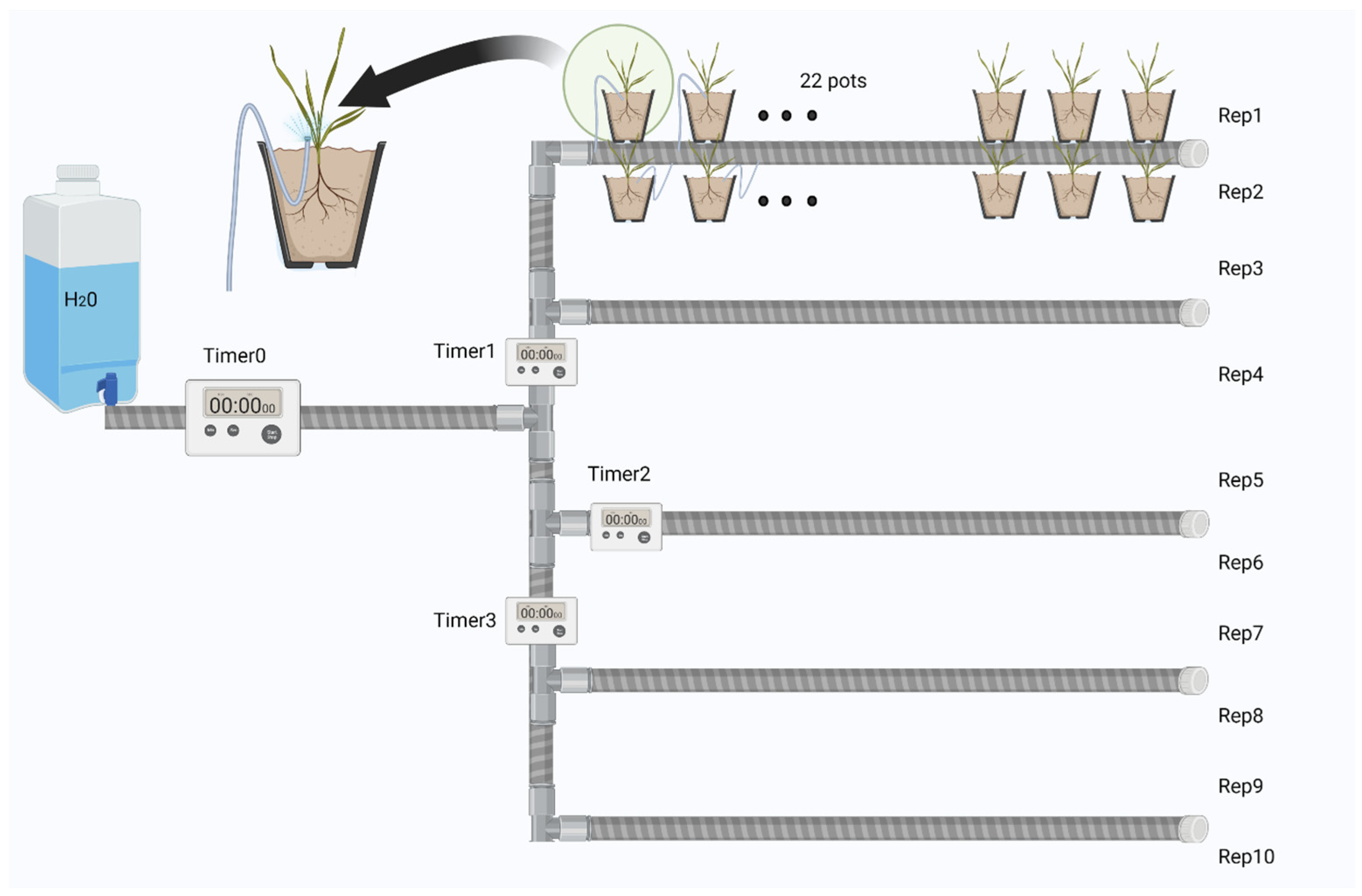
2.1. Experiment Set-Up
- (1)
- Buffel grass (G) and five legumes, including butterfly pea (B), desmanthus (D), lablab (L), Wynn cassia (W), and seca stylo (S) grown without PHY.
- (2)
- Buffel grass and each legume grown in soil treated with PHY (GP, BP, DP, LP, SP, WP).
- (3)
- Buffel grass and each legume grown together (coculture) without PHY (GB, GD, GL, GS, GW).
- (4)
- Coculture of buffel grass and each legume with application of PHY (GBP, GDP, GLP, GSP, GWP). Each treatment had ten replicates, resulting in 220 pots planted in the trial. This design allows for the independent isolation and analysis of both the effects of PHY and coculture, each with its own control group.
2.2. Data Collection and Sampling Procedure
2.3. 16S rRNA Gene Library Preparation and Sequencing
2.4. RNA Extraction and Sequencing Procedure
2.5. Data Analysis and Statistics
3. Results
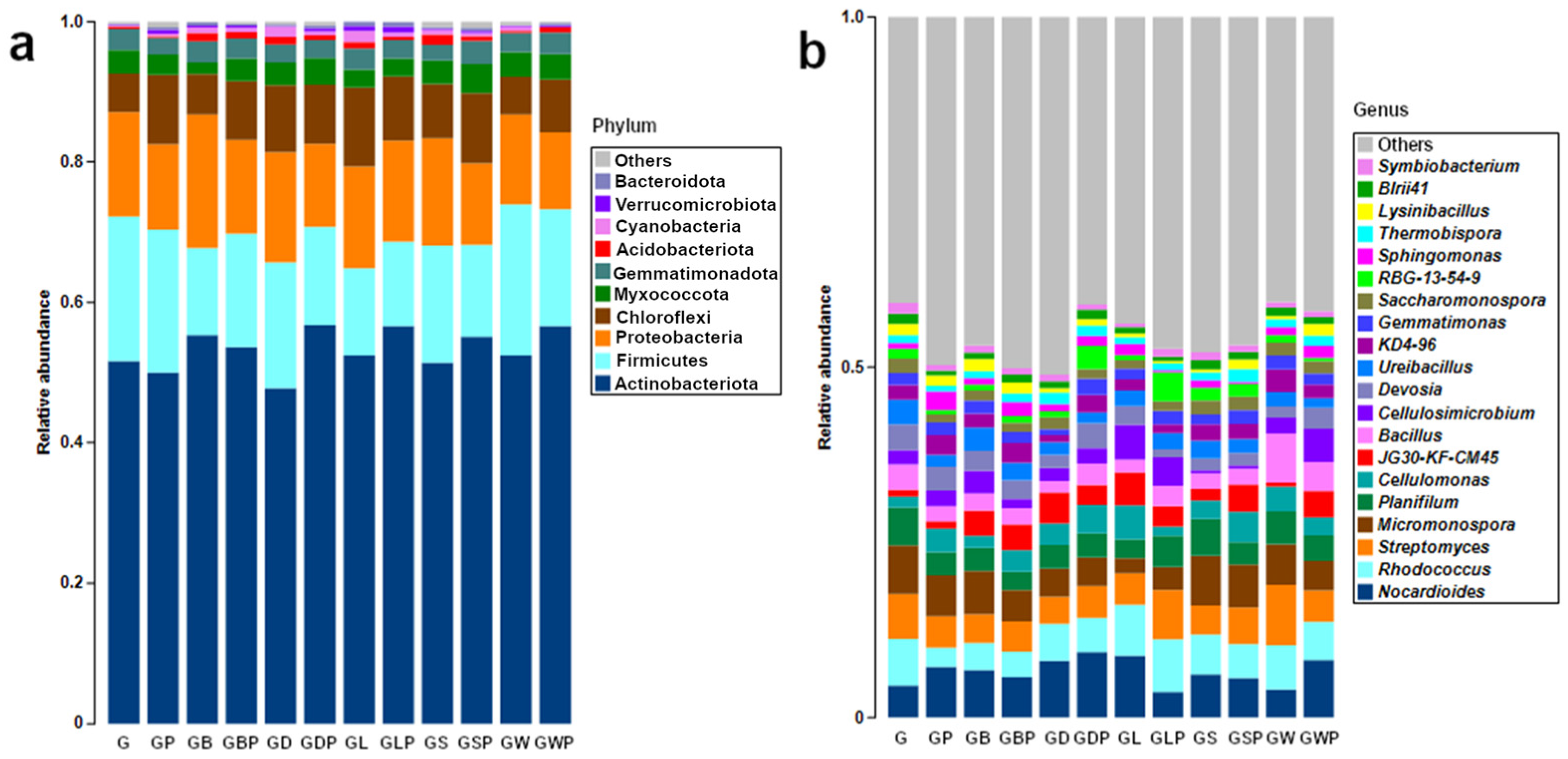
3.1. Overview of Microbial Structure
3.2. Taxa Responding to Treatment
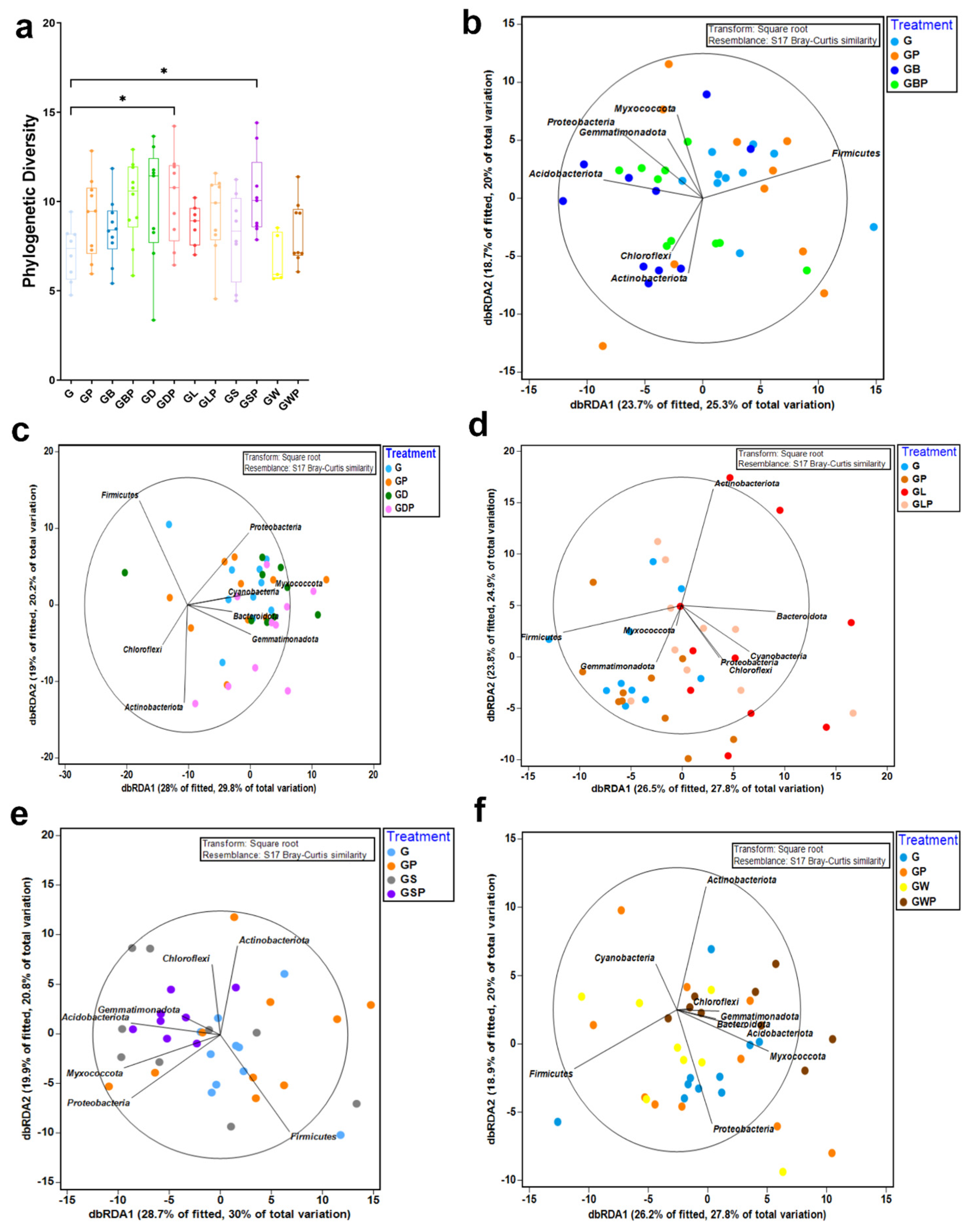
3.3. Spatial Differences in Microbial Diversity
3.4. Transcriptomic Sequencing
3.5. Differentially Expressed Genes and Functional Analysis
3.5.1. GO Enrichment Analysis—Butterfly Pea
3.5.2. GO Enrichment Analysis—Desmanthus
3.5.3. GO Enrichment Analysis—Wynn Cassia and Lablab
3.5.4. GO Enrichment Analysis—Seca Stylo
4. Discussion
4.1. Impact of Legumes on Soil Microbial Diversity and Functional Activity
4.2. Impact of Legumes on Buffel Grass Gene Expression
4.3. Limitations and Future Direction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shtark, O.Y.; Borisov, A.Y.; Zhukov, V.A.; Tikhonovich, I.A. Mutually beneficial legume symbioses with soil microbes and their potential for plant production. Symbiosis 2012, 58, 51–62. [Google Scholar] [CrossRef]
- Wei, Z.; Maxwell, T.M.R.; Robinson, B.; Dickinson, N. Legume nutrition is improved by neighbouring grasses. Plant Soil 2022, 475, 443–455. [Google Scholar] [CrossRef]
- Kebede, E. Contribution, utilization, and improvement of legumes-driven biological nitrogen fixation in agricultural systems. Front. Sustain. Food Syst. 2021, 5, 767998. [Google Scholar] [CrossRef]
- Signorelli, S.; Sainz, M.; Tabares-da Rosa, S.; Monza, J. The role of nitric oxide in nitrogen fixation by legumes. Front. Plant Sci. 2020, 11, 521. [Google Scholar] [CrossRef] [PubMed]
- Franche, C.; Lindström, K.; Elmerich, C. Nitrogen-fixing bacteria associated with leguminous and non-leguminous plants. Plant Soil 2009, 321, 35–59. [Google Scholar] [CrossRef]
- Schulte, C.C.M.; Borah, K.; Wheatley, R.M.; Terpolilli, J.J.; Saalbach, G.; Crang, N.; de Groot, D.H.; Ratcliffe, R.G.; Kruger, N.J.; Papachristodoulou, A.; et al. Metabolic control of nitrogen fixation in rhizobium-legume symbioses. Sci. Adv. 2021, 7, eabh2433. [Google Scholar] [CrossRef]
- Yu, H.; Wang, F.; Shao, M.; Huang, L.; Xie, Y.; Xu, Y.; Kong, L. Effects of rotations with legume on soil functional microbial communities involved in phosphorus transformation. Front. Microbiol. 2021, 12, 661100. [Google Scholar] [CrossRef]
- Muthuraman, Y.; Muthaiyan, P.; Pandurangan, G. Role of Legumes in Improving Soil Fertility Status. In Legume Crops; Mirza, H., Ed.; IntechOpen: Rijeka, Croatia, 2020; Chapter 2; Available online: https://www.intechopen.com/chapters/72818 (accessed on 1 December 2023).
- Makiela, S.; Harrower, K.M. Overview of the current status of buffel grass dieback. Australas. Plant Dis. Notes 2008, 3, 12. [Google Scholar] [CrossRef]
- Ren, X.; Whitton, M.M.; Yu, S.J.; Trotter, T.; Bajagai, Y.S.; Stanley, D. Application of phytogenic liquid supplementation in soil microbiome restoration in queensland pasture dieback. Microorganisms 2023, 11, 561. [Google Scholar] [CrossRef]
- Mpanza, T.D.E.; Hassen, A.; Akanmu, A.M. Evaluation of stylosanthes scabra accessions as forage source for ruminants: Growth performance, nutritive value and in vitro ruminal fermentation. Animals 2020, 10, 1939. [Google Scholar] [CrossRef]
- Seeds, S. Tropical and Subtropical Legumes. Available online: https://www.selectedseeds.com.au/pasture-legumes (accessed on 28 November 2023).
- Pathak, P.; Ramesh, C.; Bhatt, R. Stylosanthes in the reclamation and development of degraded soils in india. In High-Yielding Anthracnose Resistant Stylosanthes for Agricultural Systems; Union Offset: San Carlos, Philippines, 2004; pp. 39–51. Available online: https://www.aciar.gov.au/sites/default/files/legacy/node/568/mn111-chapters1-9.pdf (accessed on 1 December 2023).
- Hall, T.J.; Milson, J.; Hall, C. Pasture Recovery, Land Condition and Some Other Observations after the Monsoon Flooding, Chill Event in North-West Queensland in Jan-Mar 2019; Technical Report; State of Queensland: Brisbane, QLD, Australia, 2020. Available online: https://era.daf.qld.gov.au/id/eprint/7443/ (accessed on 28 November 2023).
- Jones, R.M.; Rees, M.C. Evaluation of tropical legumes on clay soils at four sites in southern inland queensland. Trop. Grassl. 1997, 31, 95–106. [Google Scholar]
- Pengelly, B.C.; Conway, M.J. Pastures on cropping soils: Which tropical pasture legume to use? Trop. Grassl. 2000, 34, 162–168. Available online: https://www.researchgate.net/publication/280020846_Pastures_on_cropping_soils_Which_tropical_pasture_legume_to_use (accessed on 29 November 2023).
- Beef, F. Legumes for Long-Term Pastures on High Fertility Soils in Queensland. Available online: https://futurebeef.com.au/resources/legumes-for-long-term-pastures-on-high-fertility-soils/ (accessed on 29 November 2023).
- Gardiner, C.; Parker, A. Steer liveweight gains on progardes desmanthus/buffelpastures in queensland. In Proceedings of the 2nd Australian and New Zealand Societies of Animal Production Joint Conference, Lincoln University, Christchurch, New Zealand, 2–5 July 2012. [Google Scholar]
- Collins, J.; Gardiner, C.; Kempe, N.; Hannah, I. Successful Pasture Development at Cungelella: A grazier, a Researcher and a Seed Company’s Perspective. Available online: https://researchonline.jcu.edu.au/45276/1/45276%20Collins%20et%20al%202016.pdf (accessed on 1 December 2023).
- Department of Primary Industries, NSW Government. Round-Leafed Cassia. Available online: https://www.dpi.nsw.gov.au/agriculture/pastures-and-rangelands/species-varieties/pf/factsheets/round-leafed-cassia (accessed on 1 December 2023).
- Clements, R.; Jones, R.; Valdes, L.; Bunch, G. Selection of chamaecrista rotundifolia by cattle. Trop. Grassl. 1996, 30, 389–394. [Google Scholar]
- Progressive Seeds. Available online: https://progressiveseeds.com.au/lablab/ (accessed on 28 November 2023).
- Ren, X.; Whitton, M.M.; Trotter, T.; Ashwath, N.; Stanley, D.; Bajagai, Y.S. Enhancing soil health and carbon sequestration through phytogenic treatment: Insights into microbial functional pathways in pasture dieback affected soil. Plant Soil 2024, 496, 1–17. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using qiime 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. Dada2: High-resolution sample inference from illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. Microeco: An r package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2020, 97, fiaa255. [Google Scholar] [CrossRef]
- Anderson, M.; Gorley, R.N.; Clarke, K. Permanova+ for Primer: Guide to Software and Statistical Methods; PRIMER-E: Plymouth, UK, 2008. [Google Scholar]
- GraphPad. Analysis Checklist: Kruskal-Wallis Test. Available online: https://www.graphpad.com/guides/prism/latest/statistics/stat_checklist_kw.htm (accessed on 28 November 2023).
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108, 4554–4561. [Google Scholar] [CrossRef]
- QIAGEN. Qiagen clc Genomics Workbench. Available online: https://digitalinsights.qiagen.com/ (accessed on 29 November 2023).
- Mi, H.; Thomas, P. Panther Pathway: An Ontology-Based Pathway Database Coupled with Data Analysis Tools; Humana Press: Totowa, NJ, USA, 2009; pp. 123–140. [Google Scholar] [CrossRef]
- Holyoake, P.; Kirkland, P.; Davis, R.; Arzey, K.; Watson, J.; Lunt, R.; Wang, J.; Wong, F.; Moloney, B.; Dunn, S. The first identified case of pandemic h1n1 influenza in pigs in australia. Aust. Vet. J. 2011, 89, 427–431. [Google Scholar] [CrossRef]
- Duchene, O.; Vian, J.-F.; Celette, F. Intercropping with legume for agroecological cropping systems: Complementarity and facilitation processes and the importance of soil microorganisms. A review. Agric. Ecosyst. Environ. 2017, 240, 148–161. [Google Scholar] [CrossRef]
- Dakora, F.D. Defining new roles for plant and rhizobial molecules in sole and mixed plant cultures involving symbiotic legumes. New Phytol. 2003, 158, 39–49. [Google Scholar] [CrossRef]
- Nalini, M.S.; Prakash, H.S. Actinobacteria: Diversity, plant interactions and biotechnology applications. In Plant Microbiomes for Sustainable Agriculture; Yadav, A.N., Singh, J., Rastegari, A.A., Yadav, N., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 199–244. [Google Scholar] [CrossRef]
- Xiao, X.; Fan, M.; Wang, E.; Chen, W.; Wei, G. Interactions of plant growth-promoting rhizobacteria and soil factors in two leguminous plants. Appl. Microbiol. Biotechnol. 2017, 101, 8485–8497. [Google Scholar] [CrossRef] [PubMed]
- Gamalero, E.; Glick, B.R. Bacterial modulation of plant ethylene levels. Plant Physiol. 2015, 169, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Speirs, L.B.M.; Rice, D.T.F.; Petrovski, S.; Seviour, R.J. The phylogeny, biodiversity, and ecology of the chloroflexi in activated sludge. Front. Microbiol. 2019, 10, 2015. [Google Scholar] [CrossRef]
- Bovio-Winkler, P.; Guerrero, L.D.; Erijman, L.; Oyarzúa, P.; Suárez-Ojeda, M.E.; Cabezas, A.; Etchebehere, C. Genome-centric metagenomic insights into the role of chloroflexi in anammox, activated sludge and methanogenic reactors. BMC Microbiol. 2023, 23, 45. [Google Scholar] [CrossRef]
- Pessi, I.S.; Viitamäki, S.; Virkkala, A.-M.; Eronen-Rasimus, E.; Delmont, T.O.; Marushchak, M.E.; Luoto, M.; Hultman, J. In-depth characterization of denitrifier communities across different soil ecosystems in the tundra. Environ. Microbiome 2022, 17, 30. [Google Scholar] [CrossRef]
- Kalam, S.; Basu, A.; Ahmad, I.; Sayyed, R.Z.; El-Enshasy, H.A.; Dailin, D.J.; Suriani, N.L. Recent understanding of soil acidobacteria and their ecological significance: A critical review. Front. Microbiol. 2020, 11, 580024. [Google Scholar] [CrossRef]
- Masson-Boivin, C.; Giraud, E.; Perret, X.; Batut, J. Establishing nitrogen-fixing symbiosis with legumes: How many rhizobium recipes? Trends Microbiol. 2009, 17, 458–466. [Google Scholar] [CrossRef]
- Hayat, R.; Ali, S.; Amara, U.; Khalid, R.; Ahmed, I. Soil beneficial bacteria and their role in plant growth promotion: A review. Ann. Microbiol. 2010, 60, 579–598. [Google Scholar] [CrossRef]
- Gu, Y.; Dong, K.; Geisen, S.; Yang, W.; Yan, Y.; Gu, D.; Liu, N.; Borisjuk, N.; Luo, Y.; Friman, V.-P. The effect of microbial inoculant origin on the rhizosphere bacterial community composition and plant growth-promotion. Plant Soil 2020, 452, 105–117. [Google Scholar] [CrossRef]
- Raman, J.; Kim, J.-S.; Choi, K.R.; Eun, H.; Yang, D.; Ko, Y.-J.; Kim, S.-J. Application of lactic acid bacteria (lab) in sustainable agriculture: Advantages and limitations. Int. J. Mol. Sci. 2022, 23, 7784. [Google Scholar] [CrossRef] [PubMed]
- Wongkiew, S.; Koottatep, T.; Polprasert, C.; Prombutara, P.; Jinsart, W.; Khanal, S.K. Bioponic system for nitrogen and phosphorus recovery from chicken manure: Evaluation of manure loading and microbial communities. Waste Manag. 2021, 125, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Schneiker, S.; Perlova, O.; Kaiser, O.; Gerth, K.; Alici, A.; Altmeyer, M.O.; Bartels, D.; Bekel, T.; Beyer, S.; Bode, E.; et al. Complete genome sequence of the myxobacterium sorangium cellulosum. Nat. Biotechnol. 2007, 25, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Malviya, M.K.; Solanki, M.K.; Li, C.-N.; Wang, Z.; Zeng, Y.; Verma, K.K.; Singh, R.K.; Singh, P.; Huang, H.-R.; Yang, L.-T.; et al. Sugarcane-legume intercropping can enrich the soil microbiome and plant growth. Front. Sustain. Food Syst. 2021, 5, 606595. [Google Scholar] [CrossRef]
- Ye, J.Y.; Tian, W.H.; Jin, C.W. Nitrogen in plants: From nutrition to the modulation of abiotic stress adaptation. Stress Biol. 2022, 2, 4. [Google Scholar] [CrossRef]
- Zayed, O.; Hewedy, O.A.; Abdelmoteleb, A.; Ali, M.; Youssef, M.S.; Roumia, A.F.; Seymour, D.; Yuan, Z.-C. Nitrogen journey in plants: From uptake to metabolism, stress response, and microbe interaction. Biomolecules 2023, 13, 1443. [Google Scholar] [CrossRef]
- Wetterhorn, K.M.; Newmister, S.A.; Caniza, R.K.; Busman, M.; McCormick, S.P.; Berthiller, F.; Adam, G.; Rayment, I. Crystal structure of os79 (os04g0206600) from Oryza sativa: A udp-glucosyltransferase involved in the detoxification of deoxynivalenol. Biochemistry 2016, 55, 6175–6186. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Dobrota, C. Energy Dependant Plant Stress Acclimation. In Life in Extreme Environments; Amils, R., Ellis-Evans, C., Hinghofer-Szalkay, H., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 277–285. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pairwise Tests | |||
|---|---|---|---|
| Groups | t | p (Perm) | Unique Perms |
| G, GP | 1.1731 | 0.218 | 991 |
| G, GB | 1.8661 | 0.01 * | 987 |
| G, GBP | 1.4441 | 0.054 | 987 |
| GB, GBP | 1.579 | 0.031 * | 993 |
| GB, GP | 2.1524 | 0.002 ** | 994 |
| GBP, GP | 1.4559 | 0.058 | 992 |
| G, GD | 1.2849 | 0.116 | 977 |
| G, GDP | 1.7931 | 0.017 * | 993 |
| GD, GDP | 1.3843 | 0.083 | 995 |
| GD, GP | 1.2172 | 0.188 | 992 |
| GDP, GP | 1.2145 | 0.175 | 994 |
| G, GL | 2.0561 | 0.003 ** | 921 |
| G, GLP | 1.7579 | 0.007 ** | 982 |
| GL, GLP | 0.90013 | 0.569 | 970 |
| GL, GP | 1.84 | 0.006 ** | 967 |
| GLP, GP | 1.5738 | 0.029 * | 996 |
| G, GS | 1.2449 | 0.146 | 911 |
| G, GSP | 1.9462 | 0.001 ** | 985 |
| GS, GSP | 1.2061 | 0.176 | 976 |
| GS, GP | 1.2994 | 0.146 | 994 |
| GSP, GP | 1.5965 | 0.036 * | 992 |
| G, GW | 0.67027 | 0.83 | 694 |
| G, GWP | 1.4618 | 0.034 * | 971 |
| GW, GWP | 1.4902 | 0.036 * | 790 |
| GW, GP | 1.0034 | 0.383 | 840 |
| GWP, GP | 1.7354 | 0.006 ** | 990 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, X.; Yu, S.J.; Brewer, P.B.; Ashwath, N.; Bajagai, Y.S.; Stanley, D.; Trotter, T. Unveiling Microbial Dynamics and Gene Expression in Legume–Buffel Grass Coculture Systems for Sustainable Agriculture. Agronomy 2024, 14, 2172. https://doi.org/10.3390/agronomy14092172
Ren X, Yu SJ, Brewer PB, Ashwath N, Bajagai YS, Stanley D, Trotter T. Unveiling Microbial Dynamics and Gene Expression in Legume–Buffel Grass Coculture Systems for Sustainable Agriculture. Agronomy. 2024; 14(9):2172. https://doi.org/10.3390/agronomy14092172
Chicago/Turabian StyleRen, Xipeng, Sung J. Yu, Philip B. Brewer, Nanjappa Ashwath, Yadav S. Bajagai, Dragana Stanley, and Tieneke Trotter. 2024. "Unveiling Microbial Dynamics and Gene Expression in Legume–Buffel Grass Coculture Systems for Sustainable Agriculture" Agronomy 14, no. 9: 2172. https://doi.org/10.3390/agronomy14092172
APA StyleRen, X., Yu, S. J., Brewer, P. B., Ashwath, N., Bajagai, Y. S., Stanley, D., & Trotter, T. (2024). Unveiling Microbial Dynamics and Gene Expression in Legume–Buffel Grass Coculture Systems for Sustainable Agriculture. Agronomy, 14(9), 2172. https://doi.org/10.3390/agronomy14092172