The G Protein-Coupled Receptor Kinases (GRKs) in Chemokine Receptor-Mediated Immune Cell Migration: From Molecular Cues to Physiopathology



Abstract
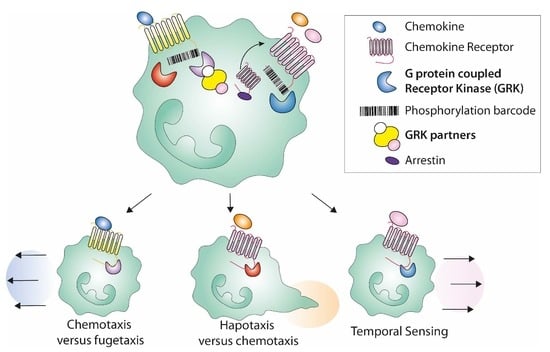
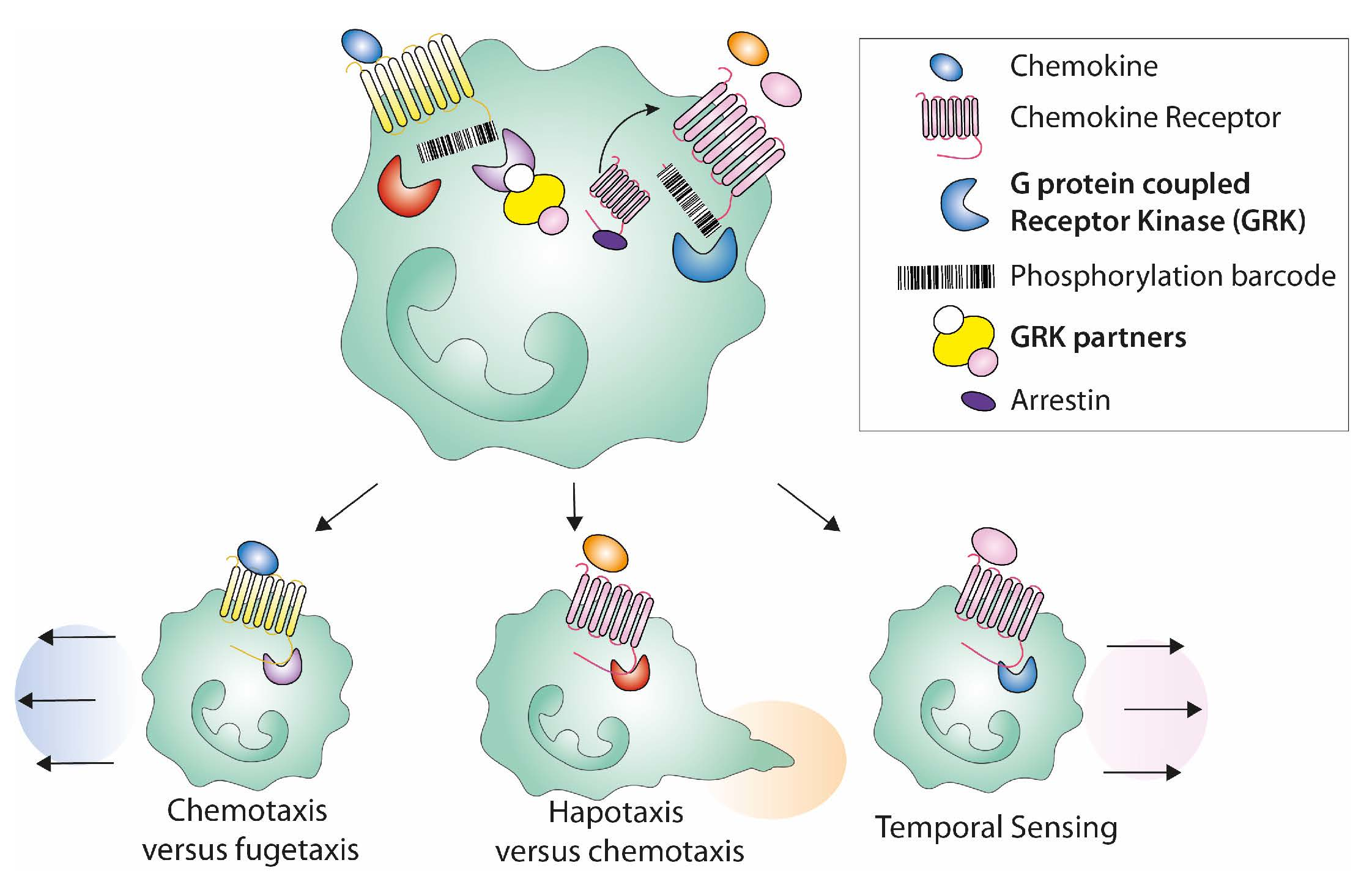
:
1. Introduction
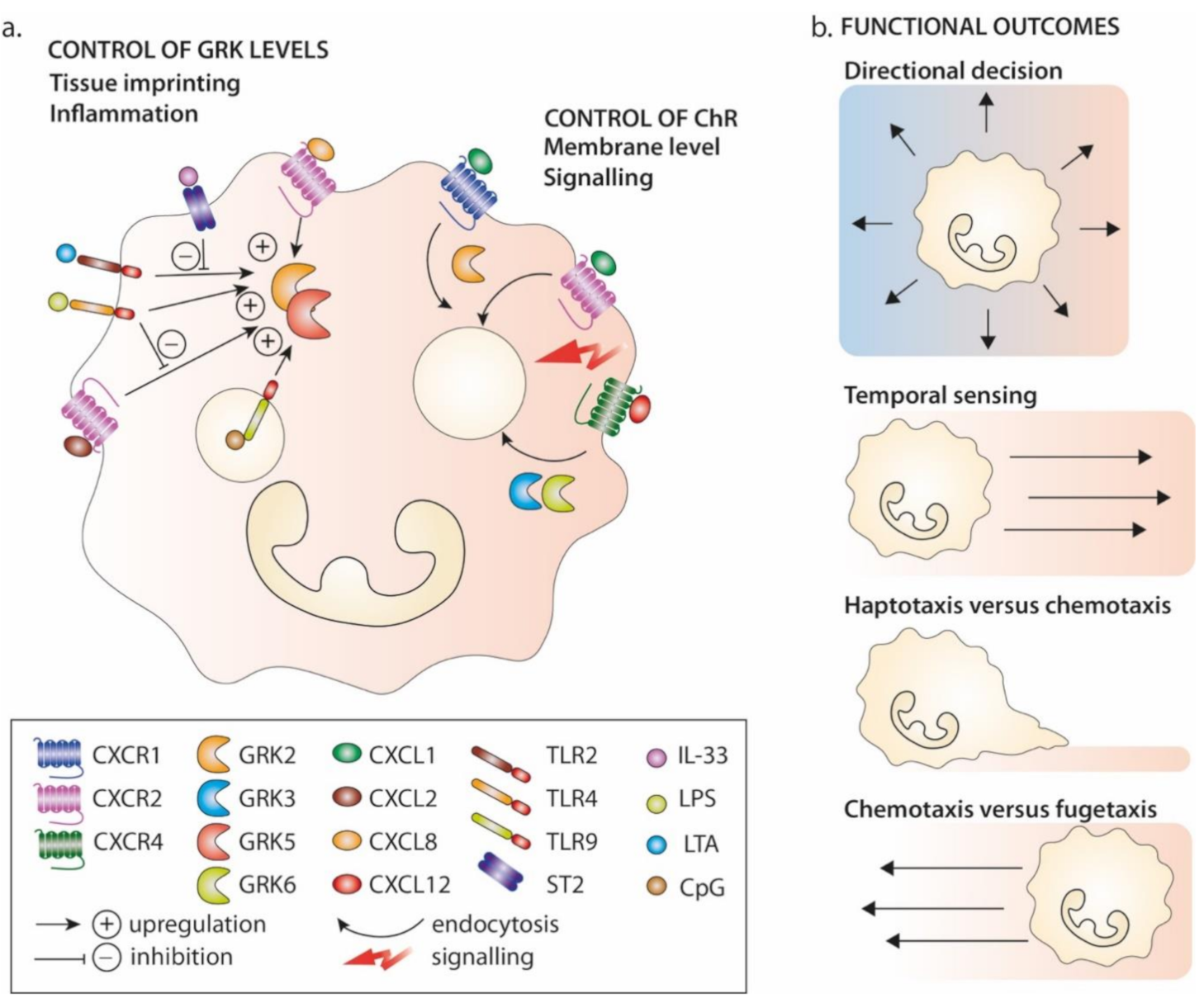
2. Linking GRK Deficiencies to Immune Dysfunction
3. Roles of GRKs in Regulating Chemokine Receptor Activation
3.1. More Than Governing Desensitization
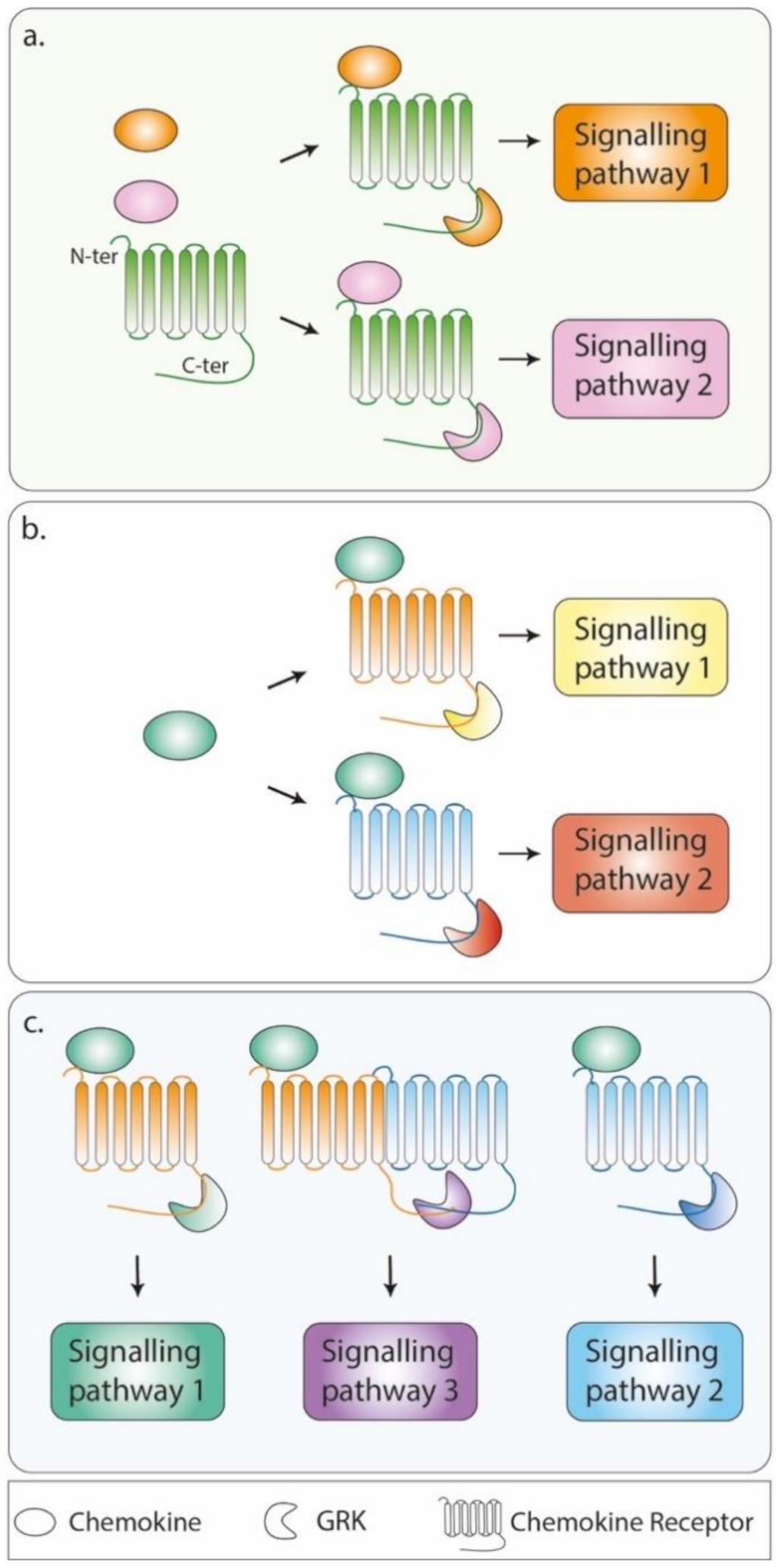
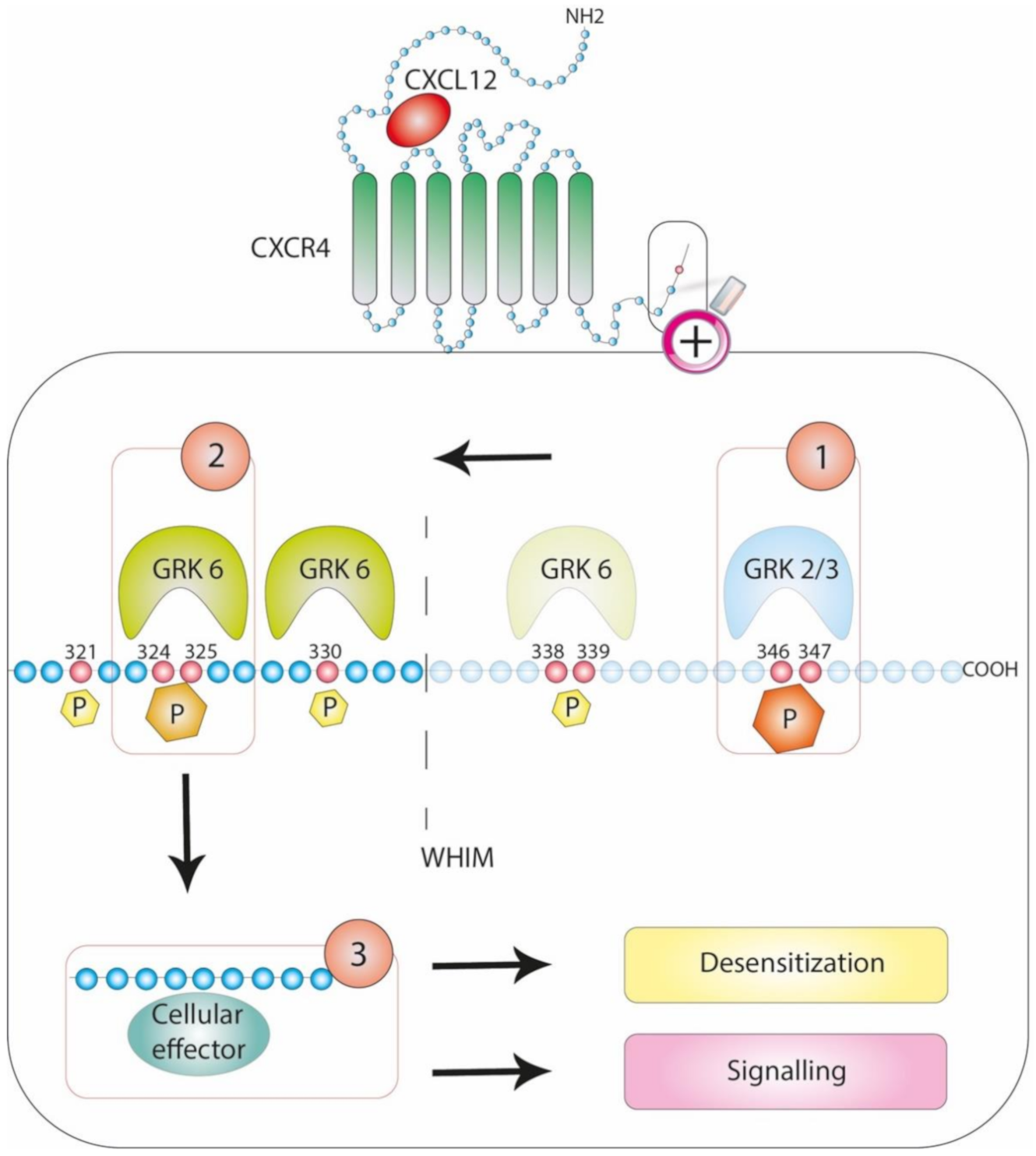
3.2. Patterning the Phosphorylation Code: Focus on CXCR4
3.3. Driving Signalling Pathways
3.4. Regulating Atypically Atypical Chemokine Receptors
3.5. Shaping the Cell Migration Mode Promoted by Chemokine Ligands
3.6. Modulating Receptor-Dependent Routing: Insights from the CXCL8 Chemokine
4. GRKs in the Immune Functions of Chemokine Receptors: Focus on Neutrophils
4.1. GRKs in Neutrophil Guidance
4.2. Modulation of GRK Expression Levels in Pathology
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gurevich, E.V.; Tesmer, J.J.G.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol. Ther. 2012, 133, 40–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saaber, F.; Schütz, D.; Miess, E.; Abe, P.; Desikan, S.; Kumar, P.A.; Balk, S.; Huang, K.; Beaulieu, J.; Schulz, S.; et al. ACKR3 Regulation of Neuronal Migration Requires ACKR3 Phosphorylation, but Not β-Arrestin. Cell Rep. 2019, 26, 1473–1488.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, X.; Zhang, L.; Zhang, L.; Hu, M.; Leng, J.; Yu, B.; Zhou, B.; Hu, Y.; Zhang, Q. The mechanism of chemo-kine receptor 9 internalization triggered by interleukin 2 and interleukin. Cell Mol. Immunol. 2009, 6, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Raghuwanshi, S.K.; Su, Y.; Singh, V.; Haynes, K.; Richmond, A.; Richardson, R.M. The Chemokine Receptors CXCR1 and CXCR2 Couple to Distinct G Protein-Coupled Receptor Kinases to Mediate and Regulate Leukocyte Functions. J. Immunol. 2012, 189, 2824–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.K.; Kim, S.D.; Kook, M.; Lee, H.Y.; Ghim, J.; Choi, Y.; Zabel, B.A.; Ryu, S.H.; Bae, Y. Phospholipase D2 drives mortality in sepsis by inhibiting neutrophil extracellular trap formation and down-regulating CXCR2. J. Exp. Med. 2015, 212, 1381–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busillo, J.M.; Armando, S.; Sengupta, R.; Meucci, O.; Bouvier, M.; Benovic, J.L. Site-specific Phosphorylation of CXCR4 Is Dynamically Regulated by Multiple Kinases and Results in Differential Modulation of CXCR4 Signaling. J. Biol. Chem. 2010, 285, 7805–7817. [Google Scholar] [CrossRef] [Green Version]
- Mueller, W.; Schütz, D.; Nagel, F.; Schulz, S.; Stumm, R. Hierarchical Organization of Multi-Site Phosphorylation at the CXCR4 C Terminus. PLoS ONE 2013, 8, e64975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, A.; Fujimoto, J.; Miyata, H.; Stumm, R.; Narazaki, M.; Schulz, S.; Baba, Y.; Kumanogoh, A.; Suzuki, K. The COMMD3/8 complex determines GRK6 specificity for chemoattractant receptors. J. Exp. Med. 2019, 216, 1630–1647. [Google Scholar] [CrossRef] [Green Version]
- Matti, C.; Salnikov, A.; Artinger, M.; D’Agostino, G.; Kindinger, I.; Uguccioni, M.; Thelen, M.; Legler, D.F. ACKR4 Recruits GRK3 Prior to beta-Arrestins but Can Scavenge Chemokines in the Absence of beta-Arrestins. Front. Immunol. 2020, 11, 720. [Google Scholar] [CrossRef] [Green Version]
- Aronin, C.E.P.; Zhao, Y.M.; Yoon, J.S.; Morgan, N.Y.; Prüstel, T.; Germain, R.N.; Meier-Schellersheim, M. Migrating Myeloid Cells Sense Temporal Dynamics of Chemoattractant Concentrations. Immunity 2017, 47, 862–874.e3. [Google Scholar] [CrossRef]
- Zidar, D.A.; Violin, J.D.; Whalen, E.J.; Lefkowitz, R.J. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 9649–9654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarrant, T.K.; Billard, M.J.; Timoshchenko, R.G.; McGinnis, M.W.; Serafin, D.S.; Foreman, O.; Esserman, D.A.; Chao, N.J.; Lento, W.E.; Lee, D.M.; et al. G protein-coupled receptor kinase-3-deficient mice exhibit WHIM syndrome features and attenuated inflammatory responses. J. Leukoc. Biol. 2013, 94, 1243–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balabanian, K.; Levoye, A.; Klemm, L.; Lagane, B.; Hermine, O.; Harriague, J.; Baleux, F.; Arenzana-Seisdedos, F.; Bachelerie, F. Leukocyte analysis from WHIM syndrome patients reveals a pivotal role for GRK3 in CXCR4 signaling. J. Clin. Investig. 2008, 118, 1074–1084. [Google Scholar] [CrossRef]
- Luo, J.; Busillo, J.M.; Stumm, R.; Benovic, J.L. G Protein-Coupled Receptor Kinase 3 and Protein Kinase C Phosphory-late the Distal C-Terminal Tail of the Chemokine Receptor CXCR4 and Mediate Recruitment of beta-Arrestin. Mol. Pharmacol. 2017, 91, 554–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.; Bierbaum, V.; Vaahtomeri, K.; Hauschild, R.; Brown, M.; de Vries, I.; Leithner, A.; Reversat, A.; Merrin, J.; Tarrant, T.; et al. Dendritic Cells Interpret Haptotactic Chemokine Gradients in a Manner Gov-erned by Signal-to-Noise Ratio and Dependent on GRK6. Curr. Biol. 2017, 27, 1314–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Métayé, T.; Gibelin, H.; Perdrisot, R.; Kraimps, J.-L. Pathophysiological roles of G-protein-coupled receptor kinases. Cell. Signal. 2005, 17, 917–928. [Google Scholar] [CrossRef]
- Jaber, M.; Koch, W.J.; Rockman, H.; Smith, B.; Bond, R.A.; Sulik, K.K.; Ross, J., Jr.; Lefkowitz, R.J.; Caron, M.G.; Giros, B. Essential role of beta-adrenergic receptor kinase 1 in cardiac development and function. Proc. Natl. Acad. Sci. USA 1996, 93, 12974–12979. [Google Scholar] [CrossRef] [Green Version]
- Peppel, K.; Boekhoff, I.; McDonald, P.; Breer, H.; Caron, M.G.; Lefkowitz, R.J. G Protein-coupled Receptor Kinase 3 (GRK3) Gene Disruption Leads to Loss of Odorant Receptor Desensitization. J. Biol. Chem. 1997, 272, 25425–25428. [Google Scholar] [CrossRef] [Green Version]
- Otten, J.J.T.; De Jager, S.C.A.; Kavelaars, A.; Seijkens, T.; Bot, I.; Wijnands, E.; Beckers, L.; Westra, M.M.; Bot, M.; Busch, M.; et al. Hematopoietic G-protein-coupled receptor kinase 2 deficiency decreases atherosclerotic lesion formation in LDL receptor-knockout mice. FASEB J. 2013, 27, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Patial, S.; Saini, Y.; Parvataneni, S.; Appledorn, D.M.; Dorn, G.W., 2nd; Lapres, J.J.; Amalfitano, A.; Senagore, P.; Parameswaran, N. Myeloid-specific GPCR kinase-2 negatively regulates NF-kappaB1p105-ERK pathway and limits endotox-emic shock in mice. J. Cell Physiol. 2011, 226, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Packiriswamy, N.; Lee, T.; Raghavendra, P.B.; Durairaj, H.; Wang, H.; Parameswaran, N. G-Protein-Coupled Receptor Kinase-5 Mediates Inflammation but Does Not Regulate Cellular Infiltration or Bacterial Load in a Polymicrobial Sepsis Model in Mice. J. Innate Immun. 2013, 5, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Fong, A.M.; Premont, R.T.; Richardson, R.M.; Yu, Y.R.; Lefkowitz, R.J.; Patel, D.D. Defective lymphocyte chem-otaxis in beta-arrestin2- and GRK6-deficient mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7478–7483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gainetdinov, R.R.; Bohn, L.M.; Sotnikova, T.D.; Cyr, M.; Laakso, A.; Macrae, A.D.; Torres, G.; Kim, K.M.; Lefkowitz, R.J.; Lefkowitz, R.J.; et al. Dopaminergic Supersensitivity in G Protein-Coupled Receptor Kinase 6-Deficient Mice. Neuron 2003, 38, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Le, Q.; Yao, W.; Chen, Y.; Yan, B.; Liu, C.; Yuan, M.; Zhou, Y.; Ma, L. GRK6 regulates ROS response and main-tains hematopoietic stem cell self-renewal. Cell Death Dis. 2016, 7, e2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chudziak, D.; Spohn, G.; Karpova, D.; Dauber, K.; Wiercinska, E.; Miettinen, J.A.; Papayannopoulou, T.; Bonig, H. Functional Consequences of Perturbed CXCL12 Signal Processing: Analyses of Immature Hematopoiesis in GRK6-Deficient Mice. Stem Cells Dev. 2015, 24, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Vroon, A.; Heijnen, C.J.; Raatgever, R.; Touw, I.P.; Ploemacher, R.E.; Premont, R.T.; Kavelaars, A. GRK6 defi-ciency is associated with enhanced CXCR4-mediated neutrophil chemotaxis in vitro and impaired responsiveness to G-CSF in vivo. J. Leukoc. Biol. 2004, 75, 698–704. [Google Scholar] [CrossRef] [Green Version]
- Zuelzer, W.W. “Myelokathexis”—A New Form of Chronic Granulocytopenia. Report of a Case. N. Engl. J. Med. 1964, 270, 699–704. [Google Scholar] [CrossRef]
- Beaussant-Cohen, S.; Fenneteau, O.; Plouvier, E.; Rohrlich, P.; Daltroff, G.; Plantier, I.; Dupuy, A.; Kérob, D.; Beaupain, B.; Bordigoni, P.; et al. Description and outcome of a cohort of 8 patients with WHIM syndrome from the French Severe Chronic Neutropenia Registry. Orphanet J. Rare Dis. 2012, 7, 71. [Google Scholar] [CrossRef] [Green Version]
- Wetzler, M.; Talpaz, M.; Kleinerman, E.S.; King, A.; Huh, Y.O.; Gutterman, J.U.; Kurzrock, R. A new familial immunodeficiency disorder characterized by severe neutropenia, a defective marrow release mechanism, and hypogammaglobulinemia. Am. J. Med. 1990, 89, 663–672. [Google Scholar] [CrossRef]
- McDermott, D.H.; Liu, Q.; Ulrick, J.; Kwatemaa, N.; Anaya-O’Brien, S.; Penzak, S.R.; Filho, J.O.; Priel, D.A.L.; Kelly, C.; Garofalo, M.; et al. The CXCR4 antagonist plerixafor corrects panleukopenia in patients with WHIM syndrome. Blood 2011, 118, 4957–4962. [Google Scholar] [CrossRef] [Green Version]
- Balabanian, K.; Brotin, E.; Biajoux, V.; Bouchet-Delbos, L.; Lainey, E.; Fenneteau, O.; Bonnet, D.; Fiette, L.; Emilie, D.; Bachelerie, F. Proper desensitization of CXCR4 is required for lymphocyte development and peripheral compartmen-talization in mice. Blood 2012, 119, 5722–5730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by beta-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.; Lefkowitz, R.J. GRKs and beta-arrestins: Roles in receptor silencing, trafficking and signaling. Trends. Endocrinol. Metab. 2006, 17, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Ribas, C.; Sánchez-Madrid, F.; Mayor, J.F. G protein-coupled receptor kinase 2 (GRK2) as a multifunctional signaling hub. Cell. Mol. Life Sci. 2019, 76, 4423–4446. [Google Scholar] [CrossRef] [Green Version]
- Murphy, P.M.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Basic and Clinical Pharmacology. LXXXIX. Update on the Extended Family of Chemokine Receptors and Introducing a New Nomenclature for Atypical Chemokine Receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef] [Green Version]
- Fonin, A.V.; Darling, A.L.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Multi-functionality of proteins in-volved in GPCR and G protein signaling: Making sense of structure-function continuum with intrinsic disorder-based proteoforms. Cell Mol. Life Sci. 2019, 76, 4461–4492. [Google Scholar] [CrossRef]
- Berzat, A.; Hall, A. Cellular responses to extracellular guidance cues. EMBO J. 2010, 29, 2734–2745. [Google Scholar] [CrossRef] [Green Version]
- Weiner, O.D.; Servant, G.; Welch, M.D.; Mitchison, T.J.; Sedat, J.W.; Bourne, H.R. Spatial control of actin polymerization during neutrophil chemotaxis. Nat. Cell Biol. 1999, 1, 75–81. [Google Scholar] [CrossRef]
- Maritzen, T.; Schachtner, H.; Legler, D.F. On the move: Endocytic trafficking in cell migration. Cell. Mol. Life Sci. 2015, 72, 2119–2134. [Google Scholar] [CrossRef] [Green Version]
- Defea, K.A. Stop that cell! Beta-arrestin-dependent chemotaxis: A tale of localized actin assembly and receptor desensitization. Annu. Rev. Physiol. 2007, 69, 535–560. [Google Scholar] [CrossRef]
- Defea, K.A. Arrestins in Actin Reorganization and Cell Migration. Prog. Mol. Biol. Transl. Sci. 2013, 118, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Liggett, S.B. Phosphorylation Barcoding as a Mechanism of Directing GPCR Signaling. Sci. Signal. 2011, 4, pe36. [Google Scholar] [CrossRef] [PubMed]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct phosphorylation sites on the be-ta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of Phosphorylation Codes for Arrestin Re-cruitment by G Protein-Coupled Receptors. Cell 2017, 170, 457–469.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baidya, M.; Kumari, P.; Dwivedi-Agnihotri, H.; Pandey, S.; Chaturvedi, M.; Stepniewski, T.M.; Kawakami, K.; Cao, Y.; Laporte, S.A.; Selent, J.; et al. Key phosphorylation sites in GPCRs orchestrate the contribution of beta-Arrestin 1 in ERK1/2 activation. EMBO Rep. 2020, 21, e49886. [Google Scholar] [CrossRef] [PubMed]
- Premont, R.T. Keys to the Kingdom: GPCR phosphorylation patterns direct beta-arrestin. EMBO Rep. 2020, 21, e51249. [Google Scholar] [CrossRef]
- Tobin, A.B. G-protein-coupled receptor phosphorylation: Where, when and by whom. Br. J. Pharmacol. 2008, 153, S167–S176. [Google Scholar] [CrossRef] [Green Version]
- Torrecilla, I.; Spragg, E.J.; Poulin, B.; McWilliams, P.J.; Mistry, S.C.; Blaukat, A.; Tobin, A.B. Phosphorylation and regulation of a G protein–coupled receptor by protein kinase CK2. J. Cell Biol. 2007, 177, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Murphy, P.M.; Heusinkveld, L. Multisystem multitasking by CXCL12 and its receptors CXCR4 and ACKR. Cytokine 2018, 109, 2–10. [Google Scholar] [CrossRef]
- Smit, M.J.; Schlecht-Louf, G.; Neves, M.; den Bor, J.V.; Penela, P.; Siderius, M.; Bachelerie, F.; Mayor, F., Jr. The CXCL12/CXCR4/ACKR3 Axis in the Tumor Microenvironment: Signaling, Crosstalk, and Therapeutic Targeting. Annu. Rev. Pharmacol. Toxicol. 2020, 61. [Google Scholar] [CrossRef]
- Fumagalli, A.; Zarca, A.; Neves, M.; Caspar, B.; Hill, S.J.; Mayor, F.; Smit, M.J.; Marin, P. CXCR4/ACKR3 Phosphorylation and Recruitment of Interacting Proteins: Key Mechanisms Regulating Their Functional Status. Mol. Pharmacol. 2019, 96, 794–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleul, C.C.; Farzan, M.; Choe, H.; Parolin, C.; Clark-Lewis, I.; Sodroski, J.; Springer, T.A. The lymphocyte chemo-attractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 1996, 382, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Oberlin, E.; Amara, A.; Bachelerie, F.; Bessia, C.; Virelizier, J.-L.; Arenzana-Seisdedos, F.; Schwartz, O.; Heard, J.-M.; Clark-Lewis, I.; Legler, D.F.; et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nat. Cell Biol. 1996, 382, 833–835. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.R.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nat. Cell Biol. 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.A.; Gorlin, R.J.; Lukens, J.N.; Taniuchi, S.; Bohinjec, J.; Francois, F.; Klotman, M.E.; Diaz, G.A. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat. Genet. 2003, 34, 70–74. [Google Scholar] [CrossRef]
- Haribabu, B.; Richardson, R.M.; Fisher, I.; Sozzani, S.; Peiper, S.C.; Horuk, R.; Ali, H.; Snyderman, R. Regula-tion of human chemokine receptors CXCRRole of phosphorylation in desensitization and internalization. J. Biol. Chem. 1997, 272, 28726–28731. [Google Scholar] [CrossRef] [Green Version]
- Signoret, N.; Oldridge, J.; Pelchen-Matthews, A.; Klasse, P.J.; Tran, T.; Brass, L.F.; Rosenkilde, M.M.; Schwartz, T.W.; Holmes, W.; Dallas, W.; et al. Phorbol Esters and SDF-1 Induce Rapid Endocytosis and Down Modulation of the Chemokine Receptor CXCR4. J. Cell Biol. 1997, 139, 651–664. [Google Scholar] [CrossRef] [Green Version]
- Orsini, M.J.; Parent, J.L.; Mundell, S.J.; Marchese, A.; Benovic, J.L. Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the c-terminal tail that mediate receptor internalization. J. Biol. Chem. 1999, 274, 31076–31086. [Google Scholar] [CrossRef] [Green Version]
- Bachelerie, F. CXCL12/CXCR4-axis dysfunctions: Markers of the rare immunodeficiency disorder WHIM syndrome. Dis. Markers 2010, 29, 189–198. [Google Scholar] [CrossRef]
- Heusinkveld, L.E.; Majumdar, S.; Gao, J.-L.; McDermott, D.H.; Murphy, P.M. WHIM Syndrome: From Pathogenesis Towards Personalized Medicine and Cure. J. Clin. Immunol. 2019, 39, 532–556. [Google Scholar] [CrossRef]
- Balabanian, K.; Lagane, B.; Pablos, J.L.; Laurent, L.; Planchenault, T.; Verola, O.; Lebbe, C.; Kerob, D.; Dupuy, A.; Hermine, O.; et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood 2005, 105, 2449–2457. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Choi, U.; Whiting-Theobald, N.L.; Linton, G.F.; Brenner, S.; Sechler, J.M.; Murphy, P.M.; Malech, H.L. Enhanced function with decreased internalization of carboxy-terminus truncated CXCR4 responsible for WHIM syndrome. Exp. Hematol. 2005, 33, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Marderstein, A.R.; Uppal, M.; Verma, A.; Bhinder, B.; Tayyebi, Z.; Mezey, J.; Clark, A.G.; Elemento, O. Demo-graphic and genetic factors influence the abundance of infiltrating immune cells in human tissues. Nat. Commun. 2020, 11, 2213. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Suzuki, K. Adrenergic control of lymphocyte trafficking and adaptive immune responses. Neurochem. Int. 2019, 130, 104320. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.J.; Zhao, J.; Sun, Y.; Hu, W.; Wu, Y.L.; Cen, B.; Wu, G.X.; Pei, G. beta-arrestin differentially regulates the chemokine receptor CXCR4-mediated signaling and receptor internalization, and this implicates multiple interaction sites be-tween beta-arrestin and CXCR4. J. Biol. Chem. 2000, 275, 2479–2485. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Cheng, Z.; Ma, L.; Pei, G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is medi-ated by its enhancement of p38 MAPK activation. J. Biol. Chem. 2002, 277, 49212–49219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagane, B.; Chow, K.Y.; Balabanian, K.; Levoye, A.; Harriague, J.; Planchenault, T.; Baleux, F.; Gunera-Saad, N.; Arenzana-Seisdedos, F.; Bachelerie, F. CXCR4 dimerization and beta-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood 2008, 112, 34–44. [Google Scholar] [CrossRef]
- McCormick, P.J.; Segarra, M.; Gasperini, P.; Gulino, A.V.; Tosato, G. Impaired recruitment of Grk6 and beta-Arrestin 2 causes delayed internalization and desensitization of a WHIM syndrome-associated CXCR4 mutant receptor. PLoS ONE 2009, 4, e8102. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Mouton, C.; Fischer, T.; Peregil, R.M.; Jiménez-Baranda, S.; Stossel, T.P.; Nakamura, F.; Mañes, S. Filamin A interaction with the CXCR4 third intracellular loop regulates endocytosis and signaling of WT and WHIM-like receptors. Blood 2015, 125, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with beta-arrestin. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Cao, Y.; Xu, L.; Yang, G.; Liu, X.; Hunter, Z.R. Somatic mutations in MYD88 and CXCR4 are determi-nants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood 2014, 123, 2791–2796. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Inserte, J.; Ramos, P.; Rodriguez-Sinovas, A.; Garcia-Dorado, D.; Mayor, J.F. Degradation of GRK2 and AKT is an early and detrimental event in myocardial ischemia/reperfusion. EBioMedicine 2019, 48, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steury, M.D.; McCabe, L.R.; Parameswaran, N. G Protein-Coupled Receptor Kinases in the Inflammatory Response and Signaling. Dev. Funct. Myeloid Subsets 2017, 136, 227–277. [Google Scholar] [CrossRef]
- Jimenez-Sainz, M.C.; Murga, C.; Kavelaars, A.; Jurado-Pueyo, M.; Krakstad, B.F.; Heijnen, C.J.; Mayor, F., Jr.; Aragay, A.M. G protein-coupled receptor kinase 2 negatively regulates chemokine signaling at a level downstream from G pro-tein subunits. Mol. Biol. Cell 2006, 17, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.D.; Pitcher, J.A. G protein-coupled receptor kinase 2 (GRK2) is a Rho-activated scaffold protein for the ERK MAP kinase cascade. Cell. Signal. 2013, 25, 2831–2839. [Google Scholar] [CrossRef] [Green Version]
- Jurado-Pueyo, M.; Campos, P.M.; Mayor, F.; Murga, C. GRK2-Dependent Desensitization Downstream of G Proteins. J. Recept. Signal. Transduct. 2008, 28, 59–70. [Google Scholar] [CrossRef]
- Penela, P.; Ribas, C.; Aymerich, I.; Eijkelkamp, N.; Barreiro, O.; Heijnen, C.J.; Kavelaars, A.; Sánchez-Madrid, F.; Mayor, J.F. G protein-coupled receptor kinase 2 positively regulates epithelial cell migration. EMBO J. 2008, 27, 1206–1218. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, N.; Pao, C.S.; Leonhard, K.S.; Kang, D.S.; Kratz, M.; Ley, S.C.; Benovic, J.L. Arrestin-2 and G protein-coupled receptor kinase 5 interact with NFkappaB1 p105 and negatively regulate lipopolysaccharide-stimulated ERK1/2 activation in macrophages. J. Biol. Chem. 2006, 281, 34159–34170. [Google Scholar] [CrossRef] [Green Version]
- Patial, S.; Luo, J.; Porter, K.J.; Benovic, J.L.; Parameswaran, N. G-protein-coupled-receptor kinases mediate TNFal-pha-induced NFkappaB signalling via direct interaction with and phosphorylation of IkappaBalpha. Biochem. J. 2009, 425, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Peregrin, S.; Jurado-Pueyo, M.; Campos, P.M.; Sanz-Moreno, V.; Ruiz-Gomez, A.; Crespo, P.; Mayor, J.F.; Murga, C. Phosphorylation of p38 by GRK2 at the Docking Groove Unveils a Novel Mechanism for Inactivating p38MAPK. Curr. Biol. 2018, 28, 2513. [Google Scholar] [CrossRef]
- Willemen, H.L.; Eijkelkamp, N.; Wang, H.; Dantzer, R.; Dorn, G.W., 2nd; Kelley, K.W.; Heijnen, C.J.; Ka-velaars, A. Microglial/macrophage GRK2 determines duration of peripheral IL-1beta-induced hyperalgesia: Contribution of spinal cord CX3CR1, p38 and IL-1 signaling. Pain 2010, 150, 550–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Premont, R.T.; Kontos, C.D.; Zhu, S.; Rockey, D.C. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat. Med. 2005, 11, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Kobayashi, T.; Takenouchi, Y.; Matsumoto, T.; Kamata, K. Angiotensin II causes endothelial dysfunction via the GRK2/Akt/eNOS pathway in aortas from a murine type 2 diabetic model. Pharmacol. Res. 2011, 64, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Cant, S.H.; Pitcher, J.A. G protein-coupled receptor kinase 2-mediated phosphorylation of ezrin is required for G pro-tein-coupled receptor-dependent reorganization of the actin cytoskeleton. Mol. Biol. Cell 2005, 16, 3088–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahsai, A.W.; Zhu, S.; Fenteany, G. G protein-coupled receptor kinase 2 activates radixin, regulating membrane protru-sion and motility in epithelial cells. Biochim. Biophys. Acta 2010, 1803, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Premont, R.T.; Claing, A.; Vitale, N.; Freeman, J.L.R.; Pitcher, J.A.; Patton, W.A.; Moss, J.; Vaughan, M.; Lefkowitz, R.J. 2-Adrenergic receptor regulation by GIT1, a G protein-coupled receptor kinase-associated ADP ribosylation factor GTPase-activating protein. Proc. Natl. Acad. Sci. USA 1998, 95, 14082–14087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafarga, V.; Aymerich, I.; Tapia, O.; Mayor, J.F.; Penela, P. A novel GRK2/HDAC6 interaction modulates cell spreading and motility. EMBO J. 2011, 31, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Benovic, J.L. G Protein-coupled Receptor Kinase Interaction with Hsp90 Mediates Kinase Maturation. J. Biol. Chem. 2003, 278, 50908–50914. [Google Scholar] [CrossRef] [Green Version]
- de Jager, S.C.; Bermudez, B.; Bot, I.; Koenen, R.R.; Bot, M.; Kavelaars, A.; de Waard, V.; Heijnen, C.J.; Mu-riana, F.J.; Weber, C.; et al. Growth differentiation factor 15 defi-ciency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J. Exp. Med. 2011, 208, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.; Cocolakis, E.; Dumas, V.M.; Posner, B.I.; Laporte, S.A.; Lebrun, J.J. The G protein-coupled receptor kinase-2 is a TGFbeta-inducible antagonist of TGFbeta signal transduction. EMBO J. 2005, 24, 3247–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, P.K.; Zhang, Y.; Coomes, A.S.; Kim, W.-J.; Stupay, R.; Lynch, L.D.; Atkinson, T.; Kim, J.I.; Nie, Z.; Daaka, Y. G Protein–Coupled Receptor Kinase GRK5 Phosphorylates Moesin and Regulates Metastasis in Prostate Cancer. Cancer Res. 2014, 74, 3489–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, B.L.; Benovic, J.L. G Protein-Coupled Receptor Kinase 5 Phosphorylation of Hip Regulates Internalization of the Chemokine Receptor CXCR4. Biochemistry 2011, 50, 6933–6941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luerman, G.C.; Powell, D.W.; Uriarte, S.M.; Cummins, T.D.; Merchant, M.L.; Ward, R.A.; McLeish, K.R. Iden-tification of phosphoproteins associated with human neutrophil granules following chemotactic peptide stimulation. Mol. Cell. Proteom. 2011, 10, M110.001552. [Google Scholar] [CrossRef] [Green Version]
- Tiedemann, R.; Zhu, Y.X.; Schmidt, J.; Yin, H.; Shi, C.-X.; Que, Q.; Basu, G.; Azorsa, D.; Perkins, L.M.; Braggio, E.; et al. Kinome-wide RNAi studies in human multiple myeloma identify vulnerable kinase targets, including a lymphoid-restricted kinase, GRK6. Blood 2010, 115, 1594–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachelerie, F.; Graham, G.J.; Locati, M.; Mantovani, A.; Murphy, P.M.; Nibbs, R.J.B.; Rot, A.; Sozzani, S.; Thelen, M. New nomenclature for atypical chemokine receptors. Nat. Immunol. 2014, 15, 207–208. [Google Scholar] [CrossRef]
- Balabanian, K.; Lagane, B.; Infantino, S.; Chow, K.Y.C.; Harriague, J.; Moepps, B.; Arenzana-Seisdedos, F.; Thelen, M.; Bachelerie, F. The Chemokine SDF-1/CXCL12 Binds to and Signals through the Orphan Receptor RDC1 in T Lymphocytes. J. Biol. Chem. 2005, 280, 35760–35766. [Google Scholar] [CrossRef] [Green Version]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.T.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef]
- Donà, E.; Barry, J.D.; Valentin, G.; Quirin, C.; Khmelinskii, A.; Kunze, A.; Durdu, S.; Newton, L.R.; Fernandez-Minan, A.; Huber, W.; et al. Directional tissue migration through a self-generated chemokine gradient. Nat. Cell Biol. 2013, 503, 285–289. [Google Scholar] [CrossRef]
- Theveneau, E.; Linker, C. Leaders in collective migration: Are front cells really endowed with a particular set of skills? F1000Research 2017, 6, 1899. [Google Scholar] [CrossRef] [Green Version]
- Abe, P.; Mueller, W.; Schütz, D.; Mackay, F.; Thelen, M.; Zhang, P.; Stumm, R. CXCR7 prevents excessive CXCL12-mediated downregulation of CXCR4 in migrating cortical interneurons. Development 2014, 141, 1857–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Alcañiz, J.A.; Haege, S.; Mueller, W.; Pla, R.; Mackay, F.; Schulz, S.; López-Bendito, G.; Stumm, R.; Marín, O. Cxcr7 Controls Neuronal Migration by Regulating Chemokine Responsiveness. Neuron 2011, 69, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, P.; Arlt, M.J.E.; Muff, R.; Campanile, C.; Gvozdenović, A.; Husmann, K.; Holzwarth, N.; Cameroni, E.; Ehrensperger, F.; Thelen, M.; et al. Expression of the Chemokine Receptor CXCR7 in CXCR4-Expressing Human 143B Osteosarcoma Cells Enhances Lung Metastasis of Intratibial Xenografts in SCID Mice. PLoS ONE 2013, 8, e74045. [Google Scholar] [CrossRef] [Green Version]
- Luker, K.E.; Lewin, S.A.; Mihalko, L.A.; Schmidt, B.T.; Winkler, J.S.; Coggins, N.L.; Thomas, D.G.; Luker, G.D. Scavenging of CXCL12 by CXCR7 promotes tumor growth and metastasis of CXCR4-positive breast cancer cells. Oncogene 2012, 31, 4750–4758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenen, J.; Bachelerie, F.; Balabanian, K.; Schlecht-Louf, G.; Gallego, C. Atypical Chemokine Receptor 3 (ACKR3): A Comprehensive Overview of its Expression and Potential Roles in the Immune System. Mol. Pharmacol. 2019, 96, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Coggins, N.L.; Trakimas, D.; Chang, S.L.; Ehrlich, A.; Ray, P.; Luker, K.E.; Linderman, J.J.; Luker, G.D. CXCR7 controls competition for recruitment of beta-arrestin 2 in cells expressing both CXCR4 and CXCR7. PLoS ONE 2014, 9, e98328. [Google Scholar] [CrossRef]
- Decaillot, F.M.; Kazmi, M.A.; Lin, Y.; Ray-Saha, S.; Sakmar, T.P.; Sachdev, P. CXCR7/CXCR4 heterodimer consti-tutively recruits beta-arrestin to enhance cell migration. J. Biol. Chem. 2011, 286, 32188–32197. [Google Scholar] [CrossRef] [Green Version]
- Levoye, A.; Balabanian, K.; Baleux, F.; Bachelerie, F.; Lagane, B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 2009, 113, 6085–6093. [Google Scholar] [CrossRef] [Green Version]
- Luker, K.E.; Gupta, M.; Luker, G.D. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2008, 23, 823–834. [Google Scholar] [CrossRef]
- Sierro, F.; Biben, C.; Martinez-Munoz, L.; Mellado, M.; Ransohoff, R.M.; Li, M.; Woehl, B.; Leung, H.; Groom, J.; Batten, M.; et al. Disrupted cardiac development but normal hem-atopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. USA 2007, 104, 14759–14764. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Tripathi, V.; Ahmad, M.; Nath, N.; Mir, R.A.; Chauhan, S.S.; Luthra, K. CXCR7 mediated Gialpha independent activation of ERK and Akt promotes cell survival and chemotaxis in T cells. Cell. Immunol. 2012, 272, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Luker, K.E.; Steele, J.M.; Mihalko, L.A.; Ray, P.; Luker, G.D. Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene 2010, 29, 4599–4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montpas, N.; St-Onge, G.; Nama, N.; Rhainds, D.; Benredjem, B.; Girard, M.; Hickson, G.; Pons, V.; Heveker, N. Ligand-specific conformational transitions and intracellular transport are required for atypical chemokine receptor 3–mediated chemokine scavenging. J. Biol. Chem. 2018, 293, 893–905. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, F.; Muller, W.; Schutz, D.; Penfold, M.E.; Wong, Y.H.; Schulz, S.; Stumm, R. Rapid uptake and deg-radation of CXCL12 depend on CXCR7 carboxyl-terminal serine/threonine residues. J. Biol. Chem. 2012, 287, 28362–28377. [Google Scholar] [CrossRef] [Green Version]
- Comerford, I.; Milasta, S.; Morrow, V.; Milligan, G.; Nibbs, R.J.B. The chemokine receptor CCX-CKR mediates effective scavenging of CCL19in vitro. Eur. J. Immunol. 2006, 36, 1904–1916. [Google Scholar] [CrossRef]
- Gosling, J.; Dairaghi, D.J.; Wang, Y.; Hanley, M.; Talbot, D.; Miao, Z.; Schall, T.J. Cutting Edge: Identification of a Novel Chemokine Receptor That Binds Dendritic Cell- and T Cell-Active Chemokines Including ELC, SLC, and TECK. J. Immunol. 2000, 164, 2851–2856. [Google Scholar] [CrossRef] [Green Version]
- Hauser, M.A.; Legler, D.F. Common and biased signaling pathways of the chemokine receptor CCR7 elicited by its ligands CCL19 and CCL21 in leukocytes. J. Leukoc. Biol. 2016, 99, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Kohout, T.A.; Nicholas, S.L.; Perry, S.J.; Reinhart, G.; Junger, S.; Struthers, R.S. Differential desensitization, re-ceptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor. J. Biol. Chem. 2004, 279, 23214–23222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Paz, J.L.; Moseman, E.A.; Noti, C.; Polito, L.; von Andrian, U.H.; Seeberger, P.H. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem. Biol. 2007, 2, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Hirose, J.; Kawashima, H.; Willis, M.S.; Springer, A.T.; Hasegawa, H.; Yoshie, O.; Miyasaka, M. Chondroitin sulfate B exerts its inhibitory effect on secondary lymphoid tissue chemokine (SLC) by binding to the C-terminus of SLC. Biochim. Biophys. Acta (BBA) Gen. Subj. 2002, 1571, 219–224. [Google Scholar] [CrossRef]
- Patel, D.D.; Koopmann, W.; Imai, T.; Whichard, L.P.; Yoshie, O.; Krangel, M.S. Chemokines Have Diverse Abilities to Form Solid Phase Gradients. Clin. Immunol. 2001, 99, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I.; Johnson, Z.; Bonvin, P.; Handel, T.M. Glycosaminoglycan Interactions with Chemokines Add Com-plexity to a Complex System. Pharmaceuticals 2017, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Förster, R.; Schubel, A.; Breitfeld, D.; Kremmer, E.; Renner-Müller, I.; Wolf, E.; Lipp, M. CCR7 Coordinates the Primary Immune Response by Establishing Functional Microenvironments in Secondary Lymphoid Organs. Cell 1999, 99, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Schumann, K.; Lämmermann, T.; Bruckner, M.; Legler, D.F.; Polleux, J.; Spatz, J.P.; Schuler, G.; Förster, R.; Lutz, M.B.; Sorokin, L.; et al. Immobilized Chemokine Fields and Soluble Chemokine Gradients Cooperatively Shape Migration Patterns of Dendritic Cells. Immunity 2010, 32, 703–713. [Google Scholar] [CrossRef] [Green Version]
- Hauser, M.A.; Schaeuble, K.; Kindinger, I.; Impellizzieri, D.; Krueger, W.A.; Hauck, C.R.; Boyman, O.; Legler, D.F. Inflammation-Induced CCR7 Oligomers Form Scaffolds to Integrate Distinct Signaling Pathways for Efficient Cell Migration. Immunity 2016, 44, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Minina, S.; Reichman-Fried, M.; Raz, E. Control of receptor internalization, signaling level, and precise arrival at the tar-get in guided cell migration. Curr. Biol. 2007, 17, 1164–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colak-Champollion, T.; Lan, L.; Jadhav, A.R.; Yamaguchi, N.; Venkiteswaran, G.; Patel, H.; Cammer, M.; Meier-Schellersheim, M.; Knaut, H. Cadherin-Mediated Cell Coupling Coordinates Chemokine Sensing across Collectively Mi-grating Cells. Curr. Biol. 2019, 29, 2570–2579.e7. [Google Scholar] [CrossRef] [PubMed]
- Mayor, R.; Etienne-Manneville, S. The front and rear of collective cell migration. Nat. Rev. Mol. Cell Biol. 2016, 17, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malet-Engra, G.; Yu, W.; Oldani, A.; Rey-Barroso, J.; Gov, N.S.; Scita, G.; Dupre, L. Collective cell motility pro-motes chemotactic prowess and resistance to chemorepulsion. Curr. Biol. 2015, 25, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, G.-H.; Lapierre, L.A.; Goldenring, J.R.; Sai, J.; Richmond, A. Rab11-Family Interacting Protein 2 and Myosin Vb Are Required for CXCR2 Recycling and Receptor-mediated Chemotaxis. Mol. Biol. Cell 2004, 15, 2456–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, R.M.; Marjoram, R.J.; Barak, L.S.; Snyderman, R. Role of the Cytoplasmic Tails of CXCR1 and CXCR2 in Mediating Leukocyte Migration, Activation, and Regulation. J. Immunol. 2003, 170, 2904–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil traf-ficking from murine bone marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Coombs, C.; Georgantzoglou, A.; Walker, H.A.; Patt, J.; Merten, N.; Poplimont, H.; Busch-Nentwich, E.M.; Williams, S.; Kotsi, C.; Kostenis, E.; et al. Author Correction: Chemokine receptor trafficking coordinates neutrophil clus-tering and dispersal at wounds in zebrafish. Nat. Commun. 2020, 11, 506. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Raghuwanshi, S.K.; Smith, N.; Rivers, E.J.; Richardson, R.M. G Protein–Coupled Receptor Kinase-6 Interacts with Activator of G Protein Signaling-3 To Regulate CXCR2-Mediated Cellular Functions. J. Immunol. 2014, 192, 2186–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lämmermann, T.; Kastenmüller, W. Concepts of GPCR—Controlled navigation in the immune system. Immunol. Rev. 2019, 289, 205–231. [Google Scholar] [CrossRef] [Green Version]
- Immunological Genome Project. ImmGen at 15. Nat. Immunol. 2020, 21, 700–703. [Google Scholar] [CrossRef]
- Ley, K.; Hoffman, H.M.; Kubes, P.; Cassatella, M.A.; Zychlinsky, A.; Hedrick, C.C.; Catz, S.D. Neutrophils: New insights and open questions. Sci. Immunol. 2018, 3, eaat4579. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Hidalgo, A.; Soehnlein, O. Neutrophil heterogeneity: Implications for homeostasis and pathogenesis. Blood 2016, 127, 2173–2181. [Google Scholar] [CrossRef] [Green Version]
- Evrard, M.; Kwok, I.W.; Chong, S.Z.; Teng, K.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e8. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, I.; Rubio-Ponce, A.; Genua, M.; Lusito, E.; Kwok, I.; Fernández-Calvo, G.; Khoyratty, T.E.; Van Grinsven, E.; González-Hernández, S.; Nicolás-Ávila, J.; et al. Co-option of Neutrophil Fates by Tissue Environments. Cell 2020, 183, 1282–1297.e18. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Acebes, M.; Nicolas-Avila, J.A.; Li, J.L.; Garcia-Silva, S.; Balachander, A.; Rubio-Ponce, A.; Weiss, L.A.; Adrover, J.M.; Burrows, K.; A-González, N.; et al. Neu-trophils instruct homeostatic and pathological states in naive tissues. J. Exp. Med. 2018, 215, 2778–2795. [Google Scholar] [CrossRef]
- Sadik, C.D.; Kim, N.D.; Luster, A.D. Neutrophils cascading their way to inflammation. Trends Immunol. 2011, 32, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.-C.; Williams, T.J.; Rankin, S.M. Chemokines Acting via CXCR2 and CXCR4 Control the Release of Neutrophils from the Bone Marrow and Their Return following Senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Walters, K.B.; Green, J.M.; Surfus, J.C.; Yoo, S.K.; Huttenlocher, A. Live imaging of neutrophil motility in a zebrafish model of WHIM syndrome. Blood 2010, 116, 2803–2811. [Google Scholar] [CrossRef] [Green Version]
- Devi, S.; Wang, Y.; Chew, W.K.; Lima, R.; A-González, N.; Mattar, C.N.; Chong, S.Z.; Schlitzer, A.; Bakocevic, N.; Chew, S.; et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J. Exp. Med. 2013, 210, 2321–2336. [Google Scholar] [CrossRef]
- McDermott, D.H.; Gao, J.-L.; Liu, Q.; Siwicki, M.; Martens, C.; Jacobs, P.; Velez, D.; Yim, E.; Bryke, C.R.; Hsu, N.; et al. Chromothriptic Cure of WHIM Syndrome. Cell 2015, 160, 686–699. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, S.; Murphy, P.M. Adaptive Immunodeficiency in WHIM Syndrome. Int. J. Mol. Sci. 2018, 20, 3. [Google Scholar] [CrossRef] [Green Version]
- Auer, P.L.; Teumer, A.; Schick, U.; O’Shaughnessy, A.; Lo, K.S.; Chami, N.; Carlson, C.; De Denus, S.; Dubé, M.-P.; Haessler, J.; et al. Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat. Genet. 2014, 46, 629–634. [Google Scholar] [CrossRef]
- De Filippo, K.; Rankin, S.M. CXCR4, the master regulator of neutrophil trafficking in homeostasis and disease. Eur. J. Clin. Investig. 2018, 48, e12949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrover, J.M.; Del Fresno, C.; Crainiciuc, G.; Cuartero, M.I.; Casanova-Acebes, M.; Weiss, L.A.; Huerga-Encabo, H.; Silvestre-Roig, C.; Rossaint, J.; Cossío, I.; et al. A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity 2019, 51, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Adrover, J.M.; Nicolás-Ávila, J.A.; Hidalgo, A. Aging: A Temporal Dimension for Neutrophils. Trends Immunol. 2016, 37, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G. Chemorepulsion and thymocyte emigration. J Clin. Investig. 2002, 109, 1011–1012. [Google Scholar] [CrossRef] [PubMed]
- Tharp, W.G.; Yadav, R.; Irimia, D.; Upadhyaya, A.; Samadani, A.; Hurtado, O.; Liu, S.Y.; Munisamy, S.; Brainard, D.M.; Mahon, M.J.; et al. Neutrophil chem-orepulsion in defined interleukin-8 gradients in vitro and in vivo. J. Leukoc. Biol. 2006, 79, 539–554. [Google Scholar] [CrossRef]
- Arraes, S.M.A.; Freitas, M.S.; Da Silva, S.V.; Neto, H.A.D.P.; Alves-Filho, J.C.; Martins, M.A.; Basile-Filho, A.; Tavares-Murta, B.M.; Barja-Fidalgo, C.; Cunha, T.M. Impaired neutrophil chemotaxis in sepsis associates with GRK expression and inhibition of actin assembly and tyrosine phosphorylation. Blood 2006, 108, 2906–2913. [Google Scholar] [CrossRef] [Green Version]
- Aves-Filho, J.C.F.; Freitas, A.; Souto, F.O.; Spiller, F.; Paula-Neto, H.; Silva, J.S.; Gazzinelli, R.T.; Teixeira, M.M.; Ferreira, S.H.; Cunha, T.M. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc. Natl. Acad. Sci. USA 2009, 106, 4018–4023. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.B.; Yan, J.J.; Kang, J.S.; Zhang, Q.; Choi, W.Y.; Kim, L.K.; Kim, Y.J. Mincle activation enhances neutro-phil migration and resistance to polymicrobial septic peritonitis. Sci. Rep. 2017, 7, 41106. [Google Scholar] [CrossRef]
- Trevelin, S.C.; Alves-Filho, J.C.; Sônego, F.; Turato, W.; Nascimento, D.C.; Souto, F.O.; Cunha, T.M.; Gazzinelli, R.T.; Cunha, F.Q. Toll-like receptor 9 activation in neutrophils impairs chemotaxis and reduces sepsis outcome*. Crit. Care Med. 2012, 40, 2631–2637. [Google Scholar] [CrossRef]
- Alves-Filho, J.C.; Sônego, F.; Souto, F.O.; Freitas, A.; Verri, W.; Auxiliadora-Martins, M.; Basile-Filho, A.; McKenzie, A.N.; Xu, D.; Cunha, F.Q.; et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 2010, 16, 708–712. [Google Scholar] [CrossRef]
- Tancevski, I.; Nairz, M.; Duwensee, K.; Auer, K.; Schroll, A.; Heim, C.; Feistritzer, C.; Hoefer, J.; Gerner, R.R.; Moschen, A.R.; et al. Fibrates ameliorate the course of bacterial sepsis by promoting neutrophil recruitment via CXCR2. EMBO Mol. Med. 2014, 6, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Malik, A.B. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nat. Med. 2003, 9, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.R.; Hummon, A.B. Mass spectrometry for the discovery of biomarkers of sepsis. Mol. BioSyst. 2017, 13, 648–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogendijk, A.J.; van Vught, L.A.; Wiewel, M.A.; Fuhler, G.M.; Belkasim-Bohoudi, H.; Horn, J.; Schultz, M.J.; Scicluna, B.P.; Peppelenbosch, M.P.; van ’t Veer, C.; et al. Kinase activity is impaired in neutrophils of sepsis patients. Haematologica 2019, 104, e233–e235. [Google Scholar] [CrossRef]
- Penela, P. Chapter Three—Ubiquitination and Protein Turnover of G-Protein-Coupled Receptor Kinases in GPCR Signaling and Cellular Regulation. Prog. Mol. Biol. Transl. Sci. 2016, 141, 85–140. [Google Scholar] [CrossRef]
- Watari, K.; Nakaya, M.; Kurose, H. Multiple functions of G protein-coupled receptor kinases. J. Mol. Signal. 2014, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Dinkel, B.A.; Kremer, K.N.; Rollins, M.R.; Medlyn, M.J.; Hedin, K.E. GRK2 mediates TCR-induced transactivation of CXCR4 and TCR-CXCR4 complex formation that drives PI3Kgamma/PREX1 signaling and T cell cytokine secretion. J. Biol. Chem. 2018, 293, 14022–14039. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GRK | Chemokine Receptor | Signalling Pathway/Cellular Response |
|---|---|---|
| GRK2 | ACKR3 | CXCL12 scavenging [2] |
| CCR9 | Receptor desensitization [3] | |
| CXCR1 | Receptor desensitization [4] | |
| CXCR2 | Receptor desensitization [5] | |
| CXCR4 | Receptor desensitization [6,7] | |
| Chemotaxis [8] | ||
| GRK3 | ACKR4 | Recruitment of arrestins [9] |
| CCR7 | Receptor desensitization [10] | |
| Arrestin signalling [11] | ||
| CXCR4 | Arrestin signalling [8] | |
| Receptor desensitization [6,7,12,13] | ||
| Recruitment of arrestins [14] | ||
| GRK 5 | ACKR3 | CXCL12 scavenging [2] |
| GRK 6 | CCR7 | Receptor desensitization [10,15] |
| Arrestin signalling [11] | ||
| CXCR1 | Receptor desensitization [4] | |
| CXCR4 | Receptor desensitization [6,7] | |
| Arrestin signalling [8] |
| GRK | Partner (s) | Partner’s Associated Signalling Pathway/Cellular Response |
|---|---|---|
| GRK2 | NF-κB p105 subunit and inhibitor (IκB-α) phosphorylation | TLR4-induced and Tumour Necrosis Factor-α (TNF-α) pathways [20,79,80] |
| p38 phosphorylation | P38 mitogen-activated protein kinases (MAPK) pathways [81,82] | |
| Raf1, MEK1, ERK2, RhoA, RKIP, GIT | Extracellular signal-regulated kinase (ERK) pathways [77] | |
| Serine-threonine kinase Akt phosphorylation | Akt-Nitric Oxide (NO) pathways [83,84] | |
| Ezrin/radixin/moesin phosphorylation | Actin cytoskeleton [85,86] | |
| ADP ribosylation factor (ARF)-specific GTPase-activating proteins (GIT) | Focal adhesion dynamic [78,87] | |
| Histone deacetylase 6 (HDAC6) phosphorylation | Microtubules network [88] | |
| Heat shock protein 90 (Hsp90) | Regulation of GRK expression [89] | |
| Receptor-regulated Smads (R-Smads) phosphorylation | Transforming growth factor beta (TGF-β) pathways [90,91] | |
| GRK3 | HSP90 | Regulation of GRK expression [89] |
| GRK5 | ERM (moesin phosphorylation) | Actin cytoskeleton [92] |
| GIT1 | Regulation of receptor endocytosis [87] | |
| HSP90, HSP70 | Regulation of GRK expression and CXCR4 endocytosis | |
| NF-κB p105 subunit and IκB-α phosphorylation | TLR4-induced and TNF-α pathways [89,93] | |
| Src Tyrosine kinase | GRK phosphorylation and neutrophils exocytosis [94] | |
| GRK6 | HSP90 | Regulation of GRK expression [89,95] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laganà, M.; Schlecht-Louf, G.; Bachelerie, F. The G Protein-Coupled Receptor Kinases (GRKs) in Chemokine Receptor-Mediated Immune Cell Migration: From Molecular Cues to Physiopathology. Cells 2021, 10, 75. https://doi.org/10.3390/cells10010075
Laganà M, Schlecht-Louf G, Bachelerie F. The G Protein-Coupled Receptor Kinases (GRKs) in Chemokine Receptor-Mediated Immune Cell Migration: From Molecular Cues to Physiopathology. Cells. 2021; 10(1):75. https://doi.org/10.3390/cells10010075
Chicago/Turabian StyleLaganà, Marta, Géraldine Schlecht-Louf, and Françoise Bachelerie. 2021. "The G Protein-Coupled Receptor Kinases (GRKs) in Chemokine Receptor-Mediated Immune Cell Migration: From Molecular Cues to Physiopathology" Cells 10, no. 1: 75. https://doi.org/10.3390/cells10010075