Acute D-Serine Co-Agonism of β-Cell NMDA Receptors Potentiates Glucose-Stimulated Insulin Secretion and Excitatory β-Cell Membrane Activity

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Subjects
2.2. In Vivo Mouse Experiments
2.3. Mouse Islet Experiments
2.4. Gene Expression
2.5. Electrophysiology
2.6. Immunostaining
2.7. Statistical Analysis
2.8. Study Approval
3. Results
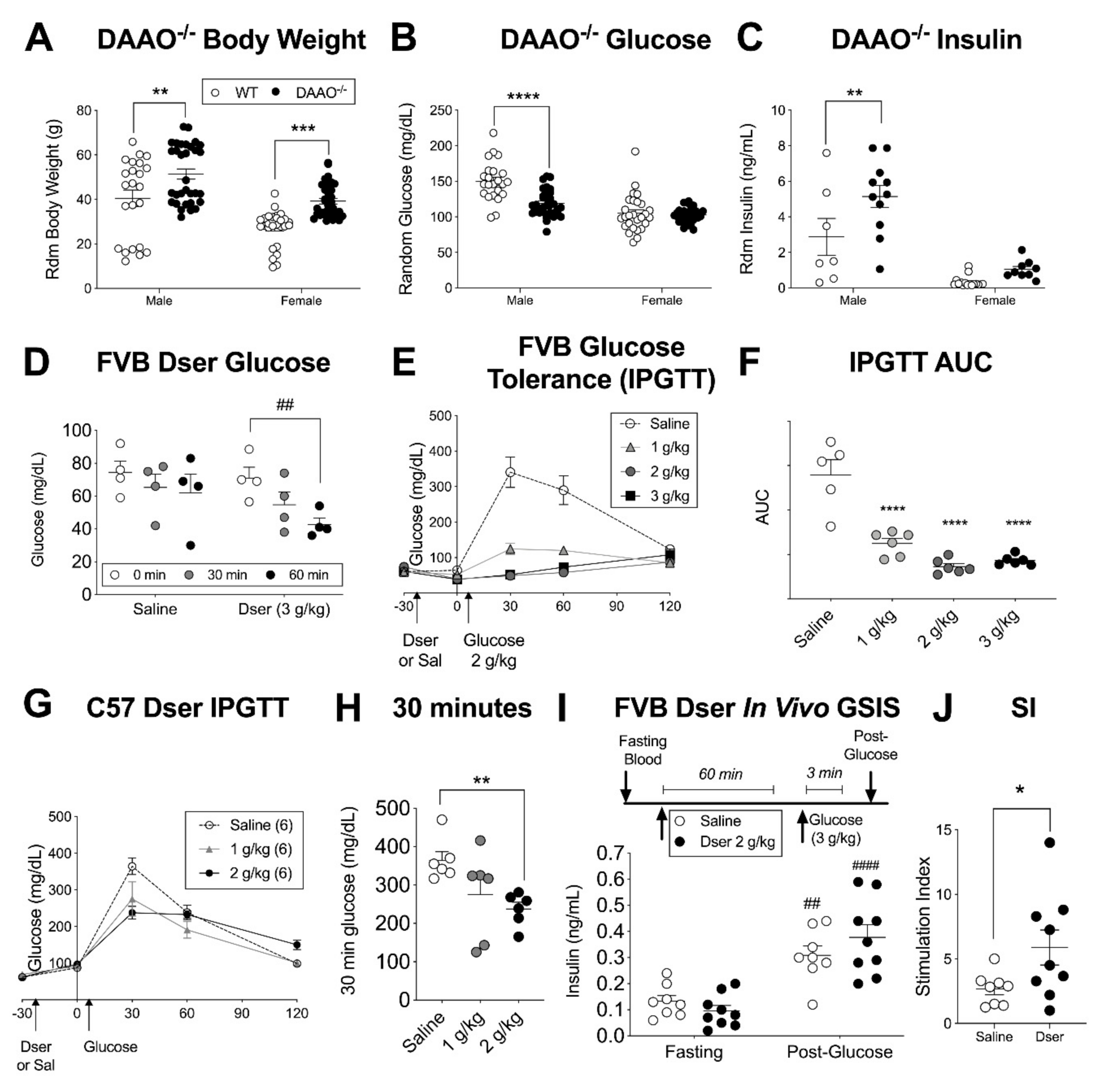
3.1. Acute Systemic D-serine Lowers Blood Glucose in Multiple Mouse Strains
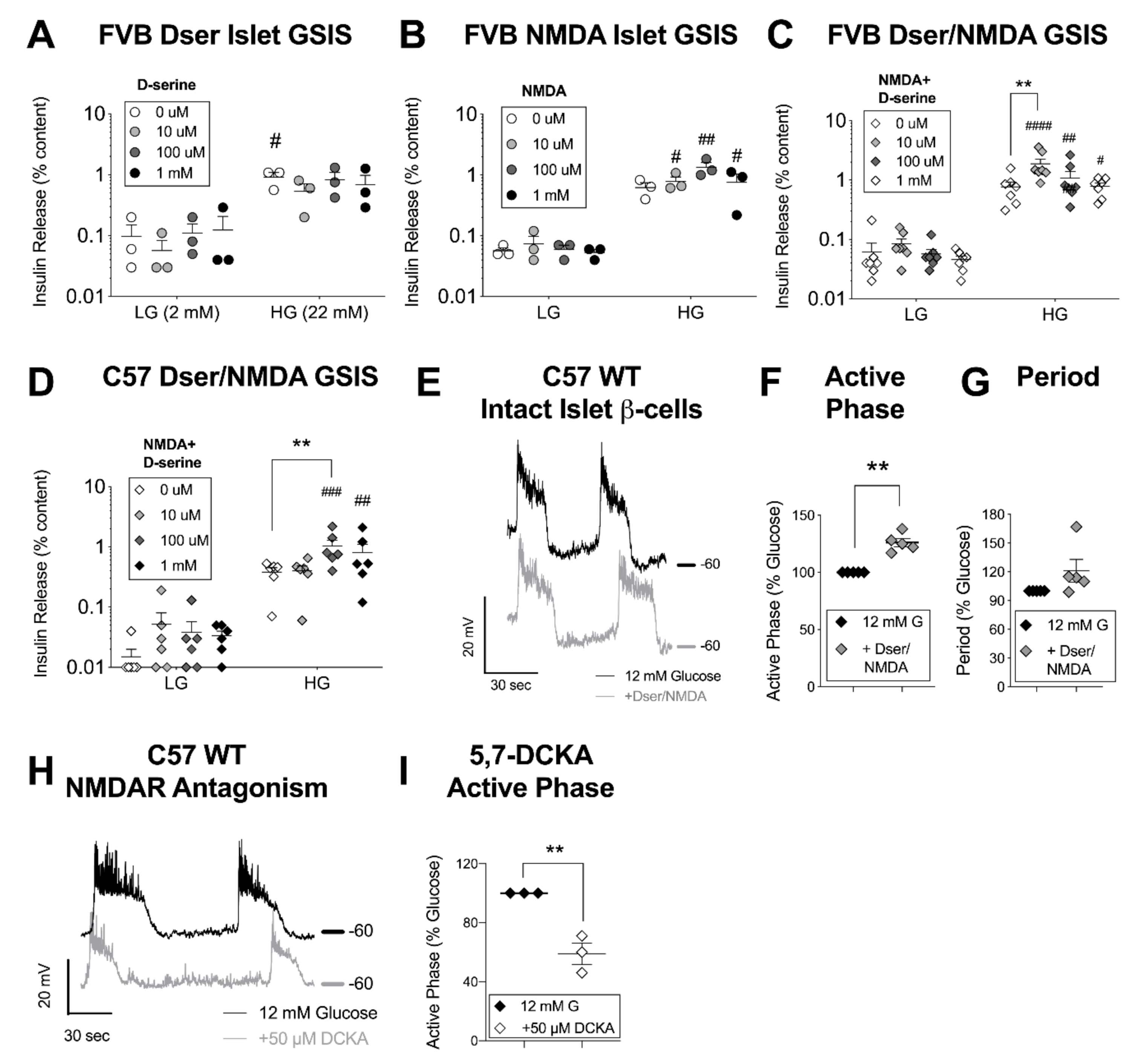
3.2. D-Serine with NMDA Potentiates Glucose-Stimulated Insulin Secretion and β-Cell Excitation
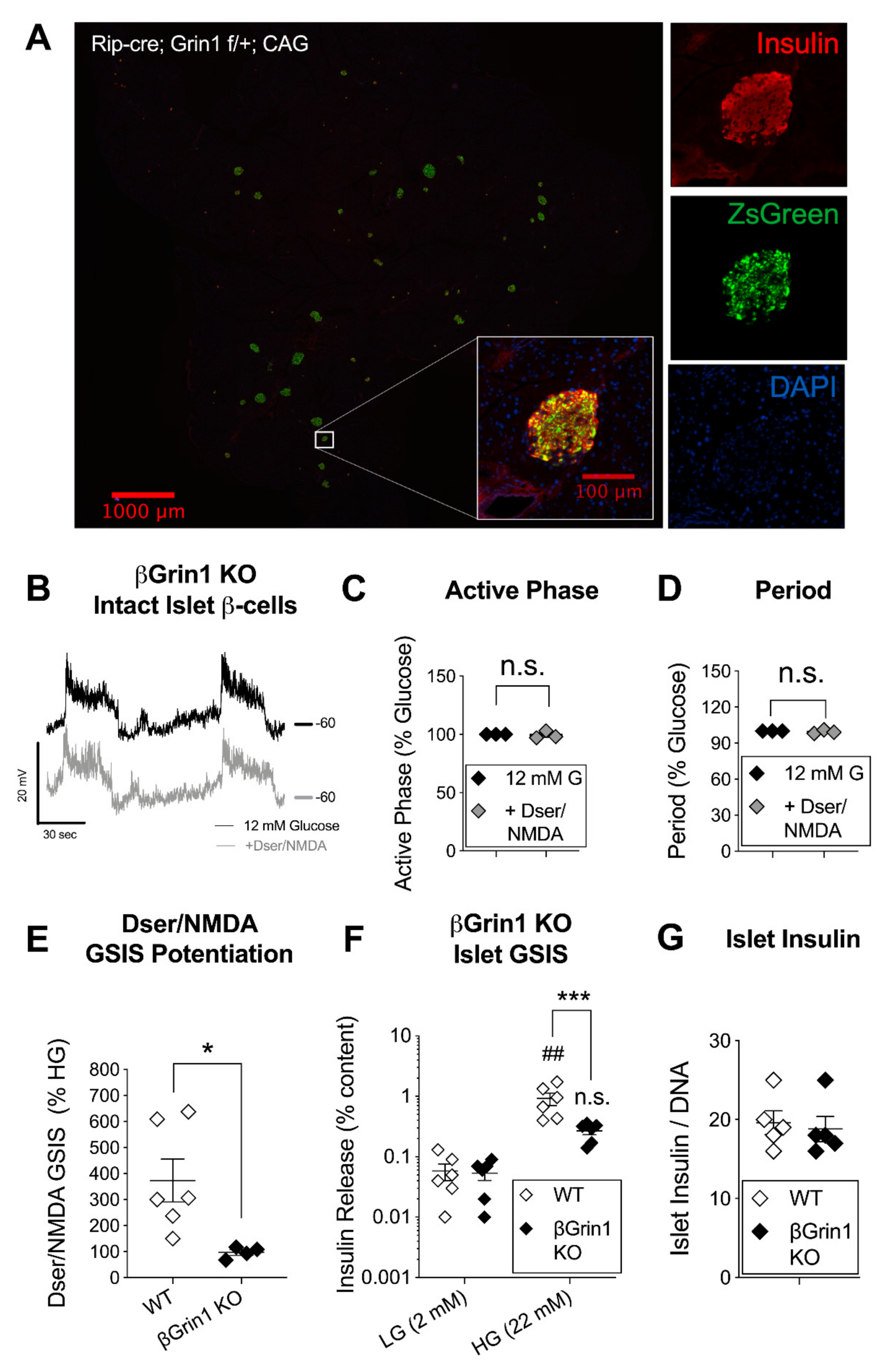
3.3. β-Cell Specific Loss of NMDAR Co-Agonist Binding Protein Negates In Vitro D-Serine/NMDA Effects
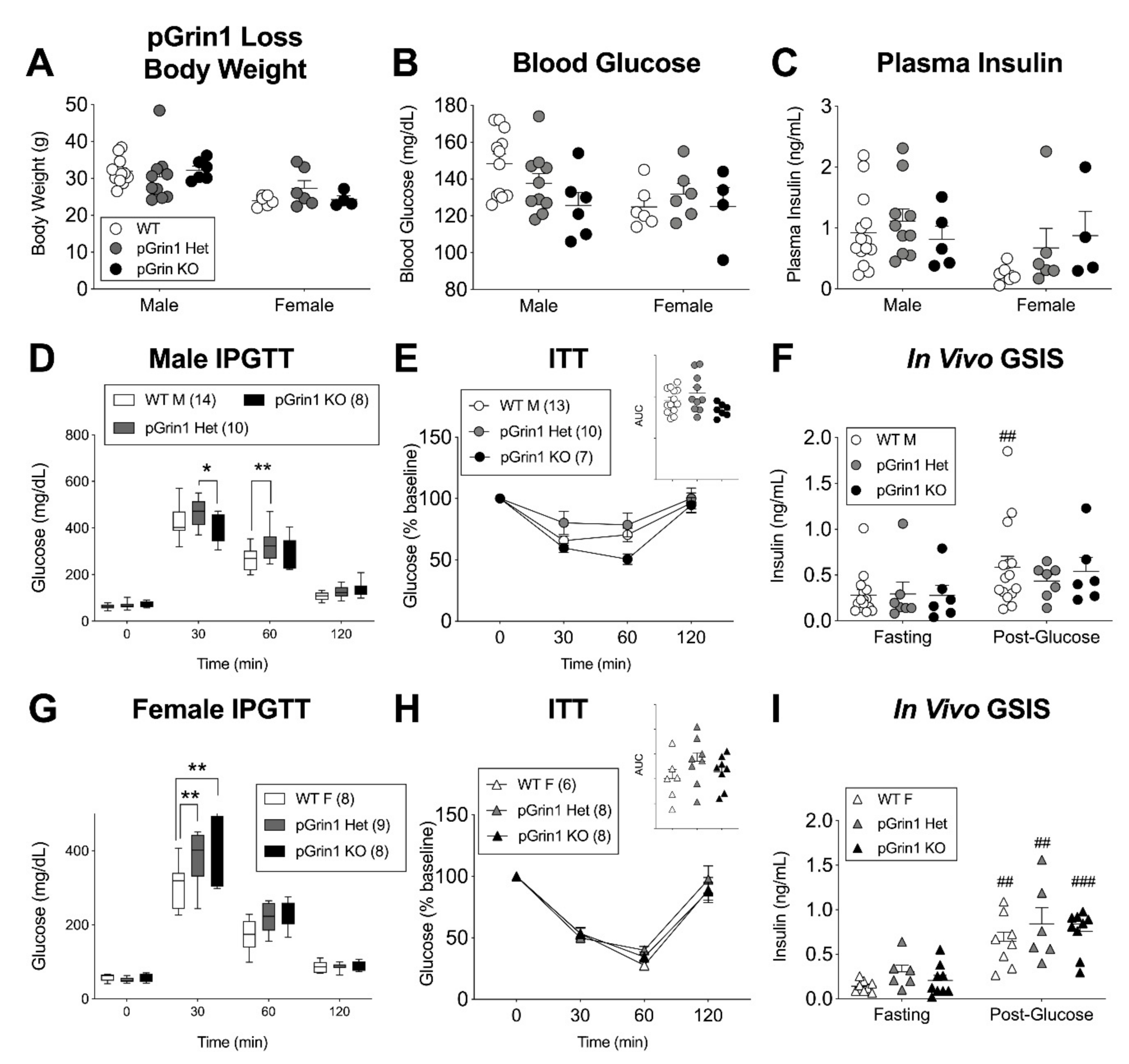
3.4. Minimal In Vivo Phenotype but Age-Related Reversal of βGrin1 KO Secretory Phenotype
3.5. Sex-Specific Glucose Intolerance in a Mouse Model with Pancreatic Deletion of GluN1
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lockridge, A.D.; Baumann, D.C.; Akhaphong, B.; Abrenica, A.; Miller, R.F.; Alejandro, E.U. Serine racemase is expressed in islets and contributes to the regulation of glucose homeostasis. Islets 2016, 8, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Nie, M.; Li, W.; Ping, F.; Hu, Y.; Ma, L.; Gao, J.; Liu, J. Association of six single nucleotide polymorphisms with gestational diabetes mellitus in a Chinese population. PLoS ONE 2011, 6, e26953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, F.J.; Yang, C.F.; Chen, C.C.; Chuang, L.M.; Lu, C.H.; Chang, C.T.; Wang, T.Y.; Chen, R.H.; Shiu, C.F.; Liu, Y.M.; et al. A genome-wide association study identifies susceptibility variants for type 2 diabetes in Han Chinese. PLoS Genet. 2010, 6, e1000847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, M.; Gong, Z.C.; Dai, X.P.; Lei, G.H.; Lu, H.B.; Fan, L.; Qu, J.; Zhou, H.H.; Liu, Z.Q. Serine racemase rs391300 G/A polymorphism influences the therapeutic efficacy of metformin in Chinese patients with diabetes mellitus type 2. Clin. Exp. Pharmacol. Physiol. 2011, 38, 824–829. [Google Scholar] [CrossRef]
- Suwandhi, L.; Hausmann, S.; Braun, A.; Gruber, T.; Heinzmann, S.S.; Galvez, E.J.C.; Buck, A.; Legutko, B.; Israel, A.; Feuchtinger, A.; et al. Chronic d-serine supplementation impairs insulin secretion. Mol. Metab. 2018, 16, 191–202. [Google Scholar] [CrossRef]
- Marquard, J.; Otter, S.; Welters, A.; Stirban, A.; Fischer, A.; Eglinger, J.; Herebian, D.; Kletke, O.; Klemen, M.S.; Stozer, A.; et al. Characterization of pancreatic NMDA receptors as possible drug targets for diabetes treatment. Nat. Med. 2015, 21, 363–372. [Google Scholar] [CrossRef]
- McKay, S.; Ryan, T.J.; McQueen, J.; Indersmitten, T.; Marwick, K.F.M.; Hasel, P.; Kopanitsa, M.V.; Baxter, P.S.; Martel, M.A.; Kind, P.C.; et al. The Developmental Shift of NMDA Receptor Composition Proceeds Independently of GluN2 Subunit-Specific GluN2 C-Terminal Sequences. Cell Rep. 2018, 25, 841–851.e844. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.K.; Liu, X.B.; Matos, M.F.; Chan, S.F.; Perez-Otano, I.; Boysen, M.; Cui, J.; Nakanishi, N.; Trimmer, J.S.; Jones, E.G.; et al. Temporal and regional expression of NMDA receptor subunit NR3A in the mammalian brain. J. Comp. Neurol. 2002, 450, 303–317. [Google Scholar] [CrossRef]
- Seif, T.; Simms, J.A.; Lei, K.; Wegner, S.; Bonci, A.; Messing, R.O.; Hopf, F.W. D-Serine and D-Cycloserine Reduce Compulsive Alcohol Intake in Rats. Neuropsychopharmacology 2015, 40, 2357–2367. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [Green Version]
- Nong, Y.; Huang, Y.Q.; Ju, W.; Kalia, L.V.; Ahmadian, G.; Wang, Y.T.; Salter, M.W. Glycine binding primes NMDA receptor internalization. Nature 2003, 422, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Gonoi, T.; Mizuno, N.; Inagaki, N.; Kuromi, H.; Seino, Y.; Miyazaki, J.; Seino, S. Functional neuronal ionotropic glutamate receptors are expressed in the non-neuronal cell line MIN6. J. Biol. Chem. 1994, 269, 16989–16992. [Google Scholar] [PubMed]
- Molnar, E.; Varadi, A.; McIlhinney, R.A.; Ashcroft, S.J. Identification of functional ionotropic glutamate receptor proteins in pancreatic beta-cells and in islets of Langerhans. FEBS Lett. 1995, 371, 253–257. [Google Scholar] [PubMed] [Green Version]
- Inagaki, N.; Kuromi, H.; Gonoi, T.; Okamoto, Y.; Ishida, H.; Seino, Y.; Kaneko, T.; Iwanaga, T.; Seino, S. Expression and role of ionotropic glutamate receptors in pancreatic islet cells. FASEB J. 1995, 9, 686–691. [Google Scholar] [CrossRef]
- Patterson, S.; Irwin, N.; Guo-Parke, H.; Moffett, R.C.; Scullion, S.M.; Flatt, P.R.; McClenaghan, N.H. Evaluation of the role of N-methyl-D-aspartate (NMDA) receptors in insulin secreting beta-cells. Eur. J. Pharmacol. 2016, 771, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Fortin, D.A.; Cochrane, V.A.; Chen, P.C.; Shyng, S.L. NMDA receptors mediate leptin signaling and regulate potassium channel trafficking in pancreatic beta-cells. J. Biol. Chem. 2017, 292, 15512–15524. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.T.; Yue, S.J.; Li, C.; Huang, Y.H.; Cheng, Q.M.; Li, X.H.; Hao, C.X.; Wang, L.Z.; Xu, J.P.; Ji, M.; et al. A Sustained Activation of Pancreatic NMDARs Is a Novel Factor of beta-Cell Apoptosis and Dysfunction. Endocrinology 2017, 158, 3900–3913. [Google Scholar] [CrossRef] [Green Version]
- Luscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [Green Version]
- Stevens, E.R.; Gustafson, E.C.; Sullivan, S.J.; Esguerra, M.; Miller, R.F. Light-evoked NMDA receptor-mediated currents are reduced by blocking D-serine synthesis in the salamander retina. Neuroreport 2010, 21, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.; Suh, J.M.; Kim, E.Y.; Dryer, S.E. Functional NMDA receptors with atypical properties are expressed in podocytes. Am. J. Physiol. Cell Physiol. 2011, 300, C22–C32. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.T.; Li, C.; Peng, X.P.; Guo, J.; Yue, S.J.; Liu, W.; Zhao, F.Y.; Han, J.Z.; Huang, Y.H.; Yang, L.; et al. An excessive increase in glutamate contributes to glucose-toxicity in beta-cells via activation of pancreatic NMDA receptors in rodent diabetes. Sci. Rep. 2017, 7, 44120. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, E.C.; Morgans, C.W.; Tekmen, M.; Sullivan, S.J.; Esguerra, M.; Konno, R.; Miller, R.F. Retinal NMDA receptor function and expression are altered in a mouse lacking D-amino acid oxidase. J. Neurophysiol. 2013, 110, 2718–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, G.E.; Lockridge, A.D.; Morgans, C.W.; Bandyopadhyay, D.; Miller, R.F. The postnatal development of D-serine in the retinas of two mouse strains, including a mutant mouse with a deficiency in D-amino acid oxidase and a serine racemase knockout mouse. ACS Chem. Neurosci. 2014, 5, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Konno, R.; Yasumura, Y. Mouse mutant deficient in D-amino acid oxidase activity. Genetics 1983, 103, 277–285. [Google Scholar] [CrossRef]
- Alejandro, E.U.; Bozadjieva, N.; Kumusoglu, D.; Abdulhamid, S.; Levine, H.; Haataja, L.; Vadrevu, S.; Satin, L.S.; Arvan, P.; Bernal-Mizrachi, E. Disruption of O-linked N-Acetylglucosamine Signaling Induces ER Stress and beta Cell Failure. Cell Rep. 2015, 13, 2527–2538. [Google Scholar] [CrossRef] [Green Version]
- Lockridge, A.; Jo, S.; Gustafson, E.; Damberg, N.; Mohan, R.; Olson, M.; Abrahante, J.E.; Alejandro, E.U. Islet O-GlcNAcylation Is Required for Lipid Potentiation of Insulin Secretion through SERCA2. Cell Rep. 2020, 31, 107609. [Google Scholar] [CrossRef]
- Lockridge, A.; Romero, G.; Harrington, J.; Newland, B.; Gong, Z.; Cameron, A.; Yuan, L.L. Timing-dependent reduction in ethanol sedation and drinking preference by NMDA receptor co-agonist d-serine. Alcohol 2012, 46, 389–400. [Google Scholar] [CrossRef]
- Zhang, G.; Byun, H.R.; Ying, Z.; Blencowe, M.; Zhao, Y.; Hong, J.; Shu, L.; Chella Krishnan, K.; Gomez-Pinilla, F.; Yang, X. Differential metabolic and multi-tissue transcriptomic responses to fructose consumption among genetically diverse mice. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165569. [Google Scholar] [CrossRef]
- Takahata, Y.; Takarada, T.; Osawa, M.; Hinoi, E.; Nakamura, Y.; Yoneda, Y. Differential regulation of cellular maturation in chondrocytes and osteoblasts by glycine. Cell Tissue Res. 2008, 333, 91–103. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Hamase, K.; Okamura, T.; Konno, R.; Kasai, N.; Tojo, Y.; Zaitsu, K. Simultaneous two-dimensional HPLC determination of free D-serine and D-alanine in the brain and periphery of mutant rats lacking D-amino-acid oxidase. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 3184–3189. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Konno, R.; Sasabe, J.; Ueno, K.; Tojo, Y.; Mita, M.; Aiso, S.; Hamase, K. Alteration of intrinsic amounts of D-serine in the mice lacking serine racemase and D-amino acid oxidase. Amino Acids 2012, 43, 1919–1931. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Hamase, K.; Tojo, Y.; Mita, M.; Konno, R.; Zaitsu, K. Determination of D-serine and D-alanine in the tissues and physiological fluids of mice with various D-amino-acid oxidase activities using two-dimensional high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 2506–2512. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.; Wallace, K.; Granger, J. Role of angiotensin II type I receptor agonistic autoantibodies (AT1-AA) in preeclampsia. Curr. Opin. Pharmacol. 2011, 11, 175–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalsbeek, A.; La Fleur, S.; Van Heijningen, C.; Buijs, R.M. Suprachiasmatic GABAergic inputs to the paraventricular nucleus control plasma glucose concentrations in the rat via sympathetic innervation of the liver. J. Neurosci. 2004, 24, 7604–7613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Wei, L.; Deveau, T.C.; Gu, X.; Yu, S.P. Expression of the NMDA receptor subunit GluN3A (NR3A) in the olfactory system and its regulatory role on olfaction in the adult mouse. Brain Struct. Funct. 2016, 221, 3259–3273. [Google Scholar] [CrossRef]
- Perez-Otano, I.; Larsen, R.S.; Wesseling, J.F. Emerging roles of GluN3-containing NMDA receptors in the CNS. Nat. Rev. Neurosci. 2016, 17, 623–635. [Google Scholar] [CrossRef]
- Horio, M.; Kohno, M.; Fujita, Y.; Ishima, T.; Inoue, R.; Mori, H.; Hashimoto, K. Levels of D-serine in the brain and peripheral organs of serine racemase (Srr) knock-out mice. Neurochem. Int. 2011, 59, 853–859. [Google Scholar] [CrossRef]
- Paredes, J.L.; Orabi, A.I.; Ahmad, T.; Benbourenane, I.; Tobita, K.; Tadros, S.; Bae, K.T.; Husain, S.Z. A non-invasive method of quantifying pancreatic volume in mice using micro-MRI. PLoS ONE 2014, 9, e92263. [Google Scholar] [CrossRef] [Green Version]
- Danysz, W.; Parsons, C.G. Glycine and N-methyl-D-aspartate receptors: Physiological significance and possible therapeutic applications. Pharmacol. Rev. 1998, 50, 597–664. [Google Scholar]
- Clark, J.P., 3rd; Kofuji, P. Stoichiometry of N-methyl-D-aspartate receptors within the suprachiasmatic nucleus. J. Neurophysiol. 2010, 103, 3448–3464. [Google Scholar] [CrossRef] [Green Version]
- Atlason, P.T.; Garside, M.L.; Meddows, E.; Whiting, P.; McIlhinney, R.A. N-Methyl-D-aspartate (NMDA) receptor subunit NR1 forms the substrate for oligomeric assembly of the NMDA receptor. J. Biol. Chem. 2007, 282, 25299–25307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, P.L. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 2000, 127, 2317–2322. [Google Scholar] [PubMed]
- Konrad, D.; Sobetzko, D.; Schmitt, B.; Schoenle, E.J. Insulin-dependent diabetes mellitus induced by the antitussive agent dextromethorphan. Diabetologia 2000, 43, 261–262. [Google Scholar] [CrossRef]
- Baumann, D.; Wong, A.; Akhaphong, B.; Jo, S.; Pritchard, S.; Mohan, R.; Chung, G.; Zhang, Y.; Alejandro, E.U. Role of nutrient-driven O-GlcNAc-posttranslational modification in pancreatic exocrine and endocrine islet development. Development 2020, 147. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar]
- Lam, C.K.; Chari, M.; Rutter, G.A.; Lam, T.K. Hypothalamic nutrient sensing activates a forebrain-hindbrain neuronal circuit to regulate glucose production in vivo. Diabetes 2011, 60, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Wicksteed, B.; Brissova, M.; Yan, W.; Opland, D.M.; Plank, J.L.; Reinert, R.B.; Dickson, L.M.; Tamarina, N.A.; Philipson, L.H.; Shostak, A.; et al. Conditional gene targeting in mouse pancreatic ss-Cells: Analysis of ectopic Cre transgene expression in the brain. Diabetes 2010, 59, 3090–3098. [Google Scholar] [CrossRef] [Green Version]
- Magnuson, M.A.; Osipovich, A.B. Pancreas-specific Cre driver lines and considerations for their prudent use. Cell Metab. 2013, 18, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Wahl, M.A.; Plehn, R.J.; Landsbeck, E.A.; Verspohl, E.J.; Ammon, H.P. Are ionic fluxes of pancreatic beta cells a target for gastric inhibitory polypeptide? Mol. Cell. Endocrinol. 1992, 90, 117–123. [Google Scholar] [CrossRef]
- Sasabe, J.; Chiba, T.; Yamada, M.; Okamoto, K.; Nishimoto, I.; Matsuoka, M.; Aiso, S. D-serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis. EMBO J. 2007, 26, 4149–4159. [Google Scholar] [CrossRef] [Green Version]
- Krug, A.W.; Volker, K.; Dantzler, W.H.; Silbernagl, S. Why is D-serine nephrotoxic and alpha-aminoisobutyric acid protective? Am. J. Physiol. Ren. Physiol. 2007, 293, F382–F390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenzen, S. Oxidative stress: The vulnerable beta-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Schulte, E.C.; Mollenhauer, B.; Zimprich, A.; Bereznai, B.; Lichtner, P.; Haubenberger, D.; Pirker, W.; Brucke, T.; Molnar, M.J.; Peters, A.; et al. Variants in eukaryotic translation initiation factor 4G1 in sporadic Parkinson‘s disease. Neurogenetics 2012, 13, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Gresch, A.; Dufer, M. Dextromethorphan and Dextrorphan Influence Insulin Secretion by Interacting with KATP and L-type Ca(2+) Channels in Pancreatic beta-Cells. J. Pharmacol. Exp. Ther. 2020, 375, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Amador, M.; Dani, J.A. MK-801 inhibition of nicotinic acetylcholine receptor channels. Synapse 1991, 7, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Hellstrom-Lindahl, E.; Grill, V. Evidence for functional nicotinic receptors on pancreatic beta cells. Metabolism 2005, 54, 247–254. [Google Scholar] [CrossRef]
- Iravani, M.M.; Muscat, R.; Kruk, Z.L. MK-801 interaction with the 5-HT transporter: A real-time study in brain slices using fast cyclic voltammetry. Synapse 1999, 32, 212–224. [Google Scholar] [CrossRef]
- Almaca, J.; Molina, J.; Menegaz, D.; Pronin, A.N.; Tamayo, A.; Slepak, V.; Berggren, P.O.; Caicedo, A. Human Beta Cells Produce and Release Serotonin to Inhibit Glucagon Secretion from Alpha Cells. Cell Rep. 2016, 17, 3281–3291. [Google Scholar] [CrossRef] [Green Version]
- Soesanto, Y.; Luo, B.; Parker, G.; Jones, D.; Cooksey, R.C.; McClain, D.A. Pleiotropic and age-dependent effects of decreased protein modification by O-linked N-acetylglucosamine on pancreatic beta-cell function and vascularization. J. Biol. Chem. 2011, 286, 26118–26126. [Google Scholar] [CrossRef] [Green Version]
- Kehoe, L.A.; Bernardinelli, Y.; Muller, D. GluN3A: An NMDA receptor subunit with exquisite properties and functions. Neural Plast. 2013, 2013, 145387. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Fukushima, T.; Santa, T.; Homma, H.; Huang, Y.; Shirao, M.; Miura, K. Whole body autoradiographic study on the distribution of 14C-D-serine administered intravenously to rats. Amino Acids 1998, 15, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, D.; Artoul, S.; Segal, A.C.; Kolodney, G.; Radzishevsky, I.; Dikopoltsev, E.; Foltyn, V.N.; Inoue, R.; Mori, H.; Billard, J.M.; et al. Neuronal D-serine and glycine release via the Asc-1 transporter regulates NMDA receptor-dependent synaptic activity. J. Neurosci. 2013, 33, 3533–3544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukasawa, Y.; Segawa, H.; Kim, J.Y.; Chairoungdua, A.; Kim, D.K.; Matsuo, H.; Cha, S.H.; Endou, H.; Kanai, Y. Identification and characterization of a Na(+)-independent neutral amino acid transporter that associates with the 4F2 heavy chain and exhibits substrate selectivity for small neutral D- and L-amino acids. J. Biol. Chem. 2000, 275, 9690–9698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakauchi, J.; Matsuo, H.; Kim, D.K.; Goto, A.; Chairoungdua, A.; Cha, S.H.; Inatomi, J.; Shiokawa, Y.; Yamaguchi, K.; Saito, I.; et al. Cloning and characterization of a human brain Na(+)-independent transporter for small neutral amino acids that transports D-serine with high affinity. Neurosci. Lett. 2000, 287, 231–235. [Google Scholar] [CrossRef]
- Fukushima, D.; Doi, H.; Fukushima, K.; Katsura, K.; Ogawa, N.; Sekiguchi, S.; Fujimori, K.; Sato, A.; Satomi, S.; Ishida, K.; et al. Glutamate exocrine dynamics augmented by plasma glutamine and the distribution of amino acid transporters of the rat pancreas. J. Physiol. Pharmacol. 2010, 61, 265–271. [Google Scholar]
- Zhou, Y.; Waanders, L.F.; Holmseth, S.; Guo, C.; Berger, U.V.; Li, Y.; Lehre, A.C.; Lehre, K.P.; Danbolt, N.C. Proteome analysis and conditional deletion of the EAAT2 glutamate transporter provide evidence against a role of EAAT2 in pancreatic insulin secretion in mice. J. Biol. Chem. 2014, 289, 1329–1344. [Google Scholar] [CrossRef] [Green Version]
- Stevens, E.R.; Gustafson, E.C.; Miller, R.F. Glycine transport accounts for the differential role of glycine vs. D-serine at NMDA receptor coagonist sites in the salamander retina. Eur. J. Neurosci. 2010, 31, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Jursky, F.; Nelson, N. Developmental expression of the glycine transporters GLYT1 and GLYT2 in mouse brain. J. Neurochem. 1996, 67, 336–344. [Google Scholar] [CrossRef]
- Nakade, Y.; Iwata, Y.; Furuichi, K.; Mita, M.; Hamase, K.; Konno, R.; Miyake, T.; Sakai, N.; Kitajima, S.; Toyama, T.; et al. Gut microbiota-derived D-serine protects against acute kidney injury. JCI Insight 2018, 3, e97957. [Google Scholar] [CrossRef] [Green Version]
- Martineau, M.; Parpura, V.; Mothet, J.P. Cell-type specific mechanisms of D-serine uptake and release in the brain. Front. Synaptic Neurosci. 2014, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Liew, C.W.; Assmann, A.; Templin, A.T.; Raum, J.C.; Lipson, K.L.; Rajan, S.; Qiang, G.; Hu, J.; Kawamori, D.; Lindberg, I.; et al. Insulin regulates carboxypeptidase E by modulating translation initiation scaffolding protein eIF4G1 in pancreatic beta cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2319–E2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Rajani, V.; Sengar, A.S.; Salter, M.W. Tripartite signalling by NMDA receptors. Mol. Brain 2020, 13, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durning, S.P.; Flanagan-Steet, H.; Prasad, N.; Wells, L. O-Linked beta-N-acetylglucosamine (O-GlcNAc) Acts as a Glucose Sensor to Epigenetically Regulate the Insulin Gene in Pancreatic Beta Cells. J. Biol. Chem. 2016, 291, 2107–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaughan, J.A.; McKnight, A.J.; Maxwell, A.P. Genetics of new-onset diabetes after transplantation. J. Am. Soc. Nephrol. 2014, 25, 1037–1049. [Google Scholar] [CrossRef] [Green Version]
- Chand, S.; McKnight, A.J.; Shabir, S.; Chan, W.; McCaughan, J.A.; Maxwell, A.P.; Harper, L.; Borrows, R. Analysis of single nucleotide polymorphisms implicate mTOR signalling in the development of new-onset diabetes after transplantation. BBA Clin. 2016, 5, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Madry, C.; Mesic, I.; Bartholomaus, I.; Nicke, A.; Betz, H.; Laube, B. Principal role of NR3 subunits in NR1/NR3 excitatory glycine receptor function. Biochem. Biophys. Res. Commun. 2007, 354, 102–108. [Google Scholar] [CrossRef]
- Chatterton, J.E.; Awobuluyi, M.; Premkumar, L.S.; Takahashi, H.; Talantova, M.; Shin, Y.; Cui, J.; Tu, S.; Sevarino, K.A.; Nakanishi, N.; et al. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 2002, 415, 793–798. [Google Scholar] [CrossRef]
- Takarada, T.; Takahata, Y.; Iemata, M.; Hinoi, E.; Uno, K.; Hirai, T.; Yamamoto, T.; Yoneda, Y. Interference with cellular differentiation by D-serine through antagonism at N-methyl-D-aspartate receptors composed of NR1 and NR3A subunits in chondrocytes. J. Cell. Physiol. 2009, 220, 756–764. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lockridge, A.; Gustafson, E.; Wong, A.; Miller, R.F.; Alejandro, E.U. Acute D-Serine Co-Agonism of β-Cell NMDA Receptors Potentiates Glucose-Stimulated Insulin Secretion and Excitatory β-Cell Membrane Activity. Cells 2021, 10, 93. https://doi.org/10.3390/cells10010093
Lockridge A, Gustafson E, Wong A, Miller RF, Alejandro EU. Acute D-Serine Co-Agonism of β-Cell NMDA Receptors Potentiates Glucose-Stimulated Insulin Secretion and Excitatory β-Cell Membrane Activity. Cells. 2021; 10(1):93. https://doi.org/10.3390/cells10010093
Chicago/Turabian StyleLockridge, Amber, Eric Gustafson, Alicia Wong, Robert F. Miller, and Emilyn U. Alejandro. 2021. "Acute D-Serine Co-Agonism of β-Cell NMDA Receptors Potentiates Glucose-Stimulated Insulin Secretion and Excitatory β-Cell Membrane Activity" Cells 10, no. 1: 93. https://doi.org/10.3390/cells10010093