
Getting Sugar Coating Right! The Role of the Golgi Trafficking Machinery in Glycosylation



Abstract
1. Introduction
2. Glycosylation Enzyme Compartmentalization
3. Membrane Trafficking Machinery
3.1. Tethers (MTCs and Coiled-Coil Tethers)
3.1.1. Coiled-Coil Tethers
3.1.2. Multisubunit Tethering Complexes
Conserved Oligomeric Golgi Complex (COG)
Golgi-Associated Retrograde Protein Complex (GARP)
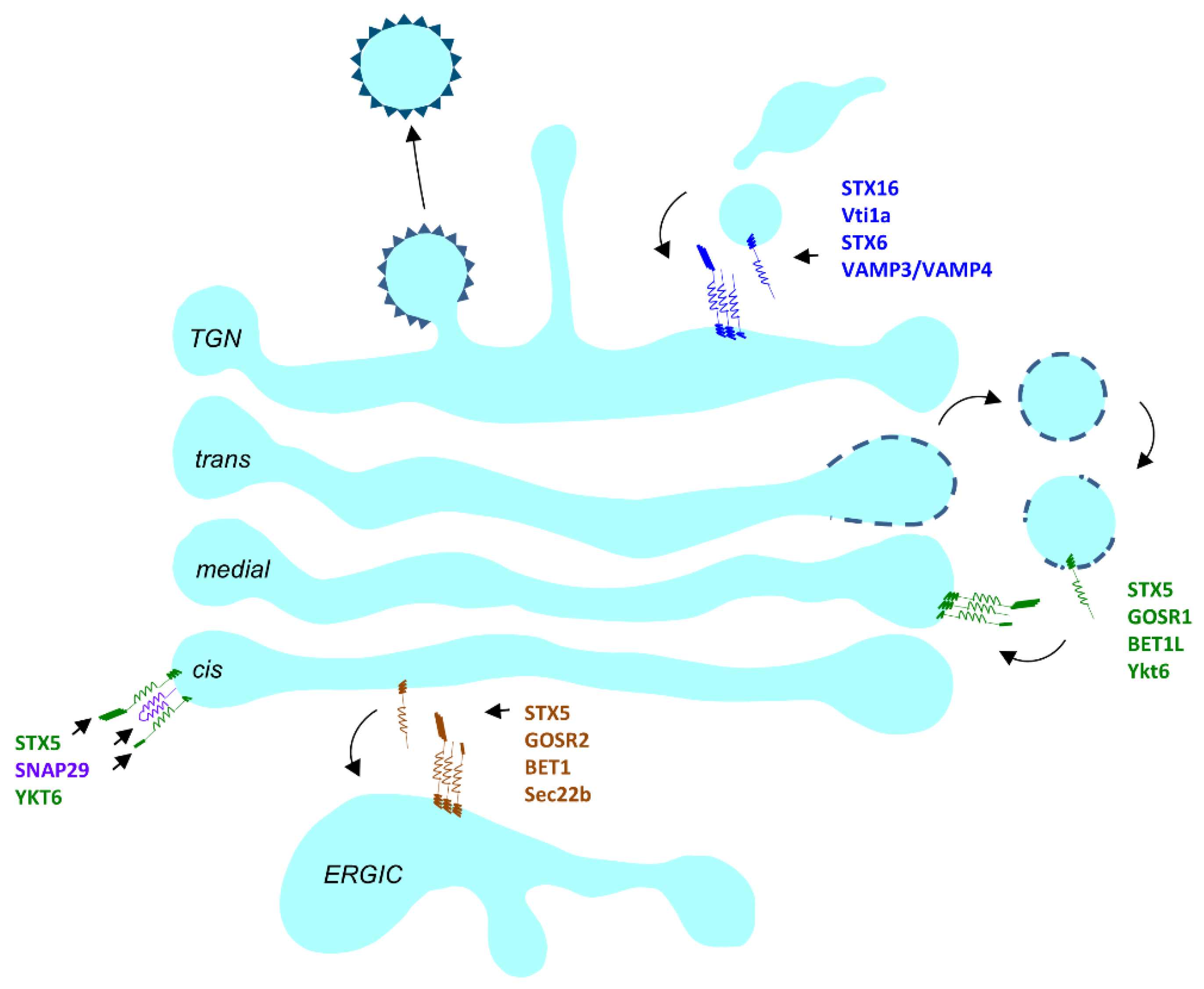
3.1.3. SNAREs
4. Conclusions

{kind=link}
{kind=link}
{kind=link}
| Gene | Alternative Names | Biological Role | Glycosylation Defects If Depleted | Known Human Mutations | References |
|---|---|---|---|---|---|
| GOLGA1 | golgin-97 | Endosome to TGN retrograde transport | No | - | [90,94] |
| GOLGA2 | GM130 | Maintenance of Golgi structure and ER to Golgi traffic | Yes (‘O’ glycosylation defects) | c.2251 C > T (p.Gln751Ter) c.629G > A (p.Arg210His) c.1266_1269del (p.Glu423Argfs*6) | [52,55,218] |
| GOLGA3 | golgin-160 | Maintain Golgi integrity and trafficking of plasma membrane protein | No | - | [219,220] |
| GOLGA4 | golgin-245 | Regulatory transport from TGN to plasma membrane | No | - | [100,221] |
| GOLGA5 | Golgin-84 | Tethering of intra-Golgi vesicles | Yes | - | [87,222] |
| GOLGA7 | GCP16 | - | No | - | [223] |
| GOLGB1 | Giantin, GCP364 | Maintaining Golgi structure and intra-Golgi retrograde trafficking | Yes (both ‘N’ and ‘O’ glycosylation defects) | (Golgb1ivs9+1G > A) | [81,85,224,225] |
| GCC1 | GCC88 | Efficient retrograde transport of cargo from the early endosomes to the TGN | [62,102] | ||
| GCC2 | GCC185 | Endosome-to-Golgi transport and maintenance of Golgi structure | [110,226] | ||
| TRIP11 | GMAP210 | Cisternal organization and anterograde, retrograde trafficking at ER-Golgi interface | Yes (‘N’ glycosylation defects) | c.5003T→A (p.L1668X) | [63] |
| TMF1 | ARA160 | Golgi organization | [118] | ||
| CUX1 | CASP | Forms a complex with Golgin 84 and tethers intra-Golgi recycling vesicles | [86,227] | ||
| USO1 | p115 | ER to Golgi trafficking | Yes | [228] | |
| GO45 | Golgin-45, BLZF1 | Golgi structure maintenance and secretion | - | [126] | |
| GORASP1 | GRASP65 | Maintaining the Golgi structure and protein trafficking, glycosylation | Yes | [229,230] | |
| GORASP2 | GRASP55 | Maintaining the Golgi structure and protein trafficking, glycosylation | Yes | [123] | |
| COG1 | KIAA1381, LDLB | Intra-Golgi retrograde trafficking | Yes (‘N’ Glycosylation defect) | 2659–2660insC (p.P888fsX900) c.1070 + 5G > A | [145,231,232] |
| COG2 | LDLC | Intra-Golgi retrograde trafficking | Yes (‘N’ and ‘O’-glycosylation defect) | c.701dup (p.Y234*), c.1900 T > G (p.W634G) | [231,233] |
| COG3 | hSec34 | Intra-Golgi retrograde trafficking | Yes | [234] | |
| COG4 | Intra-Golgi retrograde trafficking | Yes (‘N’ and ‘O’-glycosylation defect) | c.2185C > T (p.R729W) 16q22 deletion, c.697G > T (p.E233X) [de novo], c.2318 T > G (p.L773R), c.1546G > A or c.1546G > C (p.G516R) | [128,147,216,235] | |
| COG5 | GOLTC1, GTC90 | Intra-Golgi retrograde trafficking | Yes (‘N’-glycosylation defect) | c.1669-15 T > C, c.556_560delAGTAAinsCT (p.S186_K187delinsL), c.1856 T > C (p.I619T) c.95T > G (p.M32R), c.2518G > T (p.E840X), c.189delG (p.C64Vfs*6), c.2338_2340dupATT (p.I780dup), c.1780G > T (p.V594F), c.1209delG (p.M403IfsX3), c.2324C > T (p.P775L), c.330delT (p.V111Lfs*22), c.1290C > A (p.Y430X), c.2077A > C (p.T693P), c.2324C > T (p.P775L), c.1508dup (p.G505Wfs*3) | [236,237,238,239,240,241] |
| COG6 | KIAA1134 | Intra-Golgi retrograde trafficking | Yes (‘N’-glycosylation defect) | c. 1646G > T (p.G549V), c.1167-24A > G (p.G390FfsX6), c.511C > T (p.R171*), c.1746 + 2 T > G, c.1238_1239insA (p.F414Lfs*4), c.1646G > T (p.G549V), c.785A > G (p.Y262C), c.511C > T (p.R171*), c.1746 + 2 T > G | [148,242,243,244,245,246] |
| COG7 | UNQ3082/PRO10013 | Intra-Golgi retrograde trafficking | Yes (‘N’ and ‘O’ glycosylation defects) | IVS1 + 4 A → C aka c.169 + 4A > C, c.170-7A > G (p.56–57insAT) | [247,248,249,250] |
| COG8 | Intra-Golgi retrograde trafficking | Yes (‘N’ and ‘O’ glycosylation defects) | c.1611C > G (p.Y537X), IVS3 + 1G > A, TT 1687–1688, c.171dupG (p.L58Afs*29), c.1656dupC (p.A553Rfs*15), c.1583-1G > A | [251,252,253,254] | |
| VPS51 | ANG2, C11orf2, C11orf3, FFR | Retrograde transport from endosomes to TGN | Yes (‘N’ and ‘O’ glycosylation defects) | c.1468C > T, c.2232delC (c.1419_1421del; (p (Phe474del))) | [172,255] |
| VPS52 | SACM2L | Retrograde transport from endosomes to TGN | - | ||
| VPS53 | Hit1 | Retrograde transport from endosomes to TGN | Yes (‘N’ and ‘O’ glycosylation defects) | c.2084A > G c.1556+5G > A | [167] |
| VPS54 | CGP1, LUV1, RKI1, TCS3 | Retrograde transport from endosomes to TGN | Yes (‘N’ and ‘O’ glycosylation defects) | ||
| STX5 | Intra-Golgi retrograde transport | Yes (‘N’ and ‘O’ glycosylation defects) | c.163A > G (p.(Met55Val)) | [197] | |
| GOSR1 | GS28 | Intra-Golgi retrograde transport | - | - | |
| GOSR2 | GS27/membrin | ER-Golgi transport | - | c.430G > T (p.Gly144Trp) | [196,256,257] |
| YKT6 | Golgi organization and autophagy | ||||
| SNAP29 | Golgi trafficking and autophagy | - | 22q11.2 deletion | [208,210] | |
| STX16 | Endosome to TGN transport and maintaining Golgi structure | - | 3kb deletion in STX16 gene | [211] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Casares, D.; Escriba, P.V.; Rossello, C.A. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef]
- Park, K.; Ju, S.; Kim, N.; Park, S.Y. The Golgi complex: A hub of the secretory pathway. BMB Rep. 2021, 54, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, R.; Sticco, L.; Luini, A. Regulation of cargo export and sorting at the trans-Golgi network. FEBS Lett. 2019, 593, 2306–2318. [Google Scholar] [CrossRef]
- Glick, B.S.; Luini, A. Models for Golgi traffic: A critical assessment. Cold Spring Harb. Perspect. Biol. 2011, 3, a005215. [Google Scholar] [CrossRef] [PubMed]
- Luini, A. A brief history of the cisternal progression-maturation model. Cell. Logist. 2011, 1, 6–11. [Google Scholar] [CrossRef]
- Fisher, P.; Thomas-Oates, J.; Wood, A.J.; Ungar, D. The N-Glycosylation Processing Potential of the Mammalian Golgi Apparatus. Front. Cell Dev. Biol. 2019, 7, 157. [Google Scholar] [CrossRef]
- Wang, P.; Wang, H.; Gai, J.; Tian, X.; Zhang, X.; Lv, Y.; Jian, Y. Evolution of protein N-glycosylation process in Golgi apparatus which shapes diversity of protein N-glycan structures in plants, animals and fungi. Sci. Rep. 2017, 7, 40301. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P. Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 2011, 3, a005199. [Google Scholar] [CrossRef] [PubMed]
- Reynders, E.; Foulquier, F.; Annaert, W.; Matthijs, G. How Golgi glycosylation meets and needs trafficking: The case of the COG complex. Glycobiology 2011, 21, 853–863. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef]
- Ondruskova, N.; Cechova, A.; Hansikova, H.; Honzik, T.; Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129751. [Google Scholar] [CrossRef] [PubMed]
- Linders, P.T.A.; Peters, E.; Ter Beest, M.; Lefeber, D.J.; van den Bogaart, G. Sugary Logistics Gone Wrong: Membrane Trafficking and Congenital Disorders of Glycosylation. Int. J. Mol. Sci. 2020, 21, 4654. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J. Congenital disorders of glycosylation (CDG): It’s (nearly) all in it! J. Inherit. Metab. Dis. 2011, 34, 853–858. [Google Scholar] [CrossRef]
- Jaeken, J.; Hennet, T.; Matthijs, G.; Freeze, H.H. CDG nomenclature: Time for a change! Biochim. Biophys. Acta 2009, 1792, 825–826. [Google Scholar] [CrossRef]
- Lefeber, D.J.; Morava, E.; Jaeken, J. How to find and diagnose a CDG due to defective N-glycosylation. J. Inherit. Metab. Dis. 2011, 34, 849–852. [Google Scholar] [CrossRef]
- Berninsone, P.M.; Hirschberg, C.B. Nucleotide sugar transporters of the Golgi apparatus. Curr. Opin. Struct. Biol. 2000, 10, 542–547. [Google Scholar] [CrossRef]
- Maeda, Y.; Ide, T.; Koike, M.; Uchiyama, Y.; Kinoshita, T. GPHR is a novel anion channel critical for acidification and functions of the Golgi apparatus. Nat. Cell Biol. 2008, 10, 1135–1145. [Google Scholar] [CrossRef]
- Maeda, Y.; Kinoshita, T. The acidic environment of the Golgi is critical for glycosylation and transport. Methods Enzymol. 2010, 480, 495–510. [Google Scholar] [CrossRef]
- Banfield, D.K. Mechanisms of protein retention in the Golgi. Cold Spring Harb. Perspect. Biol. 2011, 3, a005264. [Google Scholar] [CrossRef]
- Tu, L.; Banfield, D.K. Localization of Golgi-resident glycosyltransferases. Cell. Mol. Life Sci. CMLS 2010, 67, 29–41. [Google Scholar] [CrossRef]
- Welch, L.G.; Munro, S. A tale of short tails, through thick and thin: Investigating the sorting mechanisms of Golgi enzymes. FEBS Lett. 2019, 593, 2452–2465. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, T.; Slusarewicz, P.; Hoe, M.H.; Warren, G. Kin recognition. A model for the retention of Golgi enzymes. FEBS Lett. 1993, 330, 1–4. [Google Scholar] [CrossRef]
- Nilsson, T.; Hoe, M.H.; Slusarewicz, P.; Rabouille, C.; Watson, R.; Hunte, F.; Watzele, G.; Berger, E.G.; Warren, G. Kin recognition between medial Golgi enzymes in HeLa cells. EMBO J. 1994, 13, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Munro, S. Sequences within and adjacent to the transmembrane segment of alpha-2,6-sialyltransferase specify Golgi retention. EMBO J. 1991, 10, 3577–3588. [Google Scholar] [CrossRef] [PubMed]
- Colley, K.J.; Lee, E.U.; Paulson, J.C. The signal anchor and stem regions of the beta-galactoside alpha 2,6-sialyltransferase may each act to localize the enzyme to the Golgi apparatus. J. Biol. Chem. 1992, 267, 7784–7793. [Google Scholar] [CrossRef]
- Khoder-Agha, F.; Harrus, D.; Brysbaert, G.; Lensink, M.F.; Harduin-Lepers, A.; Glumoff, T.; Kellokumpu, S. Assembly of B4GALT1/ST6GAL1 heteromers in the Golgi membranes involves lateral interactions via highly charged surface domains. J. Biol. Chem. 2019, 294, 14383–14393. [Google Scholar] [CrossRef] [PubMed]
- de Graffenried, C.L.; Bertozzi, C.R. The roles of enzyme localisation and complex formation in glycan assembly within the Golgi apparatus. Curr. Opin. Cell Biol. 2004, 16, 356–363. [Google Scholar] [CrossRef]
- Munro, S. An investigation of the role of transmembrane domains in Golgi protein retention. EMBO J. 1995, 14, 4695–4704. [Google Scholar] [CrossRef]
- Sharpe, H.J.; Stevens, T.J.; Munro, S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 2010, 142, 158–169. [Google Scholar] [CrossRef]
- Frappaolo, A.; Karimpour-Ghahnavieh, A.; Sechi, S.; Giansanti, M.G. The Close Relationship between the Golgi Trafficking Machinery and Protein Glycosylation. Cells 2020, 9, 2652. [Google Scholar] [CrossRef] [PubMed]
- Welch, L.G.; Peak-Chew, S.Y.; Begum, F.; Stevens, T.J.; Munro, S. GOLPH3 and GOLPH3L are broad-spectrum COPI adaptors for sorting into intra-Golgi transport vesicles. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Satpute-Krishnan, P.; Seo, A.Y.; Burnette, D.T.; Patterson, G.H.; Lippincott-Schwartz, J. ER trapping reveals Golgi enzymes continually revisit the ER through a recycling pathway that controls Golgi organization. Proc. Natl. Acad. Sci. USA 2015, 112, E6752–E6761. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.L.; Waters, M.G. Localization of a yeast early Golgi mannosyltransferase, Och1p, involves retrograde transport. J. Cell Biol. 1996, 132, 985–998. [Google Scholar] [CrossRef]
- Todorow, Z.; Spang, A.; Carmack, E.; Yates, J.; Schekman, R. Active recycling of yeast Golgi mannosyltransferase complexes through the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2000, 97, 13643–13648. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Pelham, H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987, 48, 899–907. [Google Scholar] [CrossRef]
- McMahon, H.T.; Mills, I.G. COP and clathrin-coated vesicle budding: Different pathways, common approaches. Curr. Opin. Cell Biol. 2004, 16, 379–391. [Google Scholar] [CrossRef]
- Michelsen, K.; Yuan, H.; Schwappach, B. Hide and run. Arginine-based endoplasmic-reticulum-sorting motifs in the assembly of heteromultimeric membrane proteins. EMBO Rep. 2005, 6, 717–722. [Google Scholar] [CrossRef]
- Liu, L.; Doray, B.; Kornfeld, S. Recycling of Golgi glycosyltransferases requires direct binding to coatomer. Proc. Natl. Acad. Sci. USA 2018, 115, 8984–8989. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Liu, J.; Li, S.; Setty, T.G.; Wood, C.S.; Burd, C.G.; Ferguson, K.M. Golgi localization of glycosyltransferases requires a Vps74p oligomer. Dev. Cell 2008, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Tai, W.C.; Chen, L.; Banfield, D.K. Signal-mediated dynamic retention of glycosyltransferases in the Golgi. Science 2008, 321, 404–407. [Google Scholar] [CrossRef]
- Eckert, E.S.; Reckmann, I.; Hellwig, A.; Rohling, S.; El-Battari, A.; Wieland, F.T.; Popoff, V. Golgi phosphoprotein 3 triggers signal-mediated incorporation of glycosyltransferases into coatomer-coated (COPI) vesicles. J. Biol. Chem. 2014, 289, 31319–31329. [Google Scholar] [CrossRef]
- Rizzo, R.; Russo, D.; Kurokawa, K.; Sahu, P.; Lombardi, B.; Supino, D.; Zhukovsky, M.A.; Vocat, A.; Pothukuchi, P.; Kunnathully, V.; et al. Golgi maturation-dependent glycoenzyme recycling controls glycosphingolipid biosynthesis and cell growth via GOLPH3. EMBO J. 2021, 40, e107238. [Google Scholar] [CrossRef] [PubMed]
- Kirchhausen, T. Three ways to make a vesicle. Nat. Rev. Mol. Cell Biol. 2000, 1, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Glick, B.S. The mechanisms of vesicle budding and fusion. Cell 2004, 116, 153–166. [Google Scholar] [CrossRef]
- Cevher-Keskin, B. ARF1 and SAR1 GTPases in endomembrane trafficking in plants. Int. J. Mol. Sci. 2013, 14, 18181–18199. [Google Scholar] [CrossRef]
- Zhukovsky, M.A.; Filograna, A.; Luini, A.; Corda, D.; Valente, C. Protein Amphipathic Helix Insertion: A Mechanism to Induce Membrane Fission. Front. Cell Dev. Biol. 2019, 7, 291. [Google Scholar] [CrossRef] [PubMed]
- Witkos, T.M.; Lowe, M. The Golgin Family of Coiled-Coil Tethering Proteins. Front. Cell Dev. Biol. 2015, 3, 86. [Google Scholar] [CrossRef]
- Zhou, W.; Chang, J.; Wang, X.; Savelieff, M.G.; Zhao, Y.; Ke, S.; Ye, B. GM130 is required for compartmental organization of dendritic golgi outposts. Curr. Biol. CB 2014, 24, 1227–1233. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Bennett, A.H.; Alfadhel, M.; Gupta, V.; Alkuraya, F.S. GOLGA2, encoding a master regulator of golgi apparatus, is mutated in a patient with a neuromuscular disorder. Hum. Genet. 2016, 135, 245–251. [Google Scholar] [CrossRef]
- Liu, C.; Mei, M.; Li, Q.; Roboti, P.; Pang, Q.; Ying, Z.; Gao, F.; Lowe, M.; Bao, S. Loss of the golgin GM130 causes Golgi disruption, Purkinje neuron loss, and ataxia in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, S.; Kim, M.J.; Hong, Y.; Lee, A.Y.; Lee, H.; Tran, Q.; Kim, M.; Cho, H.; Park, J.; et al. GOLGA2 loss causes fibrosis with autophagy in the mouse lung and liver. Biochem. Biophys. Res. Commun. 2018, 495, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, U.; Mistri, M.; Shah, N.; Shah, P.S.; Gupta, V.A. Bi-allelic loss of function variants in GOLGA2 are associated with a complex neurological phenotype: Report of a second family. Clin. Genet. 2021, 100, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ye, M.; Zhao, Q.; Xia, M.; Liu, D.; He, L.; Chen, G.; Peng, Y.; Liu, H. Loss of the Golgi Matrix Protein 130 Cause Aberrant IgA1 Glycosylation in IgA Nephropathy. Am. J. Nephrol. 2019, 49, 307–316. [Google Scholar] [CrossRef]
- Puthenveedu, M.A.; Bachert, C.; Puri, S.; Lanni, F.; Linstedt, A.D. GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat. Cell Biol. 2006, 8, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, A.; Ali, M.F.; Cheng, P.W. Glycosyltransferase-specific Golgi-targeting mechanisms. J. Biol. Chem. 2012, 287, 37621–37627. [Google Scholar] [CrossRef]
- Rios, R.M.; Sanchis, A.; Tassin, A.M.; Fedriani, C.; Bornens, M. GMAP-210 recruits gamma-tubulin complexes to cis-Golgi membranes and is required for Golgi ribbon formation. Cell 2004, 118, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Puri, S.; Linstedt, A.D. A primary role for Golgi positioning in directed secretion, cell polarity, and wound healing. Mol. Biol. Cell 2009, 20, 1728–1736. [Google Scholar] [CrossRef]
- Sato, K.; Roboti, P.; Mironov, A.A.; Lowe, M. Coupling of vesicle tethering and Rab binding is required for in vivo functionality of the golgin GMAP-210. Mol. Biol. Cell 2015, 26, 537–553. [Google Scholar] [CrossRef]
- Roboti, P.; Sato, K.; Lowe, M. The golgin GMAP-210 is required for efficient membrane trafficking in the early secretory pathway. J. Cell Sci. 2015, 128, 1595–1606. [Google Scholar] [CrossRef]
- Wong, M.; Gillingham, A.K.; Munro, S. The golgin coiled-coil proteins capture different types of transport carriers via distinct N-terminal motifs. BMC Biol. 2017, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Smits, P.; Bolton, A.D.; Funari, V.; Hong, M.; Boyden, E.D.; Lu, L.; Manning, D.K.; Dwyer, N.D.; Moran, J.L.; Prysak, M.; et al. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. N. Engl. J. Med. 2010, 362, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Wehrle, A.; Witkos, T.M.; Unger, S.; Schneider, J.; Follit, J.A.; Hermann, J.; Welting, T.; Fano, V.; Hietala, M.; Vatanavicharn, N.; et al. Hypomorphic mutations of TRIP11 cause odontochondrodysplasia. JCI Insight 2019, 4, e124701. [Google Scholar] [CrossRef]
- Medina, C.T.N.; Sandoval, R.; Oliveira, G.; da Costa Silveira, K.; Cavalcanti, D.P.; Pogue, R. Pathogenic variants in the TRIP11 gene cause a skeletal dysplasia spectrum from odontochondrodysplasia to achondrogenesis 1A. Am. J. Med. Genet. Part A 2020, 182, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Del Pino, M.; Sanchez-Soler, M.J.; Parron-Pajares, M.; Aza-Carmona, M.; Heath, K.E.; Fano, V. Description of four patients with TRIP11 variants expand the clinical spectrum of odontochondroplasia (ODCD) and demonstrate the existence of common variants. Eur. J. Med. Genet. 2021, 64, 104198. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Hu, G.; Chen, M.; Liu, B.; Yan, K.; Zhou, C.; Yu, Y.; Dong, M. Novel deep intronic and frameshift mutations causing a TRIP11-related disorder. Am. J. Med. Genet. Part A 2021, 185, 2482–2487. [Google Scholar] [CrossRef]
- Yadav, S.; Puthenveedu, M.A.; Linstedt, A.D. Golgin160 recruits the dynein motor to position the Golgi apparatus. Dev. Cell 2012, 23, 153–165. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- Nelson, D.S.; Alvarez, C.; Gao, Y.S.; Garcia-Mata, R.; Fialkowski, E.; Sztul, E. The membrane transport factor TAP/p115 cycles between the Golgi and earlier secretory compartments and contains distinct domains required for its localization and function. J. Cell Biol. 1998, 143, 319–331. [Google Scholar] [CrossRef]
- Alvarez, C.; Garcia-Mata, R.; Hauri, H.P.; Sztul, E. The p115-interactive proteins GM130 and giantin participate in endoplasmic reticulum-Golgi traffic. J. Biol. Chem. 2001, 276, 2693–2700. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Grabski, R.; Sztul, E.; Hay, J.C. p115-SNARE interactions: A dynamic cycle of p115 binding monomeric SNARE motifs and releasing assembled bundles. Traffic 2015, 16, 148–171. [Google Scholar] [CrossRef]
- Sapperstein, S.K.; Walter, D.M.; Grosvenor, A.R.; Heuser, J.E.; Waters, M.G. p115 is a general vesicular transport factor related to the yeast endoplasmic reticulum to Golgi transport factor Uso1p. Proc. Natl. Acad. Sci. USA 1995, 92, 522–526. [Google Scholar] [CrossRef]
- Alvarez, C.; Fujita, H.; Hubbard, A.; Sztul, E. ER to Golgi transport: Requirement for p115 at a pre-Golgi VTC stage. J. Cell Biol. 1999, 147, 1205–1222. [Google Scholar] [CrossRef]
- Allan, B.B.; Moyer, B.D.; Balch, W.E. Rab1 recruitment of p115 into a cis-SNARE complex: Programming budding COPII vesicles for fusion. Science 2000, 289, 444–448. [Google Scholar] [CrossRef]
- Shorter, J.; Beard, M.B.; Seemann, J.; Dirac-Svejstrup, A.B.; Warren, G. Sequential tethering of Golgins and catalysis of SNAREpin assembly by the vesicle-tethering protein p115. J. Cell Biol. 2002, 157, 45–62. [Google Scholar] [CrossRef]
- Shorter, J.; Warren, G. A role for the vesicle tethering protein, p115, in the post-mitotic stacking of reassembling Golgi cisternae in a cell-free system. J. Cell Biol. 1999, 146, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Sohda, M.; Misumi, Y.; Yoshimura, S.; Nakamura, N.; Fusano, T.; Ogata, S.; Sakisaka, S.; Ikehara, Y. The interaction of two tethering factors, p115 and COG complex, is required for Golgi integrity. Traffic 2007, 8, 270–284. [Google Scholar] [CrossRef]
- Sohda, M.; Misumi, Y.; Yoshimura, S.; Nakamura, N.; Fusano, T.; Sakisaka, S.; Ogata, S.; Fujimoto, J.; Kiyokawa, N.; Ikehara, Y. Depletion of vesicle-tethering factor p115 causes mini-stacked Golgi fragments with delayed protein transport. Biochem. Biophys. Res. Commun. 2005, 338, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A.; Short, B. Golgins in the structure and dynamics of the Golgi apparatus. Curr. Opin. Cell Biol. 2003, 15, 405–413. [Google Scholar] [CrossRef]
- Koreishi, M.; Gniadek, T.J.; Yu, S.; Masuda, J.; Honjo, Y.; Satoh, A. The golgin tether giantin regulates the secretory pathway by controlling stack organization within Golgi apparatus. PLoS ONE 2013, 8, e59821. [Google Scholar] [CrossRef]
- Stevenson, N.L.; Bergen, D.J.M.; Skinner, R.E.H.; Kague, E.; Martin-Silverstone, E.; Robson Brown, K.A.; Hammond, C.L.; Stephens, D.J. Giantin-knockout models reveal a feedback loop between Golgi function and glycosyltransferase expression. J. Cell Sci. 2017, 130, 4132–4143. [Google Scholar] [CrossRef]
- Lowe, M. The Physiological Functions of the Golgin Vesicle Tethering Proteins. Front. Cell Dev. Biol. 2019, 7, 94. [Google Scholar] [CrossRef]
- Stevenson, N.L.; Bergen, D.J.M.; Lu, Y.; Prada-Sanchez, M.E.; Kadler, K.E.; Hammond, C.L.; Stephens, D.J. Giantin is required for intracellular N-terminal processing of type I procollagen. J. Cell Biol. 2021, 220, e202005166. [Google Scholar] [CrossRef]
- Lan, Y.; Zhang, N.; Liu, H.; Xu, J.; Jiang, R. Golgb1 regulates protein glycosylation and is crucial for mammalian palate development. Development 2016, 143, 2344–2355. [Google Scholar] [CrossRef]
- Malsam, J.; Satoh, A.; Pelletier, L.; Warren, G. Golgin tethers define subpopulations of COPI vesicles. Science 2005, 307, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Sohda, M.; Misumi, Y.; Yamamoto, A.; Nakamura, N.; Ogata, S.; Sakisaka, S.; Hirose, S.; Ikehara, Y.; Oda, K. Interaction of Golgin-84 with the COG complex mediates the intra-Golgi retrograde transport. Traffic 2010, 11, 1552–1566. [Google Scholar] [CrossRef]
- Munro, S. The golgin coiled-coil proteins of the Golgi apparatus. Cold Spring Harb. Perspect. Biol. 2011, 3, a005256. [Google Scholar] [CrossRef]
- McGee, L.J.; Jiang, A.L.; Lan, Y. Golga5 is dispensable for mouse embryonic development and postnatal survival. Genesis 2017, 55, e23039. [Google Scholar] [CrossRef]
- Griffith, K.J.; Chan, E.K.; Lung, C.C.; Hamel, J.C.; Guo, X.; Miyachi, K.; Fritzler, M.J. Molecular cloning of a novel 97-kd Golgi complex autoantigen associated with Sjogren’s syndrome. Arthritis Rheum. 1997, 40, 1693–1702. [Google Scholar] [CrossRef]
- Shin, J.J.H.; Gillingham, A.K.; Begum, F.; Chadwick, J.; Munro, S. Author Correction: TBC1D23 is a bridging factor for endosomal vesicle capture by golgins at the trans-Golgi. Nat. Cell Biol. 2018, 20, 222. [Google Scholar] [CrossRef]
- Takatsuki, A.; Nakamura, M.; Kono, Y. Possible implication of Golgi-nucleating function for the centrosome. Biochem. Biophys. Res. Commun. 2002, 291, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; Junutula, J.R.; Wu, C.; Burden, J.; Matern, H.; Peden, A.A.; Prekeris, R. FIP1/RCP binding to Golgin-97 regulates retrograde transport from recycling endosomes to the trans-Golgi network. Mol. Biol. Cell 2010, 21, 3041–3053. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Tai, G.; Hong, W. Autoantigen Golgin-97, an effector of Arl1 GTPase, participates in traffic from the endosome to the trans-golgi network. Mol. Biol. Cell 2004, 15, 4426–4443. [Google Scholar] [CrossRef]
- Alzhanova, D.; Hruby, D.E. A host cell membrane protein, golgin-97, is essential for poxvirus morphogenesis. Virology 2007, 362, 421–427. [Google Scholar] [CrossRef]
- Yoshino, A.; Setty, S.R.; Poynton, C.; Whiteman, E.L.; Saint-Pol, A.; Burd, C.G.; Johannes, L.; Holzbaur, E.L.; Koval, M.; McCaffery, J.M.; et al. tGolgin-1 (p230, golgin-245) modulates Shiga-toxin transport to the Golgi and Golgi motility towards the microtubule-organizing centre. J. Cell Sci. 2005, 118, 2279–2293. [Google Scholar] [CrossRef]
- Kakinuma, T.; Ichikawa, H.; Tsukada, Y.; Nakamura, T.; Toh, B.H. Interaction between p230 and MACF1 is associated with transport of a glycosyl phosphatidyl inositol-anchored protein from the Golgi to the cell periphery. Exp. Cell Res. 2004, 298, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.G.; Hammond, L.A.; Houghton, F.; Gleeson, P.A.; Stow, J.L. E-cadherin transport from the trans-Golgi network in tubulovesicular carriers is selectively regulated by golgin-97. Traffic 2005, 6, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Lieu, Z.Z.; Lock, J.G.; Hammond, L.A.; La Gruta, N.L.; Stow, J.L.; Gleeson, P.A. A trans-Golgi network golgin is required for the regulated secretion of TNF in activated macrophages in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 3351–3356. [Google Scholar] [CrossRef]
- Sohda, M.; Misumi, Y.; Ogata, S.; Sakisaka, S.; Hirose, S.; Ikehara, Y.; Oda, K. Trans-Golgi protein p230/golgin-245 is involved in phagophore formation. Biochem. Biophys. Res. Commun. 2015, 456, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Lieu, Z.Z.; Derby, M.C.; Teasdale, R.D.; Hart, C.; Gunn, P.; Gleeson, P.A. The golgin GCC88 is required for efficient retrograde transport of cargo from the early endosomes to the trans-Golgi network. Mol. Biol. Cell 2007, 18, 4979–4991. [Google Scholar] [CrossRef]
- Luke, M.R.; Kjer-Nielsen, L.; Brown, D.L.; Stow, J.L.; Gleeson, P.A. GRIP domain-mediated targeting of two new coiled-coil proteins, GCC88 and GCC185, to subcompartments of the trans-Golgi network. J. Biol. Chem. 2003, 278, 4216–4226. [Google Scholar] [CrossRef]
- Cui, Y.; Yang, Z.; Teasdale, R.D. A role of GCC88 in the retrograde transport of CI-M6PR and the maintenance of lysosomal activity. Cell Biol. Int. 2019, 43, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Efimov, A.; Kharitonov, A.; Efimova, N.; Loncarek, J.; Miller, P.M.; Andreyeva, N.; Gleeson, P.; Galjart, N.; Maia, A.R.; McLeod, I.X.; et al. Asymmetric CLASP-dependent nucleation of noncentrosomal microtubules at the trans-Golgi network. Dev. Cell 2007, 12, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Espinosa, E.; Pfeffer, S.R. A syntaxin 10-SNARE complex distinguishes two distinct transport routes from endosomes to the trans-Golgi in human cells. J. Cell Biol. 2008, 180, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.V.; Burguete, A.S.; Sridevi, K.; Ganley, I.G.; Nottingham, R.M.; Pfeffer, S.R. A functional role for the GCC185 golgin in mannose 6-phosphate receptor recycling. Mol. Biol. Cell 2006, 17, 4353–4363. [Google Scholar] [CrossRef]
- Burguete, A.S.; Fenn, T.D.; Brunger, A.T.; Pfeffer, S.R. Rab and Arl GTPase family members cooperate in the localization of the golgin GCC185. Cell 2008, 132, 286–298. [Google Scholar] [CrossRef]
- Hayes, G.L.; Brown, F.C.; Haas, A.K.; Nottingham, R.M.; Barr, F.A.; Pfeffer, S.R. Multiple Rab GTPase binding sites in GCC185 suggest a model for vesicle tethering at the trans-Golgi. Mol. Biol. Cell 2009, 20, 209–217. [Google Scholar] [CrossRef]
- Houghton, F.J.; Chew, P.L.; Lodeho, S.; Goud, B.; Gleeson, P.A. The localization of the Golgin GCC185 is independent of Rab6A/A’ and Arl1. Cell 2009, 138, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Derby, M.C.; Lieu, Z.Z.; Brown, D.; Stow, J.L.; Goud, B.; Gleeson, P.A. The trans-Golgi network golgin, GCC185, is required for endosome-to-Golgi transport and maintenance of Golgi structure. Traffic 2007, 8, 758–773. [Google Scholar] [CrossRef] [PubMed]
- Siniossoglou, S.; Pelham, H.R. An effector of Ypt6p binds the SNARE Tlg1p and mediates selective fusion of vesicles with late Golgi membranes. EMBO J. 2001, 20, 5991–5998. [Google Scholar] [CrossRef] [PubMed]
- Fridmann-Sirkis, Y.; Siniossoglou, S.; Pelham, H.R. TMF is a golgin that binds Rab6 and influences Golgi morphology. BMC Cell Biol. 2004, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Yamane, J.; Kubo, A.; Nakayama, K.; Yuba-Kubo, A.; Katsuno, T.; Tsukita, S.; Tsukita, S. Functional involvement of TMF/ARA160 in Rab6-dependent retrograde membrane traffic. Exp. Cell Res. 2007, 313, 3472–3485. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.; Tsruya, R.; Levitsky, P.; Pomp, O.; Taller, M.; Weisberg, S.; Parris, W.; Kulkarni, S.; Malovani, H.; Pawson, T.; et al. TMF/ARA160 is a BC-box-containing protein that mediates the degradation of Stat3. Oncogene 2004, 23, 8908–8919. [Google Scholar] [CrossRef] [PubMed]
- Abrham, G.; Volpe, M.; Shpungin, S.; Nir, U. TMF/ARA160 downregulates proangiogenic genes and attenuates the progression of PC3 xenografts. Int. J. Cancer 2009, 125, 43–53. [Google Scholar] [CrossRef]
- Lerer-Goldshtein, T.; Bel, S.; Shpungin, S.; Pery, E.; Motro, B.; Goldstein, R.S.; Bar-Sheshet, S.I.; Breitbart, H.; Nir, U. TMF/ARA160: A key regulator of sperm development. Dev. Biol. 2010, 348, 12–21. [Google Scholar] [CrossRef]
- Elkis, Y.; Bel, S.; Lerer-Goldstein, T.; Nyska, A.; Creasy, D.M.; Shpungin, S.; Nir, U. Testosterone deficiency accompanied by testicular and epididymal abnormalities in TMF(-/-) mice. Mol. Cell. Endocrinol. 2013, 365, 52–63. [Google Scholar] [CrossRef]
- Elkis, Y.; Bel, S.; Rahimi, R.; Lerer-Goldstein, T.; Levin-Zaidman, S.; Babushkin, T.; Shpungin, S.; Nir, U. TMF/ARA160 Governs the Dynamic Spatial Orientation of the Golgi Apparatus during Sperm Development. PLoS ONE 2015, 10, e0145277. [Google Scholar] [CrossRef]
- Rahimi, R.; Malek, I.; Lerrer-Goldshtein, T.; Elkis, Y.; Shoval, I.; Jacob, A.; Shpungin, S.; Nir, U. TMF1 is upregulated by insulin and is required for a sustained glucose homeostasis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21295. [Google Scholar] [CrossRef]
- Kummel, D.; Reinisch, K.M. Structure of Golgi transport proteins. Cold Spring Harb. Perspect. Biol. 2011, 3, a007609. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bekier, M.E., 2nd; Wang, L.; Li, J.; Huang, H.; Tang, D.; Zhang, X.; Wang, Y. Knockout of the Golgi stacking proteins GRASP55 and GRASP65 impairs Golgi structure and function. Mol. Biol. Cell 2017, 28, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A.; Nakamura, N.; Warren, G. Mapping the interaction between GRASP65 and GM130, components of a protein complex involved in the stacking of Golgi cisternae. EMBO J. 1998, 17, 3258–3268. [Google Scholar] [CrossRef]
- Shorter, J.; Watson, R.; Giannakou, M.E.; Clarke, M.; Warren, G.; Barr, F.A. GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J. 1999, 18, 4949–4960. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Linstedt, A.D. GRASP: A Multitasking Tether. Front. Cell Dev. Biol. 2016, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Seemann, J. Rapid degradation of GRASP55 and GRASP65 reveals their immediate impact on the Golgi structure. J. Cell Biol. 2021, 220, e202007052. [Google Scholar] [CrossRef] [PubMed]
- Short, B.; Preisinger, C.; Korner, R.; Kopajtich, R.; Byron, O.; Barr, F.A. A GRASP55-rab2 effector complex linking Golgi structure to membrane traffic. J. Cell Biol. 2001, 155, 877–883. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhang, X.; Nix, D.B.; Katoh, T.; Aoki, K.; Tiemeyer, M.; Wang, Y. Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat. Commun. 2013, 4, 1659. [Google Scholar] [CrossRef]
- Ungar, D.; Oka, T.; Brittle, E.E.; Vasile, E.; Lupashin, V.V.; Chatterton, J.E.; Heuser, J.E.; Krieger, M.; Waters, M.G. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 2002, 157, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.A.; Yip, C.K.; Walz, T.; Hughson, F.M. Molecular organization of the COG vesicle tethering complex. Nat. Struct. Mol. Biol. 2010, 17, 1292–1297. [Google Scholar] [CrossRef]
- Blackburn, J.B.; D’Souza, Z.; Lupashin, V.V. Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS Lett. 2019, 593, 2466–2487. [Google Scholar] [CrossRef]
- D’Souza, Z.; Taher, F.S.; Lupashin, V.V. Golgi inCOGnito: From vesicle tethering to human disease. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129694. [Google Scholar] [CrossRef] [PubMed]
- Whyte, J.R.; Munro, S. The Sec34/35 Golgi transport complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev. Cell 2001, 1, 527–537. [Google Scholar] [CrossRef]
- Fotso, P.; Koryakina, Y.; Pavliv, O.; Tsiomenko, A.B.; Lupashin, V.V. Cog1p plays a central role in the organization of the yeast conserved oligomeric Golgi complex. J. Biol. Chem. 2005, 280, 27613–27623. [Google Scholar] [CrossRef] [PubMed]
- Willett, R.; Ungar, D.; Lupashin, V. The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem. Cell Biol. 2013, 140, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Ungar, D.; Oka, T.; Vasile, E.; Krieger, M.; Hughson, F.M. Subunit architecture of the conserved oligomeric Golgi complex. J. Biol. Chem. 2005, 280, 32729–32735. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.Y.; Chou, H.T.; Ungar, D.; Yip, C.K.; Walz, T.; Hughson, F.M. Molecular architecture of the complete COG tethering complex. Nat. Struct. Mol. Biol. 2016, 23, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Willett, R.; Blackburn, J.B.; Climer, L.; Pokrovskaya, I.; Kudlyk, T.; Wang, W.; Lupashin, V. COG lobe B sub-complex engages v-SNARE GS15 and functions via regulated interaction with lobe A sub-complex. Sci. Rep. 2016, 6, 29139. [Google Scholar] [CrossRef]
- Shestakova, A.; Zolov, S.; Lupashin, V. COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic 2006, 7, 191–204. [Google Scholar] [CrossRef]
- Steet, R.; Kornfeld, S. COG-7-deficient Human Fibroblasts Exhibit Altered Recycling of Golgi Proteins. Mol. Biol. Cell 2006, 17, 2312–2321. [Google Scholar] [CrossRef]
- Pokrovskaya, I.D.; Willett, R.; Smith, R.D.; Morelle, W.; Kudlyk, T.; Lupashin, V.V. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology 2011, 21, 1554–1569. [Google Scholar] [CrossRef]
- Willett, R.; Pokrovskaya, I.; Kudlyk, T.; Lupashin, V. Multipronged interaction of the COG complex with intracellular membranes. Cell. Logist. 2014, 4, e27888. [Google Scholar] [CrossRef][Green Version]
- Bailey Blackburn, J.; Pokrovskaya, I.; Fisher, P.; Ungar, D.; Lupashin, V.V. COG Complex Complexities: Detailed Characterization of a Complete Set of HEK293T Cells Lacking Individual COG Subunits. Front. Cell Dev. Biol. 2016, 4, 23. [Google Scholar] [CrossRef]
- Blackburn, J.B.; Kudlyk, T.; Pokrovskaya, I.; Lupashin, V.V. More than just sugars: Conserved oligomeric Golgi complex deficiency causes glycosylation-independent cellular defects. Traffic 2018, 19, 463–480. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, Z.; Blackburn, J.B.; Kudlyk, T.; Pokrovskaya, I.D.; Lupashin, V.V. Defects in COG-Mediated Golgi Trafficking Alter Endo-Lysosomal System in Human Cells. Front. Cell Dev. Biol. 2019, 7, 118. [Google Scholar] [CrossRef] [PubMed]
- Zeevaert, R.; Foulquier, F.; Jaeken, J.; Matthijs, G. Deficiencies in subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorders of Glycosylation. Mol. Genet. Metab. 2008, 93, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Haijes, H.A.; Jaeken, J.; Foulquier, F.; van Hasselt, P.M. Hypothesis: Lobe A (COG1-4)-CDG causes a more severe phenotype than lobe B (COG5-8)-CDG. J. Med. Genet. 2018, 55, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Reynders, E.; Foulquier, F.; Leao Teles, E.; Quelhas, D.; Morelle, W.; Rabouille, C.; Annaert, W.; Matthijs, G. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum. Mol. Genet. 2009, 18, 3244–3256. [Google Scholar] [CrossRef]
- Rymen, D.; Winter, J.; Van Hasselt, P.M.; Jaeken, J.; Kasapkara, C.; Gokcay, G.; Haijes, H.; Goyens, P.; Tokatli, A.; Thiel, C.; et al. Key features and clinical variability of COG6-CDG. Mol. Genet. Metab. 2015, 116, 163–170. [Google Scholar] [CrossRef]
- Peanne, R.; de Lonlay, P.; Foulquier, F.; Kornak, U.; Lefeber, D.J.; Morava, E.; Perez, B.; Seta, N.; Thiel, C.; Van Schaftingen, E.; et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur. J. Med. Genet. 2018, 61, 643–663. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Hierro, A. Transport according to GARP: Receiving retrograde cargo at the trans-Golgi network. Trends Cell Biol. 2011, 21, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.T.; Dukovski, D.; Chambers, M.G.; Reinisch, K.M.; Walz, T. CATCHR, HOPS and CORVET tethering complexes share a similar architecture. Nat. Struct. Mol. Biol. 2016, 23, 761–763. [Google Scholar] [CrossRef]
- Santana-Molina, C.; Gutierrez, F.; Devos, D.P. Homology and Modular Evolution of CATCHR at the Origin of the Eukaryotic Endomembrane System. Genome Biol. Evol. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Conibear, E.; Stevens, T.H. Vps52p, Vps53p, and Vps54p form a novel multisubunit complex required for protein sorting at the yeast late Golgi. Mol. Biol. Cell 2000, 11, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Siniossoglou, S.; Pelham, H.R. Vps51p links the VFT complex to the SNARE Tlg1p. J. Biol. Chem. 2002, 277, 48318–48324. [Google Scholar] [CrossRef] [PubMed]
- Conibear, E.; Cleck, J.N.; Stevens, T.H. Vps51p mediates the association of the GARP (Vps52/53/54) complex with the late Golgi t-SNARE Tlg1p. Mol. Biol. Cell 2003, 14, 1610–1623. [Google Scholar] [CrossRef]
- Brocker, C.; Engelbrecht-Vandre, S.; Ungermann, C. Multisubunit tethering complexes and their role in membrane fusion. Curr. Biol. CB 2010, 20, R943–R952. [Google Scholar] [CrossRef]
- Perez-Victoria, F.J.; Schindler, C.; Magadan, J.G.; Mardones, G.A.; Delevoye, C.; Romao, M.; Raposo, G.; Bonifacino, J.S. Ang2/fat-free is a conserved subunit of the Golgi-associated retrograde protein complex. Mol. Biol. Cell 2010, 21, 3386–3395. [Google Scholar] [CrossRef]
- Liewen, H.; Meinhold-Heerlein, I.; Oliveira, V.; Schwarzenbacher, R.; Luo, G.; Wadle, A.; Jung, M.; Pfreundschuh, M.; Stenner-Liewen, F. Characterization of the human GARP (Golgi associated retrograde protein) complex. Exp. Cell Res. 2005, 306, 24–34. [Google Scholar] [CrossRef]
- Walter, L.; Stark, S.; Helou, K.; Flugge, P.; Levan, G.; Gunther, E. Identification, characterization and cytogenetic mapping of a yeast Vps54 homolog in rat and mouse. Gene 2002, 285, 213–220. [Google Scholar] [CrossRef]
- Lobstein, E.; Guyon, A.; Ferault, M.; Twell, D.; Pelletier, G.; Bonhomme, S. The putative Arabidopsis homolog of yeast vps52p is required for pollen tube elongation, localizes to Golgi, and might be involved in vesicle trafficking. Plant Physiol. 2004, 135, 1480–1490. [Google Scholar] [CrossRef]
- Oka, T.; Krieger, M. Multi-component protein complexes and Golgi membrane trafficking. J. Biochem. 2005, 137, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Bonifacino, J.S. ARFRP1 functions upstream of ARL1 and ARL5 to coordinate recruitment of distinct tethering factors to the trans-Golgi network. J. Cell Biol. 2019, 218, 3681–3696. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Chen, Y.; Pu, J.; Guo, X.; Bonifacino, J.S. EARP is a multisubunit tethering complex involved in endocytic recycling. Nat. Cell Biol. 2015, 17, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Quenneville, N.R.; Chao, T.Y.; McCaffery, J.M.; Conibear, E. Domains within the GARP subunit Vps54 confer separate functions in complex assembly and early endosome recognition. Mol. Biol. Cell 2006, 17, 1859–1870. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Rojas, R. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 2006, 7, 568–579. [Google Scholar] [CrossRef]
- Perez-Victoria, F.J.; Mardones, G.A.; Bonifacino, J.S. Requirement of the human GARP complex for mannose 6-phosphate-receptor-dependent sorting of cathepsin D to lysosomes. Mol. Biol. Cell 2008, 19, 2350–2362. [Google Scholar] [CrossRef]
- Feinstein, M.; Flusser, H.; Lerman-Sagie, T.; Ben-Zeev, B.; Lev, D.; Agamy, O.; Cohen, I.; Kadir, R.; Sivan, S.; Leshinsky-Silver, E.; et al. VPS53 mutations cause progressive cerebello-cerebral atrophy type 2 (PCCA2). J. Med. Genet. 2014, 51, 303–308. [Google Scholar] [CrossRef]
- Frohlich, F.; Petit, C.; Kory, N.; Christiano, R.; Hannibal-Bach, H.K.; Graham, M.; Liu, X.; Ejsing, C.S.; Farese, R.V.; Walther, T.C. The GARP complex is required for cellular sphingolipid homeostasis. eLife 2015, 4, e08712. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhang, Y.Y.; Luo, J.; Wang, J.Q.; Zhou, Y.X.; Miao, H.H.; Shi, X.J.; Qu, Y.X.; Xu, J.; Li, B.L.; et al. The GARP Complex Is Involved in Intracellular Cholesterol Transport via Targeting NPC2 to Lysosomes. Cell Rep. 2017, 19, 2823–2835. [Google Scholar] [CrossRef]
- Petit, C.S.; Lee, J.J.; Boland, S.; Swarup, S.; Christiano, R.; Lai, Z.W.; Mejhert, N.; Elliott, S.D.; McFall, D.; Haque, S.; et al. Inhibition of sphingolipid synthesis improves outcomes and survival in GARP mutant wobbler mice, a model of motor neuron degeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 10565–10574. [Google Scholar] [CrossRef] [PubMed]
- Eising, S.; Thiele, L.; Frohlich, F. A systematic approach to identify recycling endocytic cargo depending on the GARP complex. eLife 2019, 8, e42837. [Google Scholar] [CrossRef] [PubMed]
- Gershlick, D.C.; Ishida, M.; Jones, J.R.; Bellomo, A.; Bonifacino, J.S.; Everman, D.B. A neurodevelopmental disorder caused by mutations in the VPS51 subunit of the GARP and EARP complexes. Hum. Mol. Genet. 2019, 28, 1548–1560. [Google Scholar] [CrossRef] [PubMed]
- Beilina, A.; Bonet-Ponce, L.; Kumaran, R.; Kordich, J.J.; Ishida, M.; Mamais, A.; Kaganovich, A.; Saez-Atienzar, S.; Gershlick, D.C.; Roosen, D.A.; et al. The Parkinson’s Disease Protein LRRK2 Interacts with the GARP Complex to Promote Retrograde Transport to the trans-Golgi Network. Cell Rep. 2020, 31, 107614. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, P.; Droce, A.; Moser, J.M.; Cuhlmann, S.; Padilla, C.O.; Heimann, P.; Bartsch, J.W.; Fuchtbauer, A.; Fuchtbauer, E.M.; Schmitt-John, T. Loss of vps54 function leads to vesicle traffic impairment, protein mis-sorting and embryonic lethality. Int. J. Mol. Sci. 2013, 14, 10908–10925. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-John, T.; Drepper, C.; Mussmann, A.; Hahn, P.; Kuhlmann, M.; Thiel, C.; Hafner, M.; Lengeling, A.; Heimann, P.; Jones, J.M.; et al. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat. Genet. 2005, 37, 1213–1215. [Google Scholar] [CrossRef]
- Perez-Victoria, F.J.; Abascal-Palacios, G.; Tascon, I.; Kajava, A.; Magadan, J.G.; Pioro, E.P.; Bonifacino, J.S.; Hierro, A. Structural basis for the wobbler mouse neurodegenerative disorder caused by mutation in the Vps54 subunit of the GARP complex. Proc. Natl. Acad. Sci.USA 2010, 107, 12860–12865. [Google Scholar] [CrossRef]
- Paiardi, C.; Pasini, M.E.; Gioria, M.; Berruti, G. Failure of acrosome formation and globozoospermia in the wobbler mouse, a Vps54 spontaneous recessive mutant. Spermatogenesis 2011, 1, 52–62. [Google Scholar] [CrossRef]
- Moser, J.M.; Bigini, P.; Schmitt-John, T. The wobbler mouse, an ALS animal model. Mol. Genet. Genom. MGG 2013, 288, 207–229. [Google Scholar] [CrossRef]
- Jockusch, H.; Holland, A.; Staunton, L.; Schmitt-John, T.; Heimann, P.; Dowling, P.; Ohlendieck, K. Pathoproteomics of testicular tissue deficient in the GARP component VPS54: The wobbler mouse model of globozoospermia. Proteomics 2014, 14, 839–852. [Google Scholar] [CrossRef]
- Schmitt-John, T. VPS54 and the wobbler mouse. Front. Neurosci. 2015, 9, 381. [Google Scholar] [CrossRef]
- Vasan, N.; Hutagalung, A.; Novick, P.; Reinisch, K.M. Structure of a C-terminal fragment of its Vps53 subunit suggests similarity of Golgi-associated retrograde protein (GARP) complex to a family of tethering complexes. Proc. Natl. Acad. Sci.USA 2010, 107, 14176–14181. [Google Scholar] [CrossRef] [PubMed]
- Khakurel, A.; Kudlyk, T.; Bonifacino, J.S.; Lupashin, V.V. The Golgi-associated retrograde protein (GARP) complex plays an essential role in the maintenance of the Golgi glycosylation machinery. Mol. Biol. Cell 2021, 32, 1594–1610. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, D.; Sutton, R.B.; Brunger, A.T.; Jahn, R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. USA 1998, 95, 15781–15786. [Google Scholar] [CrossRef]
- Bock, J.B.; Matern, H.T.; Peden, A.A.; Scheller, R.H. A genomic perspective on membrane compartment organization. Nature 2001, 409, 839–841. [Google Scholar] [CrossRef]
- Lou, X.; Shin, Y.K. SNARE zippering. Biosci. Rep. 2016, 36, e00327. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Pluhackova, K.; Bockmann, R.A. The Multifaceted Role of SNARE Proteins in Membrane Fusion. Front. Physiol. 2017, 8, 5. [Google Scholar] [CrossRef]
- Xu, D.; Joglekar, A.P.; Williams, A.L.; Hay, J.C. Subunit structure of a mammalian ER/Golgi SNARE complex. J. Biol. Chem. 2000, 275, 39631–39639. [Google Scholar] [CrossRef]
- Xu, Y.; Martin, S.; James, D.E.; Hong, W. GS15 forms a SNARE complex with syntaxin 5, GS28, and Ykt6 and is implicated in traffic in the early cisternae of the Golgi apparatus. Mol. Biol. Cell 2002, 13, 3493–3507. [Google Scholar] [CrossRef]
- Mallard, F.; Tang, B.L.; Galli, T.; Tenza, D.; Saint-Pol, A.; Yue, X.; Antony, C.; Hong, W.; Goud, B.; Johannes, L. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J. Cell Biol. 2002, 156, 653–664. [Google Scholar] [CrossRef]
- Wong, S.H.; Xu, Y.; Zhang, T.; Griffiths, G.; Lowe, S.L.; Subramaniam, V.N.; Seow, K.T.; Hong, W. GS32, a novel Golgi SNARE of 32 kDa, interacts preferentially with syntaxin 6. Mol. Biol. Cell 1999, 10, 119–134. [Google Scholar] [CrossRef][Green Version]
- Yong, C.Q.Y.; Tang, B.L. Another longin SNARE for autophagosome-lysosome fusion-how does Ykt6 work? Autophagy 2019, 15, 352–357. [Google Scholar] [CrossRef]
- Pan, S.; Nakayama, T.; Sato, N.; Izumi, Y.; Soma, M.; Aoi, N.; Ma, Y. A haplotype of the GOSR2 gene is associated with essential hypertension in Japanese men. Clin. Biochem. 2013, 46, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Lahm, H.; Jia, M.; Dressen, M.; Wirth, F.; Puluca, N.; Gilsbach, R.; Keavney, B.D.; Cleuziou, J.; Beck, N.; Bondareva, O.; et al. Congenital heart disease risk loci identified by genome-wide association study in European patients. J. Clin. Investig. 2021, 131, e141837. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Guan, G.C.; Lv, Y.; Liu, Z.W.; Liu, F.Q.; Zhang, Y.; Zhu, S.M.; Zhang, R.H.; Zhao, N.; Shi, S.; et al. G-T haplotype established by rs3785889-rs16941382 in GOSR2 gene is associated with coronary artery disease in Chinese Han population. Oncotarget 2017, 8, 82165–82173. [Google Scholar] [CrossRef]
- Corbett, M.A.; Schwake, M.; Bahlo, M.; Dibbens, L.M.; Lin, M.; Gandolfo, L.C.; Vears, D.F.; O’Sullivan, J.D.; Robertson, T.; Bayly, M.A.; et al. A mutation in the Golgi Qb-SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am. J. Hum. Genet. 2011, 88, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Henige, H.; Kaur, S.; Pappas, K. Compound heterozygous variants in GOSR2 associated with congenital muscular dystrophy: A case report. Eur. J. Med. Genet. 2021, 64, 104184. [Google Scholar] [CrossRef]
- Peter, T.A.; Linders, E.C.F.G.; Ashikov, A.; Vals, M.-A.; de Boer, R.; Revelo, N.H.; Arts, R.; Baerenfaenger, M.; Zijlstra, F.; Huijben, K.; et al. Congenital disorder of glycosylation caused by starting site-specific variant in syntaxin-5. Nat. Commun. 2021, 12, 6227. [Google Scholar] [CrossRef]
- McNew, J.A.; Sogaard, M.; Lampen, N.M.; Machida, S.; Ye, R.R.; Lacomis, L.; Tempst, P.; Rothman, J.E.; Sollner, T.H. Ykt6p, a prenylated SNARE essential for endoplasmic reticulum-Golgi transport. J. Biol. Chem. 1997, 272, 17776–17783. [Google Scholar] [CrossRef]
- Shirakawa, R.; Goto-Ito, S.; Goto, K.; Wakayama, S.; Kubo, H.; Sakata, N.; Trinh, D.A.; Yamagata, A.; Sato, Y.; Masumoto, H.; et al. A SNARE geranylgeranyltransferase essential for the organization of the Golgi apparatus. EMBO J. 2020, 39, e104120. [Google Scholar] [CrossRef] [PubMed]
- Steegmaier, M.; Yang, B.; Yoo, J.S.; Huang, B.; Shen, M.; Yu, S.; Luo, Y.; Scheller, R.H. Three novel proteins of the syntaxin/SNAP-25 family. J. Biol. Chem. 1998, 273, 34171–34179. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, A.C.; Roche, P.A. SNAP-29 is a promiscuous syntaxin-binding SNARE. Biochem. Biophys. Res. Commun. 2001, 285, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Kudlyk, T.; Willett, R.; Pokrovskaya, I.D.; Lupashin, V. COG6 interacts with a subset of the Golgi SNAREs and is important for the Golgi complex integrity. Traffic 2013, 14, 194–204. [Google Scholar] [CrossRef]
- Mastrodonato, V.; Morelli, E.; Vaccari, T. How to use a multipurpose SNARE: The emerging role of Snap29 in cellular health. Cell Stress 2018, 2, 72–81. [Google Scholar] [CrossRef]
- Sprecher, E.; Ishida-Yamamoto, A.; Mizrahi-Koren, M.; Rapaport, D.; Goldsher, D.; Indelman, M.; Topaz, O.; Chefetz, I.; Keren, H.; O’Brien, T.J.; et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am. J. Hum. Genet. 2005, 77, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Llaci, L.; Ramsey, K.; Belnap, N.; Claasen, A.M.; Balak, C.D.; Szelinger, S.; Jepsen, W.M.; Siniard, A.L.; Richholt, R.; Izat, T.; et al. Compound heterozygous mutations in SNAP29 is associated with Pelizaeus-Merzbacher-like disorder (PMLD). Hum. Genet. 2019, 138, 1409–1417. [Google Scholar] [CrossRef]
- Mastrodonato, V.; Beznoussenko, G.; Mironov, A.; Ferrari, L.; Deflorian, G.; Vaccari, T. A genetic model of CEDNIK syndrome in zebrafish highlights the role of the SNARE protein Snap29 in neuromotor and epidermal development. Sci. Rep. 2019, 9, 1211. [Google Scholar] [CrossRef] [PubMed]
- Keser, V.; Lachance, J.B.; Alam, S.S.; Lim, Y.; Scarlata, E.; Kaur, A.; Zhang, T.F.; Lv, S.; Lachapelle, P.; O’Flaherty, C.; et al. Snap29 mutant mice recapitulate neurological and ophthalmological abnormalities associated with 22q11 and CEDNIK syndrome. Commun. Biol. 2019, 2, 375. [Google Scholar] [CrossRef] [PubMed]
- Morelli, E.; Ginefra, P.; Mastrodonato, V.; Beznoussenko, G.V.; Rusten, T.E.; Bilder, D.; Stenmark, H.; Mironov, A.A.; Vaccari, T. Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in Drosophila. Autophagy 2014, 10, 2251–2268. [Google Scholar] [CrossRef]
- Xu, H.; Mohtashami, M.; Stewart, B.; Boulianne, G.; Trimble, W.S. Drosophila SNAP-29 is an essential SNARE that binds multiple proteins involved in membrane traffic. PLoS ONE 2014, 9, e91471. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Bai, Z.; Zegarek, M.H.; Grant, B.D.; Lee, J. Essential roles of snap-29 in C. elegans. Dev. Biol. 2011, 355, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Turan, S.; Ignatius, J.; Moilanen, J.S.; Kuismin, O.; Stewart, H.; Mann, N.P.; Linglart, A.; Bastepe, M.; Juppner, H. De novo STX16 deletions: An infrequent cause of pseudohypoparathyroidism type Ib that should be excluded in sporadic cases. J. Clin. Endocrinol. Metab. 2012, 97, E2314–E2319. [Google Scholar] [CrossRef][Green Version]
- Shitara, A.; Shibui, T.; Okayama, M.; Arakawa, T.; Mizoguchi, I.; Sakakura, Y.; Takuma, T. VAMP4 is required to maintain the ribbon structure of the Golgi apparatus. Mol. Cell. Biochem. 2013, 380, 11–21. [Google Scholar] [CrossRef]
- Frohlich, L.F.; Bastepe, M.; Ozturk, D.; Abu-Zahra, H.; Juppner, H. Lack of Gnas epigenetic changes and pseudohypoparathyroidism type Ib in mice with targeted disruption of syntaxin-16. Endocrinology 2007, 148, 2925–2935. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, A.J.; Rickmann, M.; Backofen, B.; Browski, S.M.; Rosenbusch, J.; Schoning, S.; Fleischmann, T.; Krieglstein, K.; Fischer von Mollard, G. Lack of the endosomal SNAREs vti1a and vti1b led to significant impairments in neuronal development. Proc. Natl. Acad. Sci. USA 2011, 108, 2575–2580. [Google Scholar] [CrossRef]
- Adolf, F.; Rhiel, M.; Hessling, B.; Gao, Q.; Hellwig, A.; Bethune, J.; Wieland, F.T. Proteomic Profiling of Mammalian COPII and COPI Vesicles. Cell Rep. 2019, 26, 250–265.e5. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Xia, Z.J.; Clement, A.; Parry, D.A.; Davids, M.; Taylan, F.; Sharma, P.; Turgeon, C.T.; Blanco-Sanchez, B.; Ng, B.G.; et al. A Recurrent De Novo Heterozygous COG4 Substitution Leads to Saul-Wilson Syndrome, Disrupted Vesicular Trafficking, and Altered Proteoglycan Glycosylation. Am. J. Hum. Genet. 2018, 103, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Sumya, F.T.; Pokrovskaya, I.D.; Lupashin, V. Development and Initial Characterization of Cellular Models for COG Complex-Related CDG-II Diseases. Front. Genet. 2021, 12, 733048. [Google Scholar] [CrossRef]
- Nakamura, N.; Rabouille, C.; Watson, R.; Nilsson, T.; Hui, N.; Slusarewicz, P.; Kreis, T.E.; Warren, G. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 1995, 131, 1715–1726. [Google Scholar] [CrossRef]
- Fritzler, M.J.; Hamel, J.C.; Ochs, R.L.; Chan, E.K. Molecular characterization of two human autoantigens: Unique cDNAs encoding 95- and 160-kD proteins of a putative family in the Golgi complex. J. Exp. Med. 1993, 178, 49–62. [Google Scholar] [CrossRef]
- Hicks, S.W.; Machamer, C.E. Isoform-specific interaction of golgin-160 with the Golgi-associated protein PIST. J. Biol. Chem. 2005, 280, 28944–28951. [Google Scholar] [CrossRef]
- Fritzler, M.J.; Lung, C.C.; Hamel, J.C.; Griffith, K.J.; Chan, E.K. Molecular characterization of Golgin-245, a novel Golgi complex protein containing a granin signature. J. Biol. Chem. 1995, 270, 31262–31268. [Google Scholar] [CrossRef]
- Bascom, R.A.; Srinivasan, S.; Nussbaum, R.L. Identification and characterization of golgin-84, a novel Golgi integral membrane protein with a cytoplasmic coiled-coil domain. J. Biol. Chem. 1999, 274, 2953–2962. [Google Scholar] [CrossRef] [PubMed]
- Ohta, E.; Misumi, Y.; Sohda, M.; Fujiwara, T.; Yano, A.; Ikehara, Y. Identification and characterization of GCP16, a novel acylated Golgi protein that interacts with GCP170. J. Biol. Chem. 2003, 278, 51957–51967. [Google Scholar] [CrossRef]
- Linstedt, A.D.; Hauri, H.P. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell 1993, 4, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Hayashi-Nishino, M.; Shakuno, T.; Masuda, J.; Koreishi, M.; Murakami, R.; Nakamura, Y.; Nakamura, T.; Abe-Kanoh, N.; Honjo, Y.; et al. The Golgin Protein Giantin Regulates Interconnections Between Golgi Stacks. Front. Cell Dev. Biol. 2019, 7, 160. [Google Scholar] [CrossRef]
- Cheung, P.Y.; Limouse, C.; Mabuchi, H.; Pfeffer, S.R. Protein flexibility is required for vesicle tethering at the Golgi. eLife 2015, 4, e12790. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, A.J.; Pohajdak, B. Getting a GRASP on CASP: Properties and role of the cytohesin-associated scaffolding protein in immunity. Immunol. Cell Biol. 2009, 87, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.G.; Clary, D.O.; Rothman, J.E. A novel 115-kD peripheral membrane protein is required for intercisternal transport in the Golgi stack. J. Cell Biol. 1992, 118, 1015–1026. [Google Scholar] [CrossRef]
- Barr, F.A.; Puype, M.; Vandekerckhove, J.; Warren, G. GRASP65, a protein involved in the stacking of Golgi cisternae. Cell 1997, 91, 253–262. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y. Nonredundant Roles of GRASP55 and GRASP65 in the Golgi Apparatus and Beyond. Trends Biochem. Sci. 2020, 45, 1065–1079. [Google Scholar] [CrossRef]
- Kingsley, D.M.; Kozarsky, K.F.; Segal, M.; Krieger, M. Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol. 1986, 102, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, F.; Vasile, E.; Schollen, E.; Callewaert, N.; Raemaekers, T.; Quelhas, D.; Jaeken, J.; Mills, P.; Winchester, B.; Krieger, M.; et al. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc. Natl. Acad. Sci. USA 2006, 103, 3764–3769. [Google Scholar] [CrossRef]
- Kodera, H.; Ando, N.; Yuasa, I.; Wada, Y.; Tsurusaki, Y.; Nakashima, M.; Miyake, N.; Saitoh, S.; Matsumoto, N.; Saitsu, H. Mutations in COG2 encoding a subunit of the conserved oligomeric golgi complex cause a congenital disorder of glycosylation. Clin. Genet. 2015, 87, 455–460. [Google Scholar] [CrossRef]
- Suvorova, E.S.; Kurten, R.C.; Lupashin, V.V. Identification of a human orthologue of Sec34p as a component of the cis-Golgi vesicle tethering machinery. J. Biol. Chem. 2001, 276, 22810–22818. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.G.; Sharma, V.; Sun, L.; Loh, E.; Hong, W.; Tay, S.K.; Freeze, H.H. Identification of the first COG-CDG patient of Indian origin. Mol. Genet. Metab. 2011, 102, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Paesold-Burda, P.; Maag, C.; Troxler, H.; Foulquier, F.; Kleinert, P.; Schnabel, S.; Baumgartner, M.; Hennet, T. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum. Mol. Genet. 2009, 18, 4350–4356. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.W.; Matthijs, G.; Sturiale, L.; Garozzo, D.; Wong, K.Y.; Wong, R.; Wong, V.; Jaeken, J. COG5-CDG with a Mild Neurohepatic Presentation. JIMD Rep. 2012, 3, 67–70. [Google Scholar] [CrossRef]
- Kim, Y.O.; Yun, M.; Jeong, J.H.; Choi, S.M.; Kim, S.K.; Yoon, W.; Park, C.; Hong, Y.; Woo, Y.J. A Mild Form of COG5 Defect Showing Early-Childhood-Onset Friedreich’s-Ataxia-Like Phenotypes with Isolated Cerebellar Atrophy. J. Korean Med. Sci. 2017, 32, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Gong, L.; Qiu, H.; Zhao, Y.; Zhang, Y.; Liu, C.; Jiang, H.; Mao, Y.; Kong, L.Y.; Liang, B.; et al. Novel compound heterozygous COG5 mutations in a Chinese male patient with severe clinical symptoms and type IIi congenital disorder of glycosylation: A case report. Exp. Ther. Med. 2019, 18, 2695–2700. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Han, L.; Wang, X.Y.; Wang, J.H.; Li, X.M.; Jin, C.H.; Wang, L. Identification of Two Novel Mutations in COG5 Causing Congenital Disorder of Glycosylation. Front. Genet. 2020, 11, 168. [Google Scholar] [CrossRef]
- Cherot, E.; Keren, B.; Dubourg, C.; Carre, W.; Fradin, M.; Lavillaureix, A.; Afenjar, A.; Burglen, L.; Whalen, S.; Charles, P.; et al. Using medical exome sequencing to identify the causes of neurodevelopmental disorders: Experience of 2 clinical units and 216 patients. Clin. Genet. 2018, 93, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Lubbehusen, J.; Thiel, C.; Rind, N.; Ungar, D.; Prinsen, B.H.; de Koning, T.J.; van Hasselt, P.M.; Korner, C. Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Hum. Mol. Genet. 2010, 19, 3623–3633. [Google Scholar] [CrossRef]
- Ciftci, H.; Celepkolo, B.; Dilmec, F.; Koksal, M.; Yeni, E.; Yagmur, I.; Gumus, K. Genetic polymorphisms of hspa1b and hspa1l in infertile men. JPMA J. Pak. Med. Assoc. 2015, 65, 701–704. [Google Scholar] [PubMed]
- Huybrechts, S.; De Laet, C.; Bontems, P.; Rooze, S.; Souayah, H.; Sznajer, Y.; Sturiale, L.; Garozzo, D.; Matthijs, G.; Ferster, A.; et al. Deficiency of Subunit 6 of the Conserved Oligomeric Golgi Complex (COG6-CDG): Second Patient, Different Phenotype. JIMD Rep. 2012, 4, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Ansari, S.; Alshammari, M.J.; Alkhalidi, H.; Alrukban, H.; Eyaid, W.; Alkuraya, F.S. A novel syndrome of hypohidrosis and intellectual disability is linked to COG6 deficiency. J. Med. Genet. 2013, 50, 431–436. [Google Scholar] [CrossRef]
- Althonaian, N.; Alsultan, A.; Morava, E.; Alfadhel, M. Secondary Hemophagocytic Syndrome Associated with COG6 Gene Defect: Report and Review. JIMD Rep. 2018, 42, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Steet, R.A.; Bohorov, O.; Bakker, J.; Newell, J.; Krieger, M.; Spaapen, L.; Kornfeld, S.; Freeze, H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004, 10, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Spaapen, L.J.; Bakker, J.A.; van der Meer, S.B.; Sijstermans, H.J.; Steet, R.A.; Wevers, R.A.; Jaeken, J. Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. J. Inherit. Metab. Dis. 2005, 28, 707–714. [Google Scholar] [CrossRef]
- Morava, E.; Zeevaert, R.; Korsch, E.; Huijben, K.; Wopereis, S.; Matthijs, G.; Keymolen, K.; Lefeber, D.J.; De Meirleir, L.; Wevers, R.A. A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur. J. Hum. Genet. EJHG 2007, 15, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Zeevaert, R.; Foulquier, F.; Cheillan, D.; Cloix, I.; Guffon, N.; Sturiale, L.; Garozzo, D.; Matthijs, G.; Jaeken, J. A new mutation in COG7 extends the spectrum of COG subunit deficiencies. Eur. J. Med. Genet. 2009, 52, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, F.; Ungar, D.; Reynders, E.; Zeevaert, R.; Mills, P.; Garcia-Silva, M.T.; Briones, P.; Winchester, B.; Morelle, W.; Krieger, M.; et al. A new inborn error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1-Cog8 interaction in COG complex formation. Hum. Mol. Genet. 2007, 16, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Kranz, C.; Ng, B.G.; Sun, L.; Sharma, V.; Eklund, E.A.; Miura, Y.; Ungar, D.; Lupashin, V.; Winkel, R.D.; Cipollo, J.F.; et al. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum. Mol. Genet. 2007, 16, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Cho, S.Y.; Jang, J.H.; Kim, J.; Kim, S.Z.; Lee, B.H.; Yoo, H.W.; Jin, D.K. Further delineation of COG8-CDG: A case with novel compound heterozygous mutations diagnosed by targeted exome sequencing. Clin. Chim. Acta Int. J. Clin. Chem. 2017, 471, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.; Puri, R.D.; Bhai, P.; Sharma, N.; Bijarnia-Mahay, S.; Dimri, N.; Baijal, A.; Saxena, R.; Verma, I. The first case of antenatal presentation in COG8-congenital disorder of glycosylation with a novel splice site mutation and an extended phenotype. Am. J. Med. Genet. Part A 2019, 179, 480–485. [Google Scholar] [CrossRef]
- Uwineza, A.; Caberg, J.H.; Hitayezu, J.; Wenric, S.; Mutesa, L.; Vial, Y.; Drunat, S.; Passemard, S.; Verloes, A.; El Ghouzzi, V.; et al. VPS51 biallelic variants cause microcephaly with brain malformations: A confirmatory report. Eur. J. Med. Genet. 2019, 62, 103704. [Google Scholar] [CrossRef]
- Fusella, A.; Micaroni, M.; Di Giandomenico, D.; Mironov, A.A.; Beznoussenko, G.V. Segregation of the Qb-SNAREs GS27 and GS28 into Golgi vesicles regulates intra-Golgi transport. Traffic 2013, 14, 568–584. [Google Scholar] [CrossRef]
- Dibbens, L.M.; Rubboli, G. GOSR2: A progressive myoclonus epilepsy gene. Epileptic Disord. Int. Epilepsy J. Videotape 2016, 18, 111–114. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Souza, Z.; Sumya, F.T.; Khakurel, A.; Lupashin, V. Getting Sugar Coating Right! The Role of the Golgi Trafficking Machinery in Glycosylation. Cells 2021, 10, 3275. https://doi.org/10.3390/cells10123275
D’Souza Z, Sumya FT, Khakurel A, Lupashin V. Getting Sugar Coating Right! The Role of the Golgi Trafficking Machinery in Glycosylation. Cells. 2021; 10(12):3275. https://doi.org/10.3390/cells10123275
Chicago/Turabian StyleD’Souza, Zinia, Farhana Taher Sumya, Amrita Khakurel, and Vladimir Lupashin. 2021. "Getting Sugar Coating Right! The Role of the Golgi Trafficking Machinery in Glycosylation" Cells 10, no. 12: 3275. https://doi.org/10.3390/cells10123275
APA StyleD’Souza, Z., Sumya, F. T., Khakurel, A., & Lupashin, V. (2021). Getting Sugar Coating Right! The Role of the Golgi Trafficking Machinery in Glycosylation. Cells, 10(12), 3275. https://doi.org/10.3390/cells10123275