
Analysis of Nucleosides and Nucleotides in Plants: An Update on Sample Preparation and LC–MS Techniques




Abstract
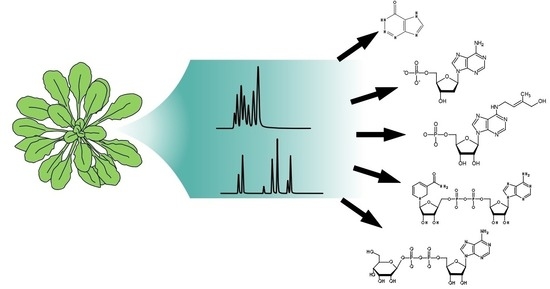
:
1. Introduction
2. Disruption of the Tissue
3. Quenching of the Sample
4. Extraction of NTs and Ns from Plant Samples
5. Solid-Phase Extraction
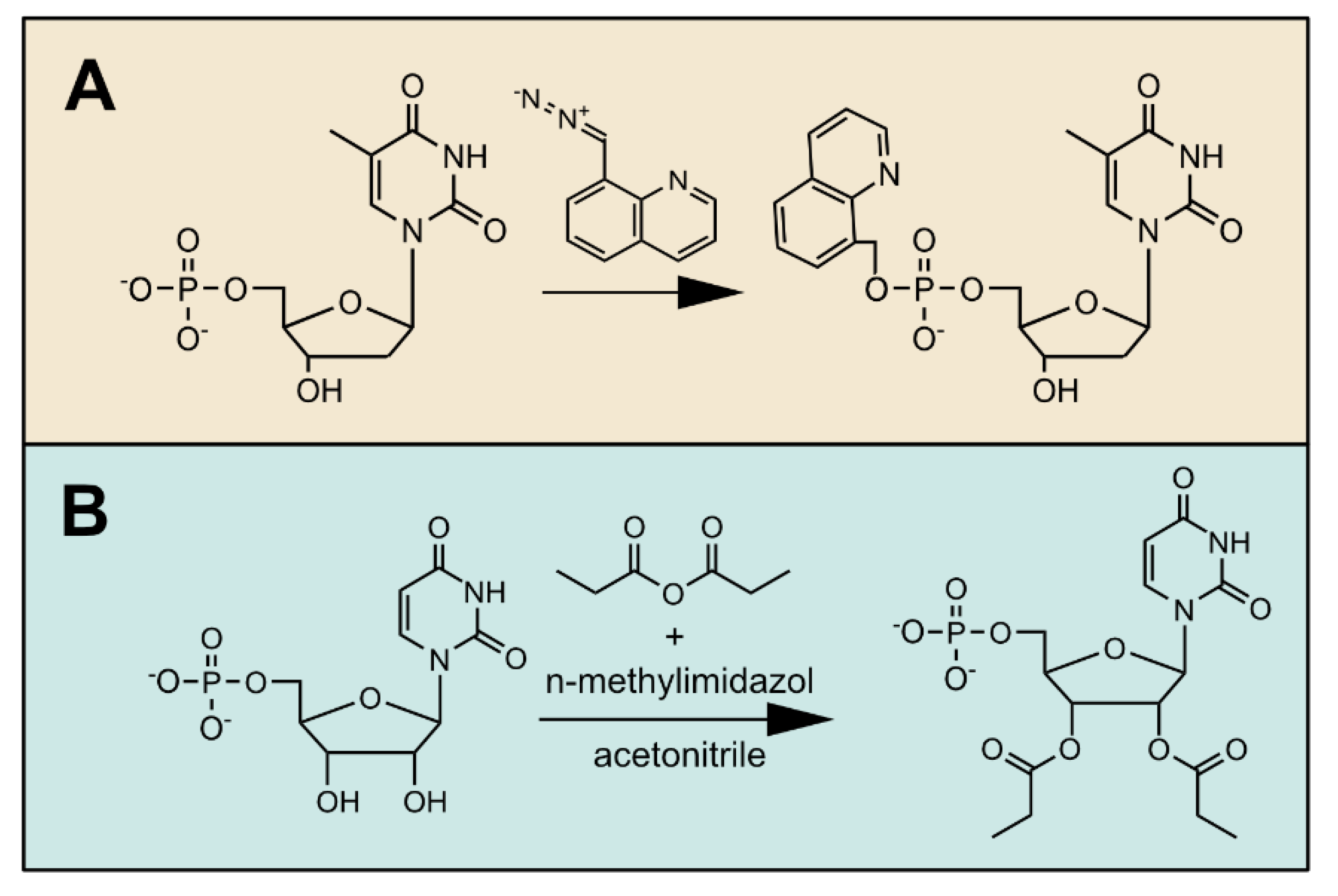
6. Derivatization
7. Reduction in Sample Volume
8. Chromatographic Separation
9. Mass Spectrometry
10. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Witte, C.-P.; Herde, M. Nucleotide metabolism in plants. Plant Physiol. 2020, 182, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Straube, H.; Niehaus, M.; Zwittian, S.; Witte, C.-P.; Herde, M. Enhanced nucleotide analysis enables the quantification of deoxynucleotides in plants and algae revealing connections between nucleoside and deoxynucleoside metabolism. Plant Cell 2020. [Google Scholar] [CrossRef]
- Hodgson, D.R.W. Physicochemical Aspects of Aqueous and Nonaqueous Approaches to the Preparation of Nucleosides, Nucleotides and Phosphate Ester Mimics. Adv. Phys. Org. 2017, 51, 187–219. [Google Scholar] [CrossRef]
- Liu, B.; Winkler, F.; Herde, M.; Witte, C.-P.; Grosshans, J. A Link between deoxyribonucleotide metabolites and embryonic cell-cycle control. Curr. Biol. 2019, 29, 1187. [Google Scholar] [CrossRef] [Green Version]
- Bielski, R.L. The problem of halting enzyme action when extracting plant tissues. Anal. Biochem. 1964, 9, 431–442. [Google Scholar] [CrossRef]
- Creydt, M.; Fischer, M. Plant metabolomics: Maximizing metabolome coverage by optimizing mobile phase additives for nontargeted mass spectrometry in positive and negative electrospray ionization mode. Anal. Chem. 2017, 89, 10474–10486. [Google Scholar] [CrossRef] [PubMed]
- Rolletschek, H.; Melkus, G.; Grafahrend-Belau, E.; Fuchs, J.; Heinzel, N.; Schreiber, F.; Jakob, P.M.; Borisjuk, L. Combined noninvasive imaging and modeling approaches reveal metabolic compartmentation in the barley endosperm. Plant Cell 2011, 23, 3041–3054. [Google Scholar] [CrossRef] [Green Version]
- De Souza, A.P.; Cocuron, J.-C.; Garcia, A.C.; Alonso, A.P.; Buckeridge, M.S. Changes in whole-plant metabolism during the grain-filling stage in sorghum grown under elevated CO2 and drought. Plant Physiol. 2015, 169, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, P. Deoxyribonucleotide pools in plant-tissue cultures. Physiol. Plant. 1972, 26, 29–33. [Google Scholar] [CrossRef]
- Khym, J.X. An analytical system for rapid separation of tissue nucleotides at low pressures on conventional anion exchangers. Clin. Chem. 1975, 21, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Dutta, I.; Dutta, P.K.; Smith, D.W.; O’Donovan, G.A. High-performance liquid-chromatography of deoxynucleoside diphosphates and triphosphates in tomato roots. J. Chromatogr. A 1991, 536, 237–243. [Google Scholar] [CrossRef]
- Pabst, M.; Grass, J.; Fischl, R.; Léonard, R.; Jin, C.; Hinterkörner, G.; Borth, N.; Altmann, F. Nucleotide and nucleotide sugar analysis by liquid chromatography-electrospray ionization-mass spectrometry on surface-conditioned porous graphitic carbon. Anal. Chem. 2010, 82, 9782–9788. [Google Scholar] [CrossRef]
- Guerard, F.; Petriacq, P.; Gakiere, B.; Tcherkez, G. Liquid chromatography/time-of-flight mass spectrometry for the analysis of plant samples: A method for simultaneous screening of common cofactors or nucleotides and application to an engineered plant line. Plant Physiol. Biochem. 2011, 49, 1117–1125. [Google Scholar] [CrossRef]
- Kopecna, M.; Blaschke, H.; Kopecny, D.; Vigouroux, A.; Koncitikova, R.; Novak, O.; Kotland, O.; Strnad, M.; Morera, S.; von Schwartzenberg, K. Structure and function of nucleoside hydrolases from physcomitrella patens and maize catalyzing the hydrolysis of purine, pyrimidine, and cytokinin ribosides. Plant Physiol. 2013, 163, 1568–1583. [Google Scholar] [CrossRef] [Green Version]
- Kuskovsky, R.; Buj, R.; Xu, P.; Hofbauer, S.; Doan, M.T.; Jiang, H.; Bostwick, A.; Mesaros, C.; Aird, K.M.; Snyder, N.W. Simultaneous isotope dilution quantification and metabolic tracing of deoxyribonucleotides by liquid chromatography high resolution mass spectrometry. Anal. Biochem. 2019, 568, 65–72. [Google Scholar] [CrossRef]
- Rodríguez-González, P.; García Alonso, J.I. Isotope Dilution Mass Spectrometry; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Ingle, J. The extraction and estimation of nucleotides and nucleic acids from plant material. Phytochemistry 1963, 2, 353–370. [Google Scholar] [CrossRef]
- Ullrich, J.; Calvin, M. Alcohol-resistant phosphatase activity in chloroplasts. Biochim. Biophys. Acta 1962, 63, 1–10. [Google Scholar] [CrossRef]
- Ullrich, J. Phosphatase action on phosphoglycolic, 3-phosphoglyceric, and phosphoenol pyruvic acids in spinach chloroplast fragments in the presence and absence of high concentrations of methanol. Biochim. Biophys. Acta 1963, 71, 589–594. [Google Scholar] [CrossRef]
- Ikuma, H.; Tetley, R.M. Possible interference by an acid-stable enzyme during extraction of nucleoside diphosphates and triphosphates from higher-plant tissues. Plant Physiol. 1976, 58, 320–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runeckles, V.C. Formation of alkyl phosphates in wheat leaves. Nature 1958, 181, 1470–1471. [Google Scholar] [CrossRef]
- Baccolini, C.; Witte, C.-P. AMP and GMP catabolism in arabidopsis converge on xanthosine, which is degraded by a nucleoside hydrolase heterocomplex. Plant Cell 2019, 31, 734–751. [Google Scholar] [CrossRef] [PubMed]
- Dietmair, S.; Timmins, N.E.; Gray, P.P.; Nielsen, L.K.; Krömer, J.O. Towards quantitative metabolomics of mammalian cells: Development of a metabolite extraction protocol. Anal. Biochem. 2010, 404, 155–164. [Google Scholar] [CrossRef]
- Dahncke, K.; Witte, C.-P. Plant purine nucleoside catabolism employs a guanosine deaminase required for the generation of xanthosine in arabidopsis. Plant Cell 2013, 25, 4101–4109. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Herde, M.; Witte, C.-P. Of the nine cytidine deaminase-like genes in arabidopsis, eight are pseudogenes and only one is required to maintain pyrimidine homeostasis in vivo. Plant Physiol. 2016, 171, 799–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chida, J.; Yamane, K.; Takei, T.; Kido, H. An efficient extraction method for quantitation of adenosine triphosphate in mammalian tissues and cells. Anal. Chim. Acta 2012, 727, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; Kimball, E. Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Anal. Chem. 2007, 79, 6167–6173. [Google Scholar] [CrossRef]
- Au, J.L.; Su, M.H.; Wientjes, M.G. Extraction of intracellular nucleosides and nucleotides with acetonitrile. Clin. Chem. 1989, 35, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, B.; Hicks, L.M.; Xiong, L. A nucleotide metabolite controls stress-responsive gene expression and plant development. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Rochfort, S. Recent progress in polar metabolite quantification in plants using liquid chromatography-mass spectrometry. J. Integr. Plant Biol. 2014, 56, 816–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zbornikova, E.; Knejzlik, Z.; Hauryliuk, V.; Krasny, L.; Rejman, D. Analysis of nucleotide pools in bacteria using HPLC-MS in HILIC mode. Talanta 2019, 205. [Google Scholar] [CrossRef]
- Riondet, C.; Morel, S.; Alcaraz, G. Determination of total ribonucleotide pool in plant materials by high-pH anion-exchange high-performance liquid chromatography following extraction with potassium hydroxide. J. Chromatogr. A 2005, 1077, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.R. Stability of nucleotide solutions on storage as determined by high-pressure liquid chromatography. Anal. Biochem. 1971, 43, 305–306. [Google Scholar] [CrossRef]
- Barnes, J.; Tian, L.; Loftis, J.; Hiznay, J.; Comhair, S.; Lauer, M.; Dweik, R. Isolation and analysis of sugar nucleotides using solid phase extraction and fluorophore assisted carbohydrate electrophoresis. MethodsX 2016, 3, 251–260. [Google Scholar] [CrossRef]
- Salem, M.A.; Yoshida, T.; de Souza, L.P.; Alseekh, S.; Bajdzienko, K.; Fernie, A.R.; Giavalisco, P. An improved extraction method enables the comprehensive analysis of lipids, proteins, metabolites and phytohormones from a single sample of leaf tissue under water-deficit stress. Plant J. 2020, 103, 1614–1632. [Google Scholar] [CrossRef] [PubMed]
- Soga, T.; Ishikawa, T.; Igarashi, S.; Sugawara, K.; Kakazu, Y.; Tomita, M. Analysis of nucleotides by pressure-assisted capillary electrophoresis-mass spectrometry using silanol mask technique. J. Chromatogr. A 2007, 1159, 125–133. [Google Scholar] [CrossRef]
- Cordell, R.L.; Hill, S.J.; Ortori, C.A.; Barrett, D.A. Quantitative profiling of nucleotides and related phosphate-containing metabolites in cultured mammalian cells by liquid chromatography tandem electrospray mass spectrometry. J. Chromatogr. B 2008, 871, 115–124. [Google Scholar] [CrossRef]
- Nieman, R.H.; Pap, D.L.; Clark, R.A. Rapid purification of plant nucleotide extracts with xad-2,polyvinyl-polypyrrolidone and charcoal. J. Chromatogr. A 1978, 161, 137–146. [Google Scholar] [CrossRef]
- Tanaka, K.; Yoshioka, A.; Tanaka, S.; Wataya, Y. An improved method for the quantitative determination deoxyribonucleoside triphosphates in cell-extracts. Anal. Biochem. 1984, 139, 35–41. [Google Scholar] [CrossRef]
- Uziel, M. Periodate oxidation and amine-catalyzed elimination of terminal nucleoside from adenylate or ribonucleic-acid products of overoxidation. Biochemistry 1973, 12, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Odmark, G.; Kihlman, B.A. Effects of chromosome-breaking purine derivates on nucleic acid synthesis and on levels of adenosine 5′-triphosphate and deoxyadenosine 5′-triphosphate in bean root tips. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1965, 2, 274–286. [Google Scholar] [CrossRef]
- Hennere, G.; Becher, F.; Pruvost, A.; Goujard, C.; Grassi, J.; Benech, H. Liquid chromatography-tandem mass spectrometry assays for intracellular deoxyribonucleotide triphosphate competitors of nucleoside antiretrovirals. J. Chromatogr. B 2003, 789, 273–281. [Google Scholar] [CrossRef]
- Ji, P. iBonD 2.0-the Most Comprehensive pKa and BDE Database so Far; Tsinghua University: Beijing, China, 2016. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Y.; Huang, L.; Yang, L.; Hong, R.; Yao, H.; Li, S. Magnetic dispersive solid-phase micro-extraction combined with high-performance liquid chromatography for determining nucleotides in anoectochilus roxburghii (Wall.) Lindl. J. Pharm. Biomed. Anal. 2019, 174, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Hennion, M.C. Graphitized carbons for solid-phase extraction. J. Chromatogr. A 2000, 885, 73–95. [Google Scholar] [CrossRef]
- Behmüller, R.; Forstenlehner, I.C.; Tenhaken, R.; Huber, C.G. Quantitative HPLC-MS analysis of nucleotide sugars in Plant Cells following off-line SPE sample preparation. Anal. Bioanal. Chem. 2014, 406, 3229–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rautengarten, C.; Heazlewood, J.L.; Ebert, B. Profiling Cell Wall Monosaccharides and Nucleotide-Sugars from Plants. Curr. Protoc. Plant Biol. 2019, 4, e20092. [Google Scholar] [CrossRef]
- Ito, J.; Herter, T.; Baidoo, E.E.K.; Lao, J.; Vega-Sanchez, M.E.; Smith-Moritz, A.M.; Adams, P.D.; Keasling, J.D.; Usadel, B.; Petzold, C.J.; et al. Analysis of plant nucleotide sugars by hydrophilic interaction liquid chromatography and tandem mass spectrometry. Anal. Biochem. 2014, 448, 14–22. [Google Scholar] [CrossRef]
- Guo, M.; Yin, D.; Han, J.; Zhang, L.; Li, X.; He, D.; Du, Y.; Tang, D. Phenylboronic acid modified solid-phase extraction column: Preparation, characterization, and application to the analysis of amino acids in sepia capsule by removing the maltose. J. Sep. Sci. 2016, 39, 3428–3435. [Google Scholar] [CrossRef]
- Xin, P.; Li, B.; Yan, J.; Chu, J. Pursuing extreme sensitivity for determination of endogenous brassinosteroids through direct fishing from plant matrices and eliminating most interferences with boronate affinity magnetic nanoparticles. Anal. Bioanal. Chem. 2018, 410, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, X.; Lu, W.; Wu, X.; Li, J. Molecular imprinting: Perspectives and applications. Chem. Soc. Rev. 2016, 45, 2137–2211. [Google Scholar] [CrossRef] [PubMed]
- Jadda, R.; Madhumanchi, S.; Suedee, R. Novel adsorptive materials by adenosine 5ʹ-triphosphate imprinted-polymer over the surface of polystyrene nanospheres for selective separation of adenosine 5ʹ-triphosphate biomarker from urine. J. Sep. Sci. 2019, 42, 3662–3678. [Google Scholar] [CrossRef] [PubMed]
- Jegourel, D.; Delepee, R.; Breton, F.; Rolland, A.; Vidal, R.; Agrofoglio, L.A. Molecularly imprinted polymer of 5-methyluridine for solid-phase extraction of pyrimidine nucleoside cancer markers in urine. Bioorg. Med. Chem. 2008, 16, 8932–8939. [Google Scholar] [CrossRef]
- Choi, J.-H.; Abe, N.; Tanaka, H.; Fushimi, K.; Nishina, Y.; Morita, A.; Kiriiwa, Y.; Motohashi, R.; Hashizume, D.; Koshino, H.; et al. Plant-growth regulator, imidazole-4-carboxamide, produced by the fairy ring forming fungus Lepista sordida. J. Agric. Food Chem. 2010, 58, 9956–9959. [Google Scholar] [CrossRef]
- Choi, J.-H.; Ohnishi, T.; Yamakawa, Y.; Takeda, S.; Sekiguchi, S.; Maruyama, W.; Yamashita, K.; Suzuki, T.; Morita, A.; Ikka, T.; et al. The Source of “Fairy Rings”: 2-Azahypoxanthine and its Metabolite Found in a Novel Purine Metabolic Pathway in Plants. Angew. Chem. 2014, 53, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.; Wu, J.; Sawada, A.; Takeda, S.; Takemura, H.; Yogosawa, K.; Hirai, H.; Kondo, M.; Sugimoto, K.; Asakawa, T.; et al. N-Glucosides of Fairy Chemicals, 2-Azahypoxanthine and 2-Aza-8-oxohypoxanthine, in Rice. Org. Lett. 2018, 20, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Stevens, W.C.; Hill, D.C. General methods for flash chromatography using disposable columns. Mol. Divers. 2009, 13, 247–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Still, W.C.; Kahn, M.; Mitra, A. Rapid chromatographic technique for preparative separations with modern resolution. J. Org. Chem. 1978, 43, 2923–2925. [Google Scholar] [CrossRef]
- Takemura, H.; Choi, J.-H.; Matsuzaki, N.; Taniguchi, Y.; Wu, J.; Hirai, H.; Motohashi, R.; Asakawa, T.; Ikeuchi, K.; Inai, M.; et al. A Fairy Chemical, Imidazole-4-carboxamide, is Produced on a Novel Purine Metabolic Pathway in Rice. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Nordstrom, A.; Tarkowski, P.; Tarkowska, D.; Dolezal, K.; Astot, C.; Sandberg, G.; Moritz, T. Derivatization for LC electrospray ionization-MS: A tool for improving reversed-phase separation and ESI responses of bases, ribosides, and intact nucleotides. Anal. Chem. 2004, 76, 2869–2877. [Google Scholar] [CrossRef]
- Jiang, H.-P.; Xiong, J.; Liu, F.-L.; Ma, C.-J.; Tang, X.-L.; Yuan, B.-F.; Feng, Y.-Q. Modified nucleoside triphosphates exist in mammals. Chem. Sci. 2018, 9, 4160–4167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.-T.; Cai, B.-D.; Jiang, H.-P.; Xiao, H.-M.; Yuan, B.-F.; Feng, Y.-Q. Sensitive analysis of trehalose-6-phosphate and related sugar phosphates in plant tissues by chemical derivatization combined with hydrophilic interaction liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2019, 1592, 82–90. [Google Scholar] [CrossRef]
- Bjorkman, P.O.; Tillberg, E. Acetylation of cytokinins and modified adenine compounds: A simple and non-destructive derivatization method for gas chromatography—Mass spectrometric analysis. Phytochem. Anal. 1996, 7, 57–68. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Li, Z.; Lam, C.W.K.; Zhu, P.; Wang, C.; Zhou, H.; Zhang, W. MTBSTFA derivatization-LC-MS/MS approach for the quantitative analysis of endogenous nucleotides in human colorectal carcinoma cells. J. Pharm. Anal. 2021. [Google Scholar] [CrossRef]
- Dudley, E.; Bond, L. Mass spectrometry analysis of nucleosides and nucleotides. Mass Spectrom. Rev. 2014, 33, 302–331. [Google Scholar] [CrossRef]
- Salvatore, F.; Paul, R.H.; Colin, F.P.; Marja-Liisa, R. (Eds.) Liquid Chromatography, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 978-0-12-805393-5. [Google Scholar]
- Sykora, D.; Rezanka, P.; Zaruba, K.; Kral, V. Recent advances in mixed-mode chromatographic stationary phases. J. Sep. Sci. 2019, 42, 89–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, N.S.; Gilroy, J.; Dolan, J.W.; Snyder, L.R. Column selectivity in reversed-phase liquid chromatography—VI. Columns with embedded or end-capping polar groups. J. Chromatogr. A 2004, 1026, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Zuvela, P.; Skoczylas, M.; Liu, J.J.; Baczek, T.; Kaliszan, R.; Wong, M.W.; Buszewski, B. Column Characterization and Selection Systems in Reversed-Phase High-Performance Liquid Chromatography. Chem. Rev. 2019, 119, 3674–3729. [Google Scholar] [CrossRef]
- Traube, F.R.; Schiffers, S.; Iwan, K.; Kellner, S.; Spada, F.; Mueller, M.; Carell, T. Isotope-dilution mass spectrometry for exact quantification of noncanonical DNA nucleosides. Nat. Protoc. 2019, 14, 283. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, S.A.; Samskog, J.; Markides, K.E.; Langstrom, B. Studies of signal suppression in liquid chromatography-electrospray ionization mass spectrometry using volatile ion-pairing reagents. J. Chromatogr. A 2001, 937, 41–47. [Google Scholar] [CrossRef]
- Annesley, T.M. Ion suppression in mass spectrometry. Clin. Chem. 2003, 49, 1041–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajin, B.; Goessler, W. Fluoroalkylamines: Novel, highly volatile, fast-equilibrating, and electrospray ionization-mass spectrometry signal-enhancing cationic ion-interaction reagents. Anal. Chem. 2020, 92, 10121–10128. [Google Scholar] [CrossRef]
- Dodbiba, E.; Breitbach, Z.S.; Wanigasekara, E.; Payagala, T.; Zhang, X.; Armstrong, D.W. Detection of nucleotides in positive-mode electrospray ionization mass spectrometry using multiply-charged cationic ion-pairing reagents. Anal. Bioanal. Chem. 2010, 398, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.J.; van de Bittner, G.C.; Kennedy, A.P.; Wei, T.-C. The use of HILIC zwitterionic phase superficially porous particles for metabolomics analysis. LC GC N. Am. 2018, 36, 30–35. [Google Scholar]
- Kong, Z.; Jia, S.; Chabes, A.L.; Appelblad, P.; Lundmark, R.; Moritz, T.; Chabes, A. Simultaneous determination of ribonucleoside and deoxyribonucleoside triphosphates in biological samples by hydrophilic interaction liquid chromatography coupled with tandem mass spectrometry. Nucleic Acids Res. 2018, 46. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, W.; Minakova, A.; Anumol, T.; Keller, A. Quantitative analysis of changes in amino acids levels for cucumber (Cucumis sativus) exposed to nano copper. Nanoimpact 2018, 12, 9–17. [Google Scholar] [CrossRef]
- Kate, P.; John, K.L. Determination of selected water-soluble vitamins (thiamine, riboflavin, nicotinamide and pyridoxine) from a food matrix using hydrophilic interaction liquid chromatography coupled with mass spectroscopy. J. Chromatogr. B 2021, 122541. [Google Scholar] [CrossRef]
- Warth, B.; Siegwart, G.; Lemmens, M.; Krska, R.; Adam, G.; Schuhmacher, R. Hydrophilic interaction liquid chromatography coupled with tandem mass spectrometry for the quantification of uridine diphosphate-glucose, uridine diphosphate-glucuronic acid, deoxynivalenol and its glucoside: In-house validation and application to wheat. J. Chromatogr. A 2015, 1423, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, J.J.; Potter, O.G.; Chu, T.-W.; Yin, H. Improved LC/MS Methods for the analysis of metal-sensitive analytes using medronic acid as a mobile phase additive. Anal. Chem. 2018, 90, 9457–9464. [Google Scholar] [CrossRef]
- Matyska, M.T.; Pesek, J.J.; Duley, J.; Zamzami, M.; Fischer, S.M. Aqueous normal phase retention of nucleotides on silica hydride-based columns: Method development strategies for analytes revelant in clinical analysis. J. Sep. Sci. 2010, 33, 930–938. [Google Scholar] [CrossRef]
- Pesek, J.J.; Matyska, M.T.; Fischer, S.M.; Sana, T.R. Analysis of hydrophilic metabolites by high-performance liquid chromatography-mass spectrometry using a silica hydride-based stationary phase. J. Chromatogr. A 2008, 1204, 48–55. [Google Scholar] [CrossRef]
- Pesek, J.J.; Matyska, M.T.; Loo, J.A.; Fischer, S.M.; Sana, T.R. Analysis of hydrophilic metabolites in physiological fluids by HPLC-MS using a silica hydride-based stationary phase. J. Sep. Sci. 2009, 32, 2200–2208. [Google Scholar] [CrossRef]
- Zhang, W.; Guled, F.; Hankemeier, T.; Ramautar, R. Profiling nucleotides in low numbers of mammalian cells by sheathless CE-MS in positive ion mode: Circumventing corona discharge. Electrophoresis 2020, 41, 360–369. [Google Scholar] [CrossRef]
- Drouin, N.; van Mever, M.; Zhang, W.; Tobolkina, E.; Ferre, S.; Servais, A.-C.; Gou, M.-J.; Nyssen, L.; Fillet, M.; Lageveen-Kammeijer, G.S.M.; et al. Capillary electrophoresis-mass spectrometry at trial by metabo-ring: Effective electrophoretic mobility for reproducible and robust compound annotation. Anal. Chem. 2020, 92, 14103–14112. [Google Scholar] [CrossRef] [PubMed]
- Stafsnes, M.H.; Rost, L.M.; Bruheim, P. Improved phosphometabolome profiling applying isotope dilution strategy and capillary ion chromatography-tandem mass spectrometry. J. Chromatogr. B 2018, 1083, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Schwaiger, M.; Rampler, E.; Hermann, G.; Miklos, W.; Berger, W.; Koellensperger, G. Anion-exchange chromatography coupled to high-resolution mass spectrometry: A powerful tool for merging targeted and non-targeted metabolomics. Anal. Chem. 2017, 89, 7667–7674. [Google Scholar] [CrossRef]
- Walsby-Tickle, J.; Gannon, J.; Hvinden, I.; Bardella, C.; Abboud, M.I.; Nazeer, A.; Hauton, D.; Pires, E.; Cadoux-Hudson, T.; Schofield, C.J.; et al. Anion-exchange chromatography mass spectrometry provides extensive coverage of primary metabolic pathways revealing altered metabolism in IDH1 mutant cells. Commun. Biol. 2020, 3. [Google Scholar] [CrossRef]
- Cech, N.B.; Enke, C.G. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 2001, 20, 362–387. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-B.; Liu, D.-B.; Guo, X.M.; Yu, S.-J.; Yu, P. Improvement of sugar analysis sensitivity using anion-exchange chromatography-electrospray ionization mass spectrometry with sheath liquid interface. J. Chromatogr. A 2014, 1366, 65–72. [Google Scholar] [CrossRef]
- Shi, G.; Wu, J.T.; Li, Y.; Geleziunas, R.; Gallagher, K.; Emm, T.; Olah, T.; Unger, S. Novel direct detection method for quantitative determination of intracellular nucleoside triphosphates using weak anion exchange liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2002, 16, 1092–1099. [Google Scholar] [CrossRef]
- Cech, N.B.; Enke, C.G. Relating electrospray ionization response to nonpolar character of small peptides. Anal. Chem. 2000, 72, 2717–2723. [Google Scholar] [CrossRef] [PubMed]
- Bonfiglio, R.; King, R.C.; Olah, T.V.; Merkle, K. The effects of sample preparation methods on the variability of the electrospray ionization response for model drug compounds. Rapid Commun. Mass Spectrom. 1999, 13, 1175–1185. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Havlikova, L.; Vlckova, H.; Solich, P.; Novakova, L. HILIC UHPLC-MS/MS for fast and sensitive bioanalysis: Accounting for matrix effects in method development. Bioanalysis 2013, 5, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Strzelecka, D.; Chmielinski, S.; Bednarek, S.; Jemielity, J.; Kowalska, J. Analysis of mononucleotides by tandem mass spectrometry: Investigation of fragmentation pathways for phosphate- and ribose-modified nucleotide analogues. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, C.G.; Krajete, A. Sheath liquid effects in capillary high-performance liquid chromatography-electrospray mass spectrometry of oligonucleotides. J. Chromatogr. A 2000, 870, 413–424. [Google Scholar] [CrossRef]
- Apffel, A.; Chakel, J.A.; Fischer, S.; Lichtenwalter, K.; Hancock, W.S. Analysis of oligonucleotides by HPLC-electrospray ionization mass spectrometry. Anal. Chem. 1997, 69, 1320–1325. [Google Scholar] [CrossRef]
- Griffey, R.H.; Greig, M.J.; Gaus, H.J.; Liu, K.; Monteith, D.; Winniman, M.; Cummins, L.L. Characterization of oligonucleotide metabolism in vivo via liquid chromatography electrospray tandem mass spectrometry with a quadrupole ion trap mass spectrometer. J. Mass Spectrom. 1997, 32, 305–313. [Google Scholar] [CrossRef]
- Wu, Z.; Gao, W.; Phelps, M.A.; Wu, D.; Miller, D.D.; Dalton, J.T. Favorable effects of weak acids on negative-ion electrospray ionization mass spectrometry. Anal. Chem. 2004, 76, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Megherbi, M.; Jordheim, L.P.; Lefebvre, I.; Perigaud, C.; Dumontet, C.; Guitton, J. Simultaneous analysis of eight nucleoside triphosphates in cell lines by liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B 2009, 877, 3831–3840. [Google Scholar] [CrossRef]
- Guo, S.; Duan, J.; Qian, D.; Wang, H.; Tang, Y.; Qian, Y.; Wu, D.; Su, S.; Shang, E. Hydrophilic interaction ultra-high performance liquid chromatography coupled with triple quadrupole mass spectrometry for determination of nucleotides, nucleosides and nucleobases in Ziziphus plants. J. Chromatogr. A 2013, 1301, 147–155. [Google Scholar] [CrossRef]
- Zhou, S.L.; Cook, K.D. Protonation in electrospray mass spectrometry: Wrong-way-round or right-way-round? J. Am. Soc. Mass Spectrom. 2000, 11, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Amad, M.H.; Cech, N.B.; Jackson, G.S.; Enke, C.G. Importance of gas-phase proton affinities in determining the electrospray ionization response for analytes and solvents. J. Mass Spectrom. 2000, 35, 784–789. [Google Scholar] [CrossRef]
- Green-Church, K.B.; Limbach, P.A. Mononucleotide gas-phase proton affinities as determined by the kinetic method. J. Am. Soc. Mass Spectrom. 2000, 11, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Birdsall, R.E.; Gilar, M.; Shion, H.; Yu, Y.Q.; Chen, W. Reduction of metal adducts in oligonucleotide mass spectra in ion-pair reversed-phase chromatography/mass spectrometry analysis. Rapid Commun. Mass Spectrom. 2016, 30, 1667–1679. [Google Scholar] [CrossRef]
- Schug, K.; McNair, H.M. Adduct formation in electrospray ionization mass spectrometry II. Benzoic acid derivatives. J. Chromatogr. A 2003, 985, 531–539. [Google Scholar] [CrossRef]
- Harvey, D.J. Collision-induced fragmentation of underivatized N-linked carbohydrates ionized by electrospray. J. Mass Spectrom. 2000, 35, 1178–1190. [Google Scholar] [CrossRef]
- Ernst, M.; Silva, D.B.; Silva, R.R.; Vencio, R.Z.N.; Lopes, N.P. Mass spectrometry in plant metabolomics strategies: From analytical platforms to data acquisition and processing. Nat. Prod. Rep. 2014, 31, 784–806. [Google Scholar] [CrossRef]
- Erngren, I.; Haglof, J.; Engskog, M.K.R.; Nestor, M.; Hedeland, M.; Arvidsson, T.; Pettersson, C. Adduct formation in electrospray ionisation-mass spectrometry with hydrophilic interaction liquid chromatography is strongly affected by the inorganic ion concentration of the samples. J. Chromatogr. A 2019, 1600, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Vessecchi, R.; Crotti, A.E.M.; Guaratini, T.; Colepicolo, P.; Galembeck, S.E.; Lopes, N.P. Radical ion generation processes of organic compounds in electrospray ionization mass spectrometry. Mini Rev. Org. Chem. 2007, 4, 75–87. [Google Scholar] [CrossRef]
- Studzinska, S.; Siecinska, L.; Buszewski, B. On-line electrochemistry/electrospray ionization mass spectrometry (EC-ESI-MS) system for the study of nucleosides and nucleotides oxidation products. J. Pharm. Biomed. Anal. 2018, 158, 416–424. [Google Scholar] [CrossRef]
- Weissberg, A.; Dagan, S. Interpretation of ESI(+)-MS-MS spectra—Towards the identification of “unknowns”. Int. J. Mass Spectrom. 2011, 299, 158–168. [Google Scholar] [CrossRef]
- Xu, Y.-F.; Lu, W.; Rabinowitz, J.D. Avoiding misannotation of in-source fragmentation products as cellular metabolites in liquid chromatography-mass spectrometry-based metabolomics. Anal. Chem. 2015, 87, 2273–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondrat, R.W.; McClusky, G.A.; Cooks, R.G. Multiple reaction monitoring in mass spectrometry/mass spectrometry for direct analysis of complex mixtures. Anal. Chem. 1978, 50, 2017–2021. [Google Scholar] [CrossRef]
- Moerlein, S.; Schuster, C.; Paal, M.; Vogeser, M. Collision energy-breakdown curves—An additional tool to characterize MS/MS methods. Clin. Mass Spectrom. 2020, 18, 48–53. [Google Scholar] [CrossRef]
- Glauser, G.; Veyrat, N.; Rochat, B.; Wolfender, J.-L.; Turlings, T.C.J. Ultra-high pressure liquid chromatography-mass spectrometry for plant metabolomics: A systematic comparison of high-resolution quadrupole-time-of-flight and single stage Orbitrap mass spectrometers. J. Chromatogr. A 2013, 1292, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A. High-resolution mass spectrometry for bioanalytical applications: Is this the new gold standard? J. Mass Spectrom. 2020, 55. [Google Scholar] [CrossRef]
- Chen, M.; Witte, C.-P. A kinase and a glycosylase catabolize pseudouridine in the peroxisome to prevent toxic pseudouridine monophosphate accumulation. Plant Cell 2020, 32, 722–739. [Google Scholar] [CrossRef]
- Chen, M.; Urs, M.J.; Sanchez-Gonzalez, I.; Olayioye, M.A.; Herde, M.; Witte, C.-P. m(6)A RNA degradation products are catabolized by an evolutionarily conserved N-6-Methyl-AMP deaminase in plant and mammalian cells. Plant Cell 2018, 30, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galperin, M.Y.; Moroz, O.V.; Wilson, K.S.; Murzin, A.G. House cleaning, a part of good housekeeping. Mol. Microbiol. 2006, 59, 5–19. [Google Scholar] [CrossRef]
- Senkler, J.; Rugen, N.; Eubel, H.; Hegermann, J.; Braun, H.-P. Absence of complex I implicates rearrangement of the respiratory chain in european mistletoe. Curr. Biol. 2018, 28, 1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaiger, M.; Schoeny, H.; El Abiead, Y.; Hermann, G.; Rampler, E.; Koellensperger, G. Merging metabolomics and lipidomics into one analytical run. Analyst 2019, 144, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voon, C.P.; Guan, X.; Sun, Y.; Sahu, A.; Chan, M.N.; Gardestrom, P.; Wagner, S.; Fuchs, P.; Nietzel, T.; Versaw, W.K.; et al. ATP compartmentation in plastids and cytosol of Arabidopsis thaliana revealed by fluorescent protein sensing. Proc. Natl. Acad. Sci. USA 2018, 115, E10778–E10787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbeck, J.; Fuchs, P.; Negroni, Y.L.; Elsaesser, M.; Lichtenauer, S.; Stockdreher, Y.; Feitosa-Araujo, E.; Kroll, J.B.; Niemeier, J.-O.; Humberg, C.; et al. In Vivo NADH/NAD+ Biosensing Reveals the Dynamics of Cytosolic Redox Metabolism in Plants. Plant Cell 2020, 32, 3324–3345. [Google Scholar] [CrossRef] [PubMed]
- Arrivault, S.; Guenther, M.; Florian, A.; Encke, B.; Feil, R.; Vosloh, D.; Lunn, J.E.; Sulpice, R.; Fernie, A.R.; Stitt, M. Dissecting the subcellular compartmentation of proteins and metabolites in arabidopsis leaves using non-aqueous fractionation. Mol. Cell. Proteom. 2014, 13, 2246–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuertauer, L.; Kuestner, L.; Weckwerth, W.; Heyer, A.G.; Naegele, T. Resolving subcellular plant metabolism. Plant J. 2019, 100, 438–455. [Google Scholar] [CrossRef] [Green Version]
- Niehaus, M.; Straube, H.; Kuenzler, P.; Rugen, N.; Hegermann, J.; Giavalisco, P.; Eubel, H.; Witte, C.-P.; Herde, M. Rapid affinity purification of tagged plant mitochondria (Mito-AP) for metabolome and proteome analyses. Plant Physiol. 2020, 182, 1194–1210. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Technique | Achievement | Reference |
|---|---|---|---|
| 1964 | Different extraction methods compared by enzymatic assays | Identification of plant-specific problems during the extraction of phosphorylated metabolites | [5] |
| 1972 | Acid quenching combined with thin-layer chromatography | Comprehensive identification of radiolabeled nucleotide triphosphates in plants | [9] |
| 1975 | Acid quenching combined with ion-exchange LC a coupled to a UV b-detector | Establishment of a workflow that allows the proper quenching of enzyme activity by acidic conditions coupled with a liquid–liquid extraction removing the acid from the extractant, allowing for chromatographic separation by ion-exchange LC | [10] |
| 1991 | Acid quenching combined with β-elimination and LC–UV | Analysis of ribo- and deoxyribonucleotides in plants by LC–UV | [11] |
| 2010 | Graphitized carbon SPE c combined with porous graphitized carbon chromatography MS d | Comprehensive analysis of ribonucleotides and nucleotide sugars in plants utilizing porous graphitized carbon chromatography | [12] |
| 2011 | SPE combined with liquid chromatography and time-of-flight MS | Simultaneous analysis of 23 nucleotides and cofactors | [13] |
| 2013 | SPE combined with LC–MS | Comprehensive analysis of nucleosides and nucleobases | [14] |
| 2020 | SPE combined with LC–MS | Comprehensive analysis of nucleotides and nucleosides | [2] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Straube, H.; Witte, C.-P.; Herde, M. Analysis of Nucleosides and Nucleotides in Plants: An Update on Sample Preparation and LC–MS Techniques. Cells 2021, 10, 689. https://doi.org/10.3390/cells10030689
Straube H, Witte C-P, Herde M. Analysis of Nucleosides and Nucleotides in Plants: An Update on Sample Preparation and LC–MS Techniques. Cells. 2021; 10(3):689. https://doi.org/10.3390/cells10030689
Chicago/Turabian StyleStraube, Henryk, Claus-Peter Witte, and Marco Herde. 2021. "Analysis of Nucleosides and Nucleotides in Plants: An Update on Sample Preparation and LC–MS Techniques" Cells 10, no. 3: 689. https://doi.org/10.3390/cells10030689