
AGMO Inhibitor Reduces 3T3-L1 Adipogenesis

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microsome Preparation
2.2. Alkylglycerol Monooxygenase Activity Assay and Inhibitor Screening
2.3. Fatty Aldehyde Dehydrogenase Activity Assay
2.4. Phenylalanine Hydroxylase Activity Assay
2.5. Nitric Oxide Synthase Activity Assay
2.6. Culture and Differentiation of Mouse 3T3-L1 Preadipocytes
2.7. RAW264.7 Cell Lines and Culture
2.8. Culture and Free Fatty Acid Induced Cytotoxicity in HepG2 Cells
2.9. Cytotoxicity Assays
2.10. Oil-Red O Staining
2.11. Cell Cycle Analysis Using Flow Cytometry
2.12. Quantitative Real-Time PCR
2.13. Immunofluorescence
2.14. Animals
2.15. Statistics
3. Results
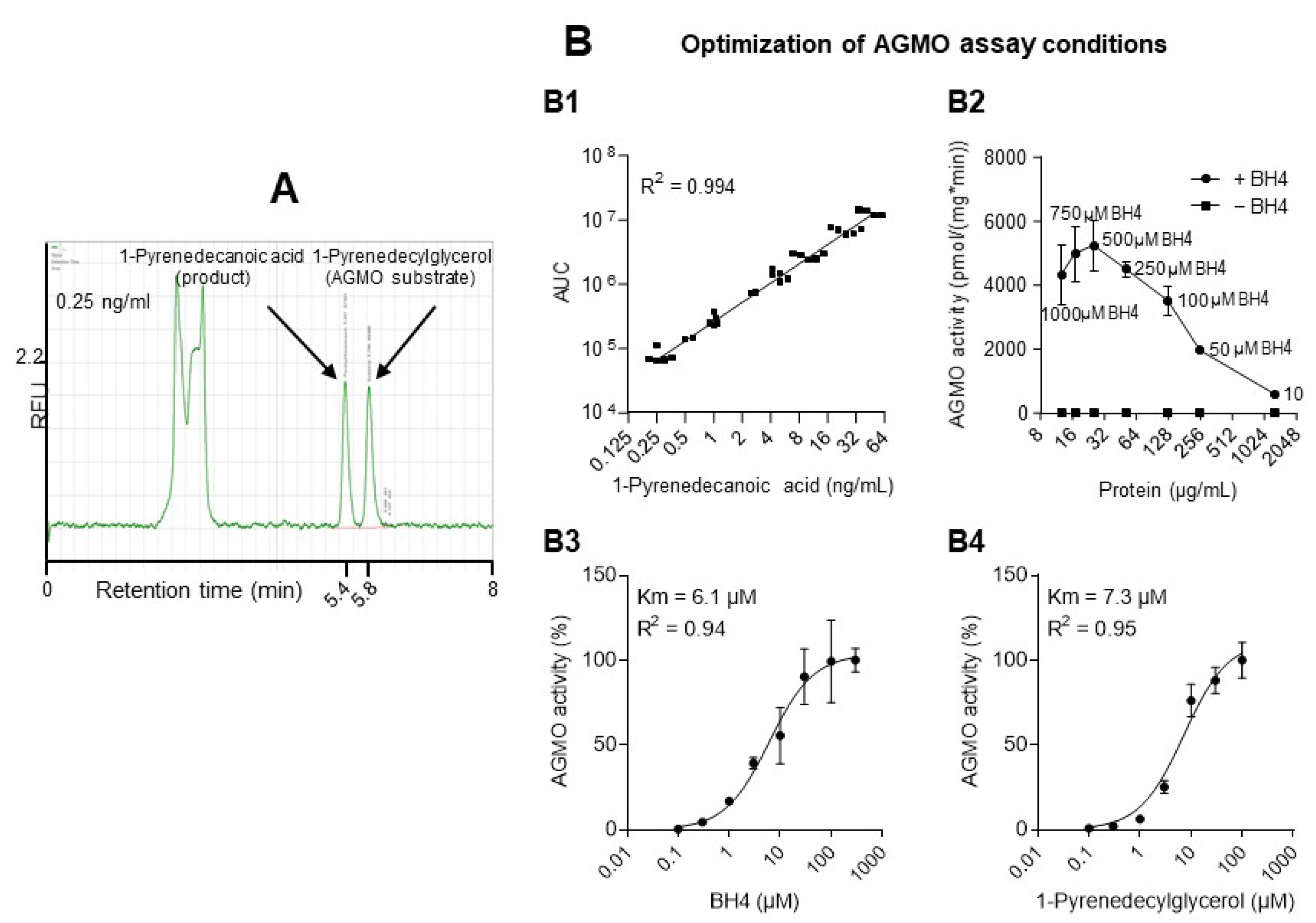
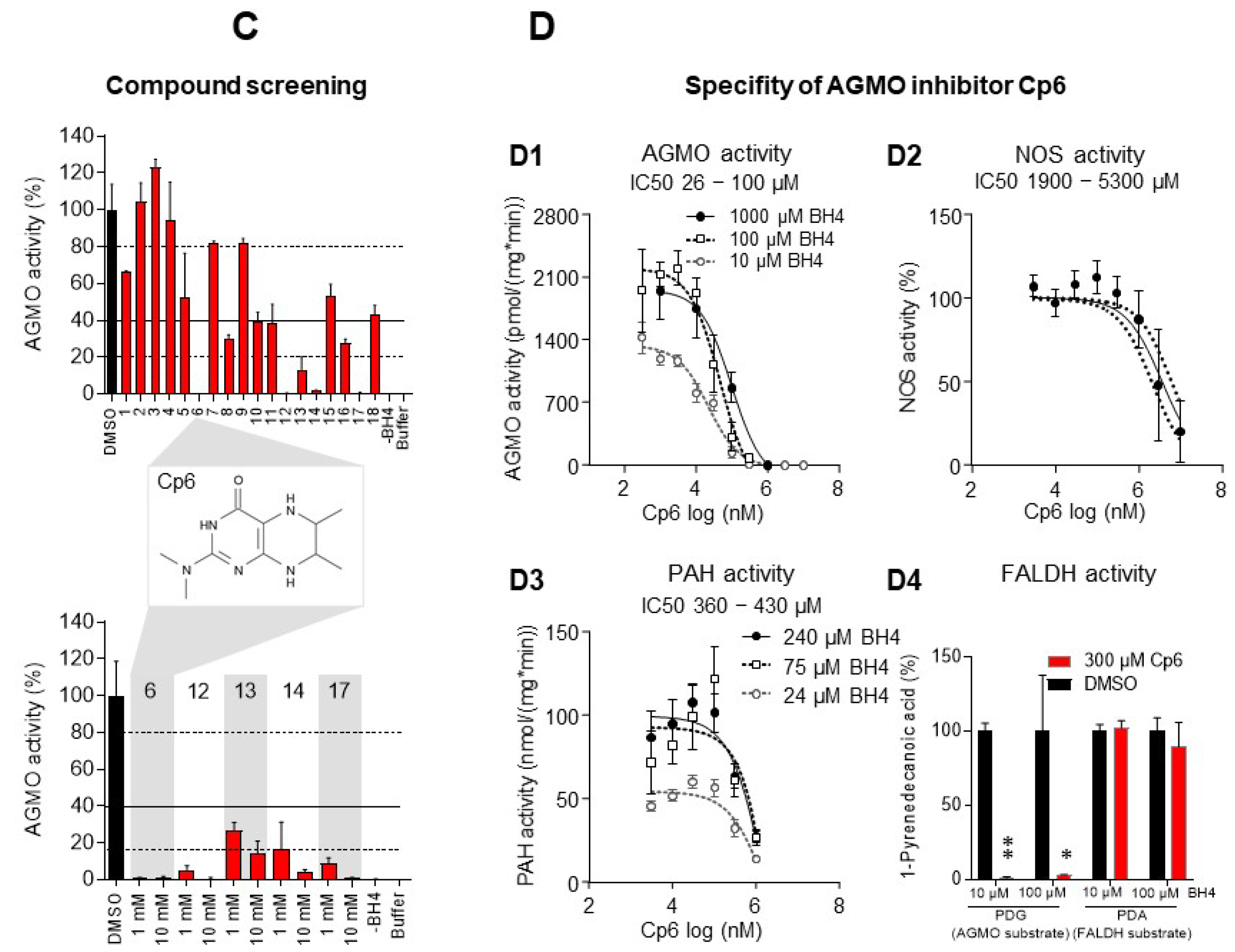
3.1. AGMO Activity Assay and Compound Screen
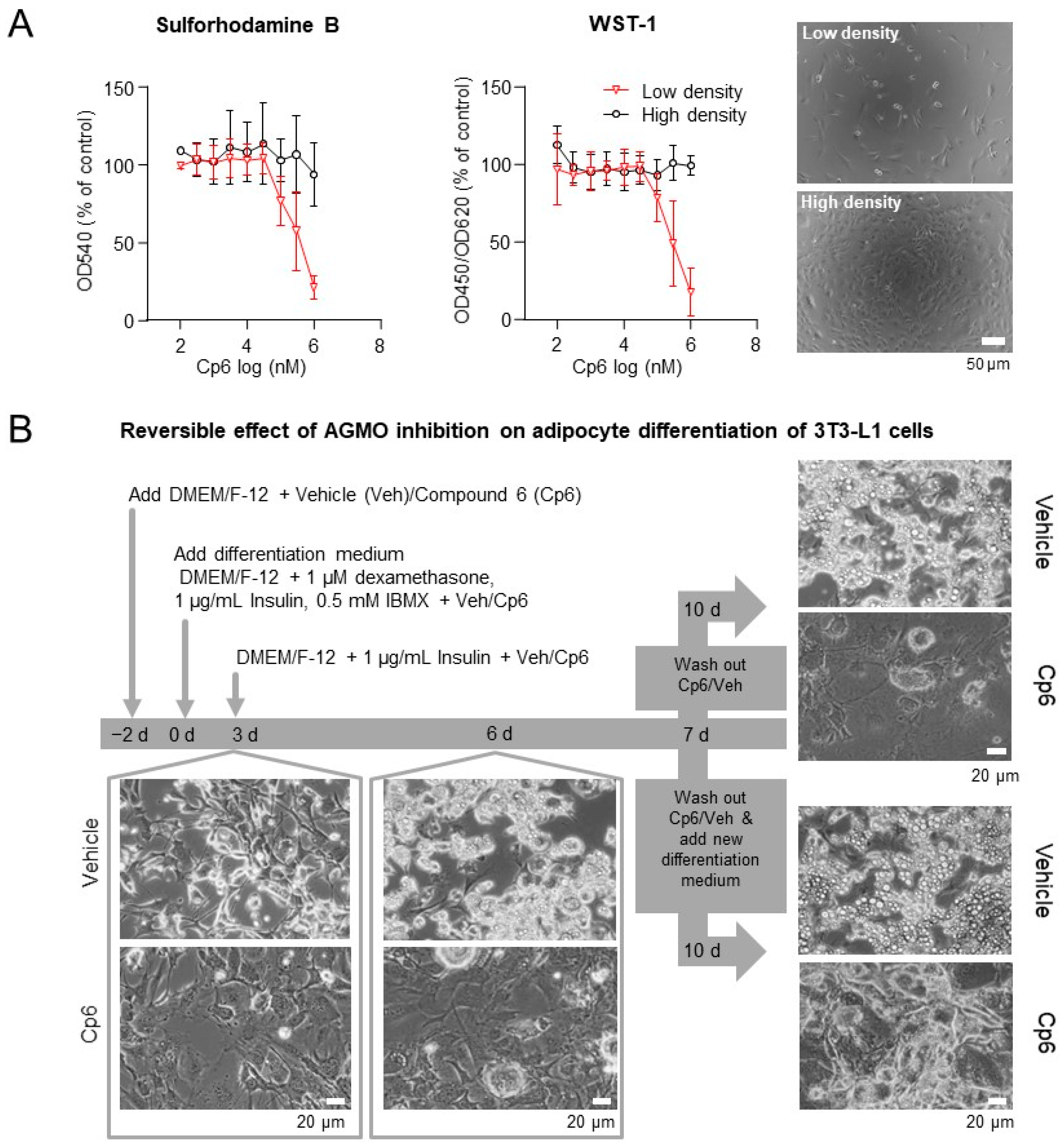
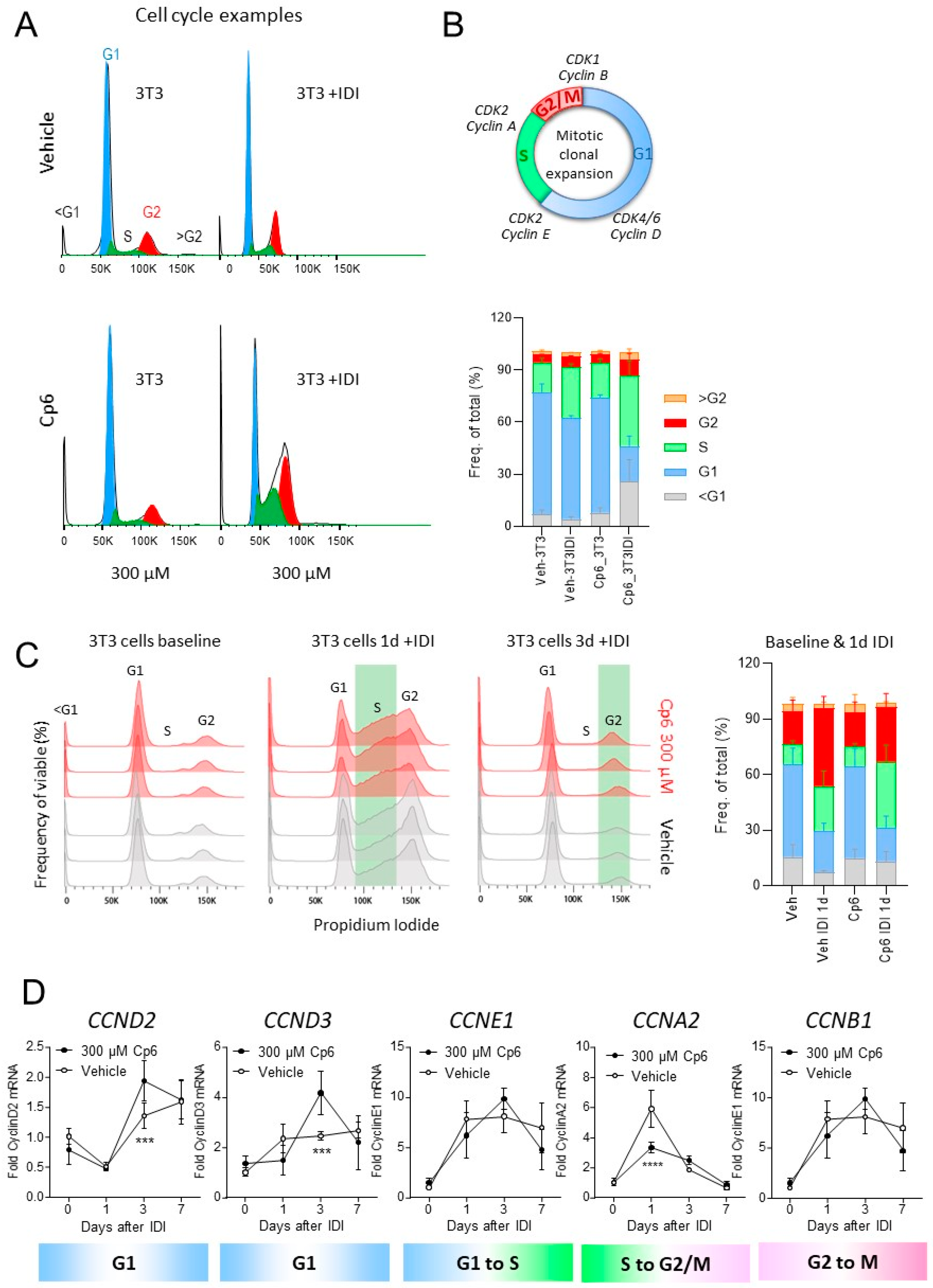
3.2. Cp6 Inhibits Adipogenesis in 3T3-L1 Cells
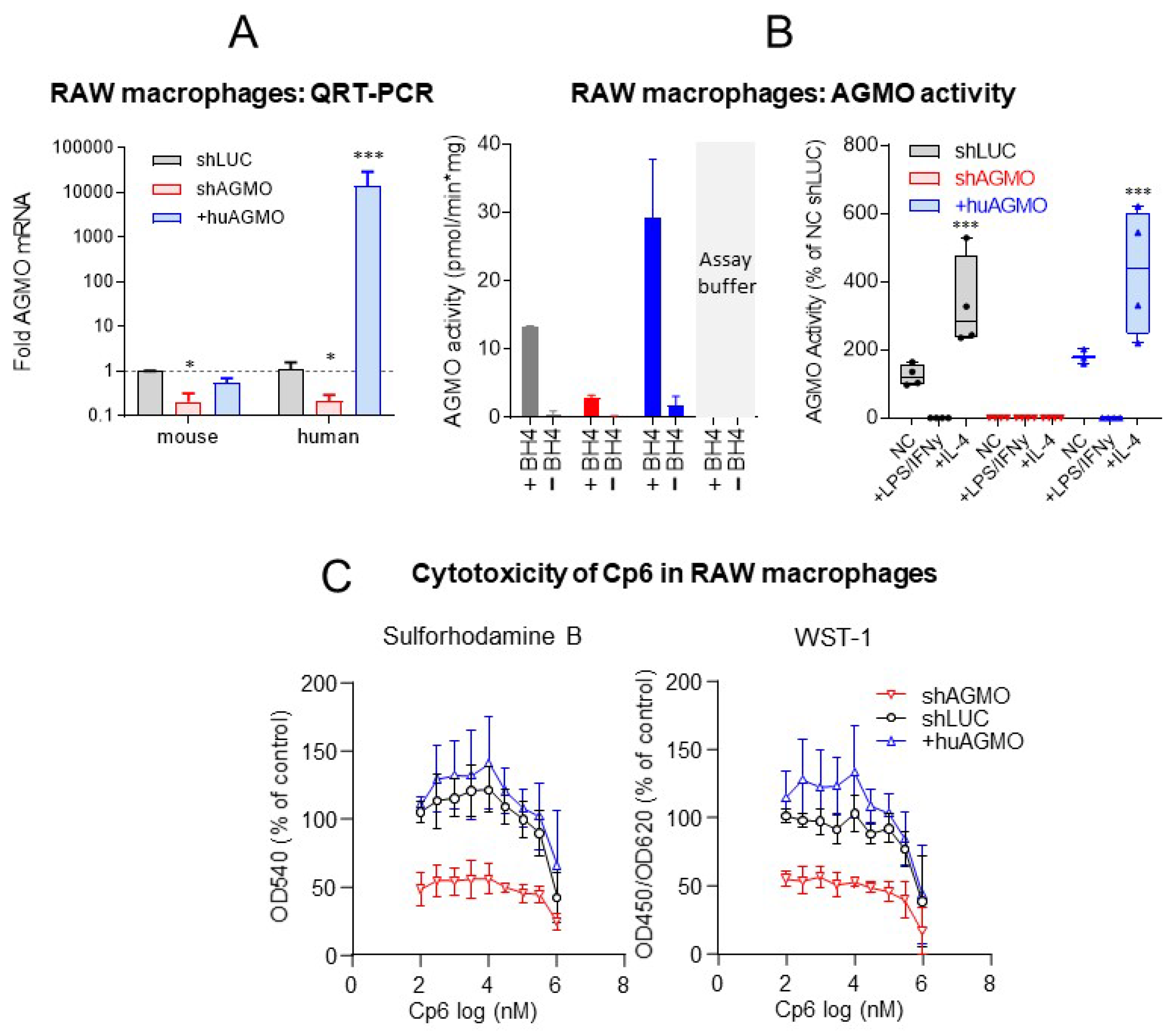
3.3. Cp6 Does Not Affect Macrophage Proliferation but Blocks the M1 to M2 Polarization
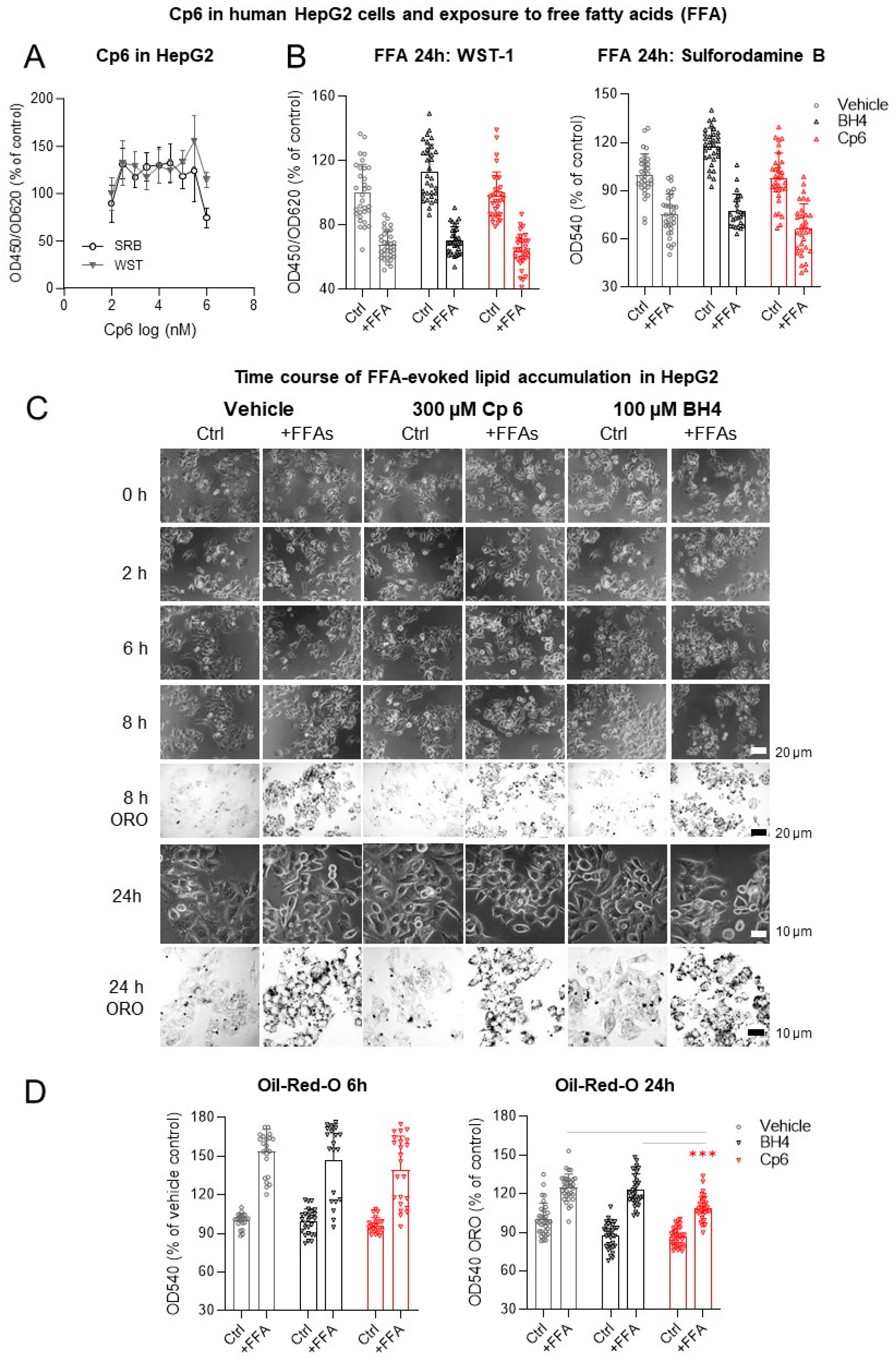
3.4. AGMO Is Upregulated in the Liver under HFD but Cp6 Does Not Protect against Free Fatty Acid-Induced Toxicity in Hepatocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Lohner, K. Is the high propensity of ethanolamine plasmalogens to form non-lamellar lipid structures manifested in the properties of biomembranes? Chem. Phys. Lipids 1996, 81, 167–184. [Google Scholar] [CrossRef]
- Paltauf, F. Ether lipids in biomembranes. Chem. Phys. Lipids 1994, 74, 101–139. [Google Scholar] [CrossRef]
- Pike, L.J.; Han, X.; Chung, K.N.; Gross, R.W. Lipid rafts are enriched in arachidonic acid and plasmenylethanolamine and their composition is independent of caveolin-1 expression: A quantitative electrospray ionization/mass spectrometric analysis. Biochemistry 2002, 41, 2075–2088. [Google Scholar] [CrossRef]
- Benjamin, D.I.; Cozzo, A.; Ji, X.; Roberts, L.S.; Louie, S.M.; Mulvihill, M.M.; Luo, K.; Nomura, D.K. Ether lipid generating enzyme agps alters the balance of structural and signaling lipids to fuel cancer pathogenicity. Proc. Natl. Acad. Sci. USA 2013, 110, 14912–14917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, C.J.; Thukkani, A.K.; Heuertz, R.M.; Slungaard, A.; Hazen, S.L.; Ford, D.A. Eosinophil peroxidase-derived reactive brominating species target the vinyl ether bond of plasmalogens generating a novel chemoattractant, alpha-bromo fatty aldehyde. J. Biol. Chem. 2003, 278, 8942–8950. [Google Scholar] [CrossRef] [Green Version]
- Brodde, A.; Teigler, A.; Brugger, B.; Lehmann, W.D.; Wieland, F.; Berger, J.; Just, W.W. Impaired neurotransmission in ether lipid-deficient nerve terminals. Hum. Mol. Genet. 2012, 21, 2713–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, T.F.; Eira, J.; Lopes, A.T.; Malheiro, A.R.; Sousa, V.; Luoma, A.; Avila, R.L.; Wanders, R.J.; Just, W.W.; Kirschner, D.A.; et al. Peripheral nervous system plasmalogens regulate schwann cell differentiation and myelination. J. Clin. Investig. 2014, 124, 2560–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdlenbruch, B.; Jendrossek, V.; Eibl, H.; Lakomek, M. Transient and controllable opening of the blood-brain barrier to cytostatic and antibiotic agents by alkylglycerols in rats. Exp. Brain Res. 2000, 135, 417–422. [Google Scholar] [PubMed]
- Fakhro, K.A.; Choi, M.; Ware, S.M.; Belmont, J.W.; Towbin, J.A.; Lifton, R.P.; Khokha, M.K.; Brueckner, M. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc. Natl. Acad. Sci. USA 2011, 108, 2915–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, P.L.; Mankidy, R.; Ritchie, S.; Heath, D.; Wood, J.A.; Flax, J.; Goodenowe, D.B. Circulating plasmalogen levels and alzheimer disease assessment scale-cognitive scores in alzheimer patients. J. Psychiatry Neurosci. 2010, 35, 59–62. [Google Scholar] [CrossRef] [Green Version]
- Meikle, P.J.; Wong, G.; Barlow, C.K.; Weir, J.M.; Greeve, M.A.; MacIntosh, G.L.; Almasy, L.; Comuzzie, A.G.; Mahaney, M.C.; Kowalczyk, A.; et al. Plasma lipid profiling shows similar associations with prediabetes and type 2 diabetes. PLoS ONE 2013, 8, e74341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Chen, H.; Dai, M.; Ai, J.; Li, Y.; Mahon, B.; Dai, S.; Deng, Y. Plasma lipidomics profiling identified lipid biomarkers in distinguishing early-stage breast cancer from benign lesions. Oncotarget 2016, 7, 36622–36631. [Google Scholar] [CrossRef]
- Piano, V.; Benjamin, D.I.; Valente, S.; Nenci, S.; Marrocco, B.; Mai, A.; Aliverti, A.; Nomura, D.K.; Mattevi, A. Discovery of inhibitors for the ether lipid-generating enzyme agps as anti-cancer agents. ACS Chem. Biol. 2015, 10, 2589–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiroz, A.; Pinto, I.F.D.; Lima, M.; Giovanetti, M.; De Jesus, J.G.; Xavier, J.; Barreto, F.K.; Canuto, G.A.B.; Do Amaral, H.R.; De Filippis, A.M.B.; et al. Lipidomic analysis reveals serum alteration of plasmalogens in patients infected with zika virus. Front. Microbiol. 2019, 10, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Rojo, N.; Riezman, H. On the road to unraveling the molecular functions of ether lipids. FEBS Lett. 2019, 593, 2378–2389. [Google Scholar] [CrossRef] [Green Version]
- Jean Beltran, P.M.; Cook, K.C.; Hashimoto, Y.; Galitzine, C.; Murray, L.A.; Vitek, O.; Cristea, I.M. Infection-induced peroxisome biogenesis is a metabolic strategy for herpesvirus replication. Cell Host Microbe 2018, 24, 526–541.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cara, F.; Sheshachalam, A.; Braverman, N.E.; Rachubinski, R.A.; Simmonds, A.J. Peroxisome-mediated metabolism is required for immune response to microbial infection. Immunity 2017, 47, 93–106.e107. [Google Scholar] [CrossRef] [Green Version]
- Zschiebsch, K.; Fischer, C.; Pickert, G.; Haussler, A.; Radeke, H.; Grosch, S.; Ferreiros, N.; Geisslinger, G.; Werner, E.R.; Tegeder, I. Tetrahydrobiopterin attenuates dss-evoked colitis in mice by rebalancing redox and lipid signalling. J. Crohns Colitis 2016, 10, 965–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuoka, S.M.; Kita, Y.; Shindou, H.; Shimizu, T. Alkylglycerol monooxygenase as a potential modulator for paf synthesis in macrophages. Biochem. Biophys. Res. Commun. 2013, 436, 306–312. [Google Scholar] [CrossRef]
- Engeli, S.; Bohnke, J.; Feldpausch, M.; Gorzelniak, K.; Janke, J.; Batkai, S.; Pacher, P.; Harvey-White, J.; Luft, F.C.; Sharma, A.M.; et al. Activation of the peripheral endocannabinoid system in human obesity. Diabetes 2005, 54, 2838–2843. [Google Scholar] [CrossRef] [Green Version]
- Gonthier, M.P.; Hoareau, L.; Festy, F.; Matias, I.; Valenti, M.; Bes-Houtmann, S.; Rouch, C.; Robert-Da Silva, C.; Chesne, S.; Lefebvre d’Hellencourt, C.; et al. Identification of endocannabinoids and related compounds in human fat cells. Obesity 2007, 15, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, J.L.; Joseph, B.K.; Ruckle, M.B.; Mayeux, P.R.; Prather, P.L. The endocannabinoid noladin ether acts as a full agonist at human cb2 cannabinoid receptors. J. Pharmacol. Exp. Ther. 2005, 314, 868–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, K.; Mangels, N.; Haussler, A.; Ferreiros, N.; Fleming, I.; Tegeder, I. Pro-inflammatory obesity in aged cannabinoid-2 receptor-deficient mice. Int. J. Obes. 2015, 25, 169. [Google Scholar] [CrossRef] [PubMed]
- Watschinger, K.; Keller, M.A.; Golderer, G.; Hermann, M.; Maglione, M.; Sarg, B.; Lindner, H.H.; Hermetter, A.; Werner-Felmayer, G.; Konrat, R.; et al. Identification of the gene encoding alkylglycerol monooxygenase defines a third class of tetrahydrobiopterin-dependent enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 13672–13677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renstrom, F.; Shungin, D.; Johansson, I.; Investigators, M.; Florez, J.C.; Hallmans, G.; Hu, F.B.; Franks, P.W. Genetic predisposition to long-term nondiabetic deteriorations in glucose homeostasis: Ten-year follow-up of the glacier study. Diabetes 2011, 60, 345–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boesgaard, T.W.; Grarup, N.; Jorgensen, T.; Borch-Johnsen, K.; Meta-Analysis of, G.; Insulin-Related Trait, C.; Hansen, T.; Pedersen, O. Variants at dgkb/tmem195, adra2a, glis3 and c2cd4b loci are associated with reduced glucose-stimulated beta cell function in middle-aged danish people. Diabetologia 2010, 53, 1647–1655. [Google Scholar] [CrossRef] [Green Version]
- Goodarzi, M.O.; Guo, X.; Cui, J.; Jones, M.R.; Haritunians, T.; Xiang, A.H.; Chen, Y.D.; Taylor, K.D.; Buchanan, T.A.; Hsueh, W.A.; et al. Systematic evaluation of validated type 2 diabetes and glycaemic trait loci for association with insulin clearance. Diabetologia 2013, 56, 1282–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, E.; Chen, G.; Shriner, D.; Doumatey, A.; Gerry, N.P.; Herbert, A.; Huang, H.; Zhou, J.; Christman, M.F.; Adeyemo, A.; et al. Replication of genome-wide association studies (gwas) loci for fasting plasma glucose in african-americans. Diabetologia 2011, 54, 783–788. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.R. Three classes of tetrahydrobiopterin-dependent enzymes. Pteridines 2013, 24, 7–11. [Google Scholar] [CrossRef]
- Werner, E.R.; Hermetter, A.; Prast, H.; Golderer, G.; Werner-Felmayer, G. Widespread occurrence of glyceryl ether monooxygenase activity in rat tissues detected by a novel assay. J. Lipid Res. 2007, 48, 1422–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotting, J.; Unger, C.; Eibl, H. Substrate specificity of o-alkylglycerol monooxygenase (e.C. 1.14.16.5), solubilized from rat liver microsomes. Lipids 1987, 22, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Hofer, D.C.; Pessentheiner, A.R.; Pelzmann, H.J.; Schlager, S.; Madreiter-Sokolowski, C.T.; Kolb, D.; Eichmann, T.O.; Rechberger, G.; Bilban, M.; Graier, W.F.; et al. Critical role of the peroxisomal protein pex16 in white adipocyte development and lipid homeostasis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 358–368. [Google Scholar] [CrossRef]
- Pietilainen, K.H.; Rog, T.; Seppanen-Laakso, T.; Virtue, S.; Gopalacharyulu, P.; Tang, J.; Rodriguez-Cuenca, S.; Maciejewski, A.; Naukkarinen, J.; Ruskeepaa, A.L.; et al. Association of lipidome remodeling in the adipocyte membrane with acquired obesity in humans. PLoS Biol. 2011, 9, e1000623. [Google Scholar] [CrossRef] [Green Version]
- Homan, E.A.; Kim, Y.G.; Cardia, J.P.; Saghatelian, A. Monoalkylglycerol ether lipids promote adipogenesis. J. Am. Chem. Soc. 2011, 133, 5178–5181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppe, A.A.; Carey, G.B. Polybrominated diphenyl ethers as endocrine disruptors of adipocyte metabolism. Obesity 2007, 15, 2942–2950. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cao, S.; Arhatte, M.; Li, D.; Shi, Y.; Kurz, S.; Hu, J.; Wang, L.; Shao, J.; Atzberger, A.; et al. Adipocyte piezo1 mediates obesogenic adipogenesis through the fgf1/fgfr1 signaling pathway in mice. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Fujiwara, K.; Mori, K.; Kaneko, Y.S.; Nakashima, A.; Nagasaka, A.; Itoh, M.; Ota, A. Tetrahydrobiopterin biosynthesis in white and brown adipose tissues is enhanced following intraperitoneal administration of bacterial lipopolysaccharide. Biochim. Biophys. Acta 2004, 1670, 181–198. [Google Scholar] [CrossRef]
- Hill, A.A.; Reid Bolus, W.; Hasty, A.H. A decade of progress in adipose tissue macrophage biology. Immunol. Rev. 2014, 262, 134–152. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Watschinger, K.; Keller, M.A.; McNeill, E.; Alam, M.T.; Lai, S.; Sailer, S.; Rauch, V.; Patel, J.; Hermetter, A.; Golderer, G.; et al. Tetrahydrobiopterin and alkylglycerol monooxygenase substantially alter the murine macrophage lipidome. Proc. Natl. Acad. Sci. USA 2015, 112, 2431–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, E.R.; Habisch, H.J.; Gorren, A.C.; Schmidt, K.; Canevari, L.; Werner-Felmayer, G.; Mayer, B. Contrasting effects of n5-substituted tetrahydrobiopterin derivatives on phenylalanine hydroxylase, dihydropteridine reductase and nitric oxide synthase. Biochem. J. 2000, 348 Pt 3, 579–583. [Google Scholar] [CrossRef]
- Kraus, N.A.; Ehebauer, F.; Zapp, B.; Rudolphi, B.; Kraus, B.J.; Kraus, D. Quantitative assessment of adipocyte differentiation in cell culture. Adipocyte 2016, 5, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.V.; Chambers, S.H.; Smith, P.J. A pragmatic approach to the analysis of DNA histograms with a definable g1 peak. Cytometry 1987, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Watschinger, K.; Fuchs, J.E.; Yarov-Yarovoy, V.; Keller, M.A.; Golderer, G.; Hermetter, A.; Werner-Felmayer, G.; Hulo, N.; Werner, E.R. First insights into structure-function relationships of alkylglycerol monooxygenase. Pteridines 2013, 24. [Google Scholar] [CrossRef] [Green Version]
- Strack, E.; Rolfe, P.A.; Fink, A.F.; Bankov, K.; Schmid, T.; Solbach, C.; Savai, R.; Sha, W.; Pradel, L.; Hartmann, S.; et al. Identification of tumor-associated macrophage subsets that are associated with breast cancer prognosis. Clin. Transl. Med. 2020, 10, e239. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Turkowski, K.; Mora, J.; Brüne, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, A.; Tobe, K. M2-like macrophages serve as a niche for adipocyte progenitors in adipose tissue. J. Diabetes Investig. 2019, 10, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Petkova, A.P.; Granneman, J.G. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 2013, 18, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.K.; Barnard, R.J.; Sindhu, R.K.; Jurczak, M.; Ehdaie, A.; Vaziri, N.D. A high-fat, refined-carbohydrate diet induces endothelial dysfunction and oxidant/antioxidant imbalance and depresses nos protein expression. J. Appl. Physiol. 2005, 98, 203–210. [Google Scholar] [CrossRef]
- Sansbury, B.E.; Cummins, T.D.; Tang, Y.; Hellmann, J.; Holden, C.R.; Harbeson, M.A.; Chen, Y.; Patel, R.P.; Spite, M.; Bhatnagar, A.; et al. Overexpression of endothelial nitric oxide synthase prevents diet-induced obesity and regulates adipocyte phenotype. Circ. Res. 2012, 111, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Gori, M.; Simonelli, M.C.; Giannitelli, S.M.; Businaro, L.; Trombetta, M.; Rainer, A. Investigating nonalcoholic fatty liver disease in a liver-on-a-chip microfluidic device. PLoS ONE 2016, 11, e0159729. [Google Scholar] [CrossRef]
- Geng, Y.; Hernández Villanueva, A.; Oun, A.; Buist-Homan, M.; Blokzijl, H.; Faber, K.N.; Dolga, A.; Moshage, H. Protective effect of metformin against palmitate-induced hepatic cell death. Biochim. Biophys. Acta Mol. Basis. Dis. 2020, 1866, 165621. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Tzieply, N.; Von Knethen, A.; Johann, A.M.; Schmidt, H.; Geisslinger, G.; Brune, B. Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol. Biol. Cell 2007, 18, 3810–3819. [Google Scholar] [CrossRef] [Green Version]
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and its microenvironment: A synergistic interplay. Semin. Cancer Biol. 2013, 23, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Dilbaz, S.; Cossmann, J.; Hoang, A.C.; Diedrich, V.; Herwig, A.; Harauma, A.; Hoshi, Y.; Moriguchi, T.; Landgraf, K.; et al. Breast milk alkylglycerols sustain beige adipocytes through adipose tissue macrophages. J. Clin. Investig. 2019, 129, 2485–2499. [Google Scholar] [CrossRef] [Green Version]
- Okuno, A.; Tamemoto, H.; Tobe, K.; Ueki, K.; Mori, Y.; Iwamoto, K.; Umesono, K.; Akanuma, Y.; Fujiwara, T.; Horikoshi, H.; et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese zucker rats. J. Clin. Investig. 1998, 101, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Sato, D.; Oda, K.; Kusunoki, M.; Nishina, A.; Takahashi, K.; Feng, Z.; Tsutsumi, K.; Nakamura, T. Pparγ activation alters fatty acid composition in adipose triglyceride, in addition to proliferation of small adipocytes, in insulin resistant high-fat fed rats. Eur. J. Pharmacol. 2016, 773, 71–77. [Google Scholar] [CrossRef]
- Pol, A.; Gross, S.P.; Parton, R.G. Review: Biogenesis of the multifunctional lipid droplet: Lipids, proteins, and sites. J. Cell Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brites, P.; Ferreira, A.S.; Da Silva, T.F.; Sousa, V.F.; Malheiro, A.R.; Duran, M.; Waterham, H.R.; Baes, M.; Wanders, R.J. Alkyl-glycerol rescues plasmalogen levels and pathology of ether-phospholipid deficient mice. PLoS ONE 2011, 6, e28539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sailer, S.; Coassin, S.; Lackner, K.; Fischer, C.; McNeill, E.; Streiter, G.; Kremser, C.; Maglione, M.; Green, C.M.; Moralli, D.; et al. When the genome bluffs: A tandem duplication event during generation of a novel agmo knockout mouse model fools routine genotyping. Cell Biosci. 2021, 11, 54. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, C.; Wilken-Schmitz, A.; Hernandez-Olmos, V.; Proschak, E.; Stark, H.; Fleming, I.; Weigert, A.; Thurn, M.; Hofmann, M.; Werner, E.R.; et al. AGMO Inhibitor Reduces 3T3-L1 Adipogenesis. Cells 2021, 10, 1081. https://doi.org/10.3390/cells10051081
Fischer C, Wilken-Schmitz A, Hernandez-Olmos V, Proschak E, Stark H, Fleming I, Weigert A, Thurn M, Hofmann M, Werner ER, et al. AGMO Inhibitor Reduces 3T3-L1 Adipogenesis. Cells. 2021; 10(5):1081. https://doi.org/10.3390/cells10051081
Chicago/Turabian StyleFischer, Caroline, Annett Wilken-Schmitz, Victor Hernandez-Olmos, Ewgenij Proschak, Holger Stark, Ingrid Fleming, Andreas Weigert, Manuela Thurn, Martine Hofmann, Ernst R. Werner, and et al. 2021. "AGMO Inhibitor Reduces 3T3-L1 Adipogenesis" Cells 10, no. 5: 1081. https://doi.org/10.3390/cells10051081