
The Roadmap of RANKL/RANK Pathway in Cancer



Abstract
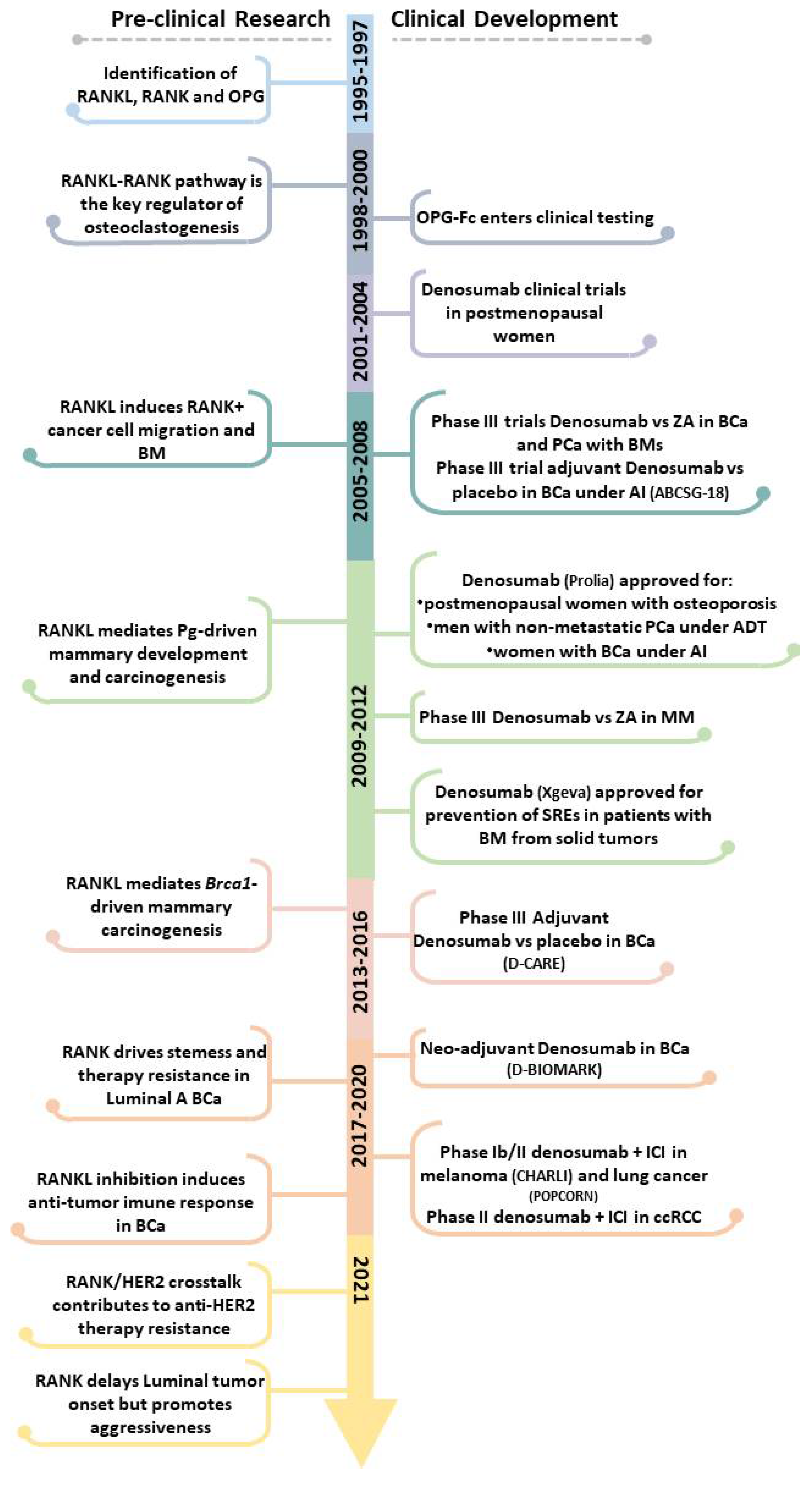
:1. Introduction
2. The Role of RANKL/RANK Pathway in Bone Health and Disease
2.1. Osteoclasts
2.2. (In)Direct Anti-Tumor Effects
2.3. Bone Pre-Metastatic Niche
2.4. Anti-RANKL Therapy in BM: Discovery and Current and Future Perspectives
2.4.1. Bone Metastatic Disease
2.4.2. Prevention of BMs
Breast Cancer
Prostate Cancer
3. RANKL/RANK Pathway in Breast: Friend and Foe
3.1. Breast Carcinogenesis
3.2. Prognosis
3.3. Aggressiveness
3.4. Therapeutic Perspectives of RANKL Inhibition in Early BC beyond BM Prevention
3.4.1. Prevention (BRCA-Mutated BCa)
3.4.2. Neoadjuvant Treatment
4. RANKL/RANK Pathway as a Mediator of Systemic and Tumor Microenvironment (Innate and Acquired) Immunity
4.1. Breast Cancer
4.2. Melanoma and Non-Small Cell Lung Cancer
4.3. (Immuno)Therapeutic Perspectives of RANKL Inhibition
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIs | aromatase inhibitors |
| ASCO | American Society of Clinical Oncology |
| BCa | Breast cancer |
| BM | bone metastases |
| BMFS | bone metastases-free survival |
| BPs | bisphosphonates |
| BTA | bone-targeted agent |
| CRPC | castration-resistant prostate cancer |
| CTCs | circulating cancer cells |
| DC | dendritic cells |
| EBCTCG | Early Breast Cancer Trialists’ Collaborative Group |
| EMT | epithelial-to-mesenchymal transition |
| ER | estrogen receptor |
| ESMO | European Society for Medical Oncology |
| ET | endocrine therapy |
| HR | hormone receptor |
| ICI | immune checkpoint inhibitor |
| IGFs | insulin-like growth factors |
| IHC | immunohistochemistry |
| MM | multiple myeloma |
| MPA | medroxy-progesterone acetate |
| NSCLC | non-small cell lung cancer |
| OPG | osteoprotegerin |
| ORR | objective response rate |
| OS | overall survival |
| PCa | prostate cancer |
| PFS | progression-free survival |
| PR | progesterone receptor |
| RANKL | receptor activator of nuclear factor-κB ligand |
| RCC | renal cell carcinoma |
| RCTs | randomized clinical trials |
| SREs | skeletal-related events |
| TAMs | tumor-associated macrophages |
| TILs | tumor infiltrating lymphocytes |
| TKIs | tyrosine kinase inhibitors |
| TNBCa | triple-negative breast cancer |
| ZA | zoledronate |
References
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef]
- Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; Yasuda, H.; Yano, K.; Morinaga, T.; Higashio, K. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem. Biophys. Res. Commun. 1998, 253, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Schramek, D.; Leibbrandt, A.; Sigl, V.; Kenner, L.; Pospisilik, J.A.; Lee, H.J.; Hanada, R.; Joshi, P.A.; Aliprantis, A.; Glimcher, L.; et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 2010, 468, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Tanos, T.; Sflomos, G.; Echeverria, P.C.; Ayyanan, A.; Gutierrez, M.; Delaloye, J.F.; Raffoul, W.; Fiche, M.; Dougall, W.; Schneider, P.; et al. Progesterone/RANKL is a major regulatory axis in the human breast. Sci. Transl. Med. 2013, 5, 182ra155. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Suarez, E.; Jacob, A.P.; Jones, J.; Miller, R.; Roudier-Meyer, M.P.; Erwert, R.; Pinkas, J.; Branstetter, D.; Dougall, W.C. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010, 468, 103–107. [Google Scholar] [CrossRef]
- Afzal, M.Z.; Shirai, K. Immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy alone versus immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy in combination with anti-RANKL denosumuab in malignant melanoma: A retrospective analysis at a tertiary care center. Melanoma Res. 2018, 28, 341–347. [Google Scholar] [CrossRef]
- Angela, Y.; Haferkamp, S.; Weishaupt, C.; Ugurel, S.; Becker, J.C.; Oberndorfer, F.; Alar, V.; Satzger, I.; Gutzmer, R. Combination of denosumab and immune checkpoint inhibition: Experience in 29 patients with metastatic melanoma and bone metastases. Cancer Immunol. Immunother. 2019, 68, 1187–1194. [Google Scholar] [CrossRef]
- Gomez-Aleza, C.; Gonzalez-Suarez, E. Inhibition of RANK signaling as a potential immunotherapy in breast cancer. Oncoimmunology 2021, 10, 1923156. [Google Scholar] [CrossRef]
- Wong, B.R.; Rho, J.; Arron, J.; Robinson, E.; Orlinick, J.; Chao, M.; Kalachikov, S.; Cayani, E.; Bartlett, F.S., 3rd; Frankel, W.N.; et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J. Biol. Chem. 1997, 272, 25190–25194. [Google Scholar] [CrossRef] [Green Version]
- Wong, B.R.; Josien, R.; Lee, S.Y.; Sauter, B.; Li, H.L.; Steinman, R.M.; Choi, Y. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J. Exp. Med. 1997, 186, 2075–2080. [Google Scholar] [CrossRef] [PubMed]
- Hikita, A.; Yana, I.; Wakeyama, H.; Nakamura, M.; Kadono, Y.; Oshima, Y.; Nakamura, K.; Seiki, M.; Tanaka, S. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappaB ligand. J. Biol. Chem. 2006, 281, 36846–36855. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Ikeda, T.; Kuroyama, H.; Seki, S.; Kasai, M.; Utsuyama, M.; Tatsumi, M.; Uematsu, H.; Hirokawa, K. Regulation of osteoclastogenesis by three human RANKL isoforms expressed in NIH3T3 cells. Biochem. Biophys. Res. Commun. 2004, 314, 1021–1027. [Google Scholar] [CrossRef]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Mochizuki, S.I.; Yano, K.; Fujise, N.; Sato, Y.; Goto, M.; Yamaguchi, K.; Kuriyama, M.; et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): A mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 1998, 139, 1329–1337. [Google Scholar] [CrossRef]
- Capparelli, C.; Morony, S.; Warmington, K.; Adamu, S.; Lacey, D.; Dunstan, C.R.; Stouch, B.; Martin, S.; Kostenuik, P.J. Sustained antiresorptive effects after a single treatment with human recombinant osteoprotegerin (OPG): A pharmacodynamic and pharmacokinetic analysis in rats. J. Bone Miner. Res. 2003, 18, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A.; Goessl, C. Clinical development of anti-RANKL therapies for treatment and prevention of bone metastasis. Bone 2011, 48, 96–99. [Google Scholar] [CrossRef]
- Lacey, D.L.; Boyle, W.J.; Simonet, W.S.; Kostenuik, P.J.; Dougall, W.C.; Sullivan, J.K.; San Martin, J.; Dansey, R. Bench to bedside: Elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat. Rev. Drug Discov. 2012, 11, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E.; Croucher, P.I.; Padhani, A.R.; Clezardin, P.; Chow, E.; Fallon, M.; Guise, T.; Colangeli, S.; Capanna, R.; Costa, L. Bone metastases. Nat. Rev. Dis. Primers 2020, 6, 83. [Google Scholar] [CrossRef]
- Coleman, R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Mundy, G.R. Mechanisms of bone metastasis. Cancer 1997, 80, 1546–1556. [Google Scholar] [CrossRef]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006, 12, 6213s–6216s. [Google Scholar] [CrossRef] [Green Version]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef]
- Yin, J.J.; Mohammad, K.S.; Kakonen, S.M.; Harris, S.; Wu-Wong, J.R.; Wessale, J.L.; Padley, R.J.; Garrett, I.R.; Chirgwin, J.M.; Guise, T.A. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc. Natl. Acad. Sci. USA 2003, 100, 10954–10959. [Google Scholar] [CrossRef] [Green Version]
- de Groot, A.F.; Appelman-Dijkstra, N.M.; van der Burg, S.H.; Kroep, J.R. The anti-tumor effect of RANKL inhibition in malignant solid tumors—A systematic review. Cancer Treat. Rev. 2018, 62, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Lipton, A.; Fizazi, K.; Stopeck, A.T.; Henry, D.H.; Brown, J.E.; Yardley, D.A.; Richardson, G.E.; Siena, S.; Maroto, P.; Clemens, M.; et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: A combined analysis of 3 pivotal, randomised, phase 3 trials. Eur. J. Cancer 2012, 48, 3082–3092. [Google Scholar] [CrossRef]
- Burgess, T.L.; Qian, Y.; Kaufman, S.; Ring, B.D.; Van, G.; Capparelli, C.; Kelley, M.; Hsu, H.; Boyle, W.J.; Dunstan, C.R.; et al. The ligand for osteoprotegerin (OPGL) directly activates mature osteoclasts. J. Cell Biol. 1999, 145, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Lacey, D.L.; Tan, H.L.; Lu, J.; Kaufman, S.; Van, G.; Qiu, W.; Rattan, A.; Scully, S.; Fletcher, F.; Juan, T.; et al. Osteoprotegerin ligand modulates murine osteoclast survival in vitro and in vivo. Am. J. Pathol. 2000, 157, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Fleisch, H. Development of bisphosphonates. Breast Cancer Res. 2002, 4, 30–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopeck, A.T.; Lipton, A.; Body, J.J.; Steger, G.G.; Tonkin, K.; de Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: A randomized, double-blind study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef] [Green Version]
- Fizazi, K.; Carducci, M.; Smith, M.; Damiao, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Henry, D.H.; Costa, L.; Goldwasser, F.; Hirsh, V.; Hungria, V.; Prausova, J.; Scagliotti, G.V.; Sleeboom, H.; Spencer, A.; Vadhan-Raj, S. Randomized, double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. J. Clin. Oncol. 2011, 29, 1125–1132. [Google Scholar] [CrossRef] [Green Version]
- Fournier, P.G.; Stresing, V.; Ebetino, F.H.; Clezardin, P. How do bisphosphonates inhibit bone metastasis in vivo? Neoplasia 2010, 12, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Stresing, V.; Daubine, F.; Benzaid, I.; Monkkonen, H.; Clezardin, P. Bisphosphonates in cancer therapy. Cancer Lett. 2007, 257, 16–35. [Google Scholar] [CrossRef]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.P.; Miller, R.E.; Jones, J.C.; Zhang, J.; Keller, E.T.; Dougall, W.C. RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate 2008, 68, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Casimiro, S.; Mohammad, K.S.; Pires, R.; Tato-Costa, J.; Alho, I.; Teixeira, R.; Carvalho, A.; Ribeiro, S.; Lipton, A.; Guise, T.A.; et al. RANKL/RANK/MMP-1 molecular triad contributes to the metastatic phenotype of breast and prostate cancer cells in vitro. PLoS ONE 2013, 8, e63153. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Le Goff, B.; Charrier, C.; Battaglia, S.; Heymann, D.; Redini, F. DU145 human prostate cancer cells express functional receptor activator of NFkappaB: New insights in the prostate cancer bone metastasis process. Bone 2007, 40, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Le Goff, B.; Berreur, M.; Riet, A.; Moreau, A.; Blanchard, F.; Chevalier, C.; Guisle-Marsollier, I.; Leger, J.; Guicheux, J.; et al. Human osteosarcoma cells express functional receptor activator of nuclear factor-kappa B. J. Pathol. 2007, 211, 555–562. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhou, H.; Brennan, K.; Blair, J.M.; Modzelewski, J.R.; Seibel, M.J.; Dunstan, C.R. Inhibition of bone resorption, rather than direct cytotoxicity, mediates the anti-tumour actions of ibandronate and osteoprotegerin in a murine model of breast cancer bone metastasis. Bone 2007, 40, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Blake, M.L.; Tometsko, M.; Miller, R.; Jones, J.C.; Dougall, W.C. RANK expression on breast cancer cells promotes skeletal metastasis. Clin. Exp. Metastasis 2014, 31, 233–245. [Google Scholar] [CrossRef]
- Lu, X.; Wang, Q.; Hu, G.; Van Poznak, C.; Fleisher, M.; Reiss, M.; Massague, J.; Kang, Y. ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes Dev. 2009, 23, 1882–1894. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Bryant, R.; Roudier, M.; Branstetter, D.G.; Dougall, W.C. RANKL inhibition combined with tamoxifen treatment increases anti-tumor efficacy and prevents tumor-induced bone destruction in an estrogen receptor-positive breast cancer bone metastasis model. Breast Cancer Res. Treat. 2012, 135, 771–780. [Google Scholar] [CrossRef]
- Gomes, I.; de Almeida, B.P.; Damaso, S.; Mansinho, A.; Correia, I.; Henriques, S.; Cruz-Duarte, R.; Vilhais, G.; Felix, P.; Alves, P.; et al. Expression of receptor activator of NFkB (RANK) drives stemness and resistance to therapy in ER+HER2- breast cancer. Oncotarget 2020, 11, 1714–1728. [Google Scholar] [CrossRef]
- Zhang, J.; Dai, J.; Qi, Y.; Lin, D.L.; Smith, P.; Strayhorn, C.; Mizokami, A.; Fu, Z.; Westman, J.; Keller, E.T. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J. Clin. Investig. 2001, 107, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Yonou, H.; Kanomata, N.; Goya, M.; Kamijo, T.; Yokose, T.; Hasebe, T.; Nagai, K.; Hatano, T.; Ogawa, Y.; Ochiai, A. Osteoprotegerin/osteoclastogenesis inhibitory factor decreases human prostate cancer burden in human adult bone implanted into nonobese diabetic/severe combined immunodeficient mice. Cancer Res. 2003, 63, 2096–2102. [Google Scholar] [PubMed]
- Zhang, J.; Dai, J.; Yao, Z.; Lu, Y.; Dougall, W.; Keller, E.T. Soluble receptor activator of nuclear factor kappaB Fc diminishes prostate cancer progression in bone. Cancer Res. 2003, 63, 7883–7890. [Google Scholar]
- Kiefer, J.A.; Vessella, R.L.; Quinn, J.E.; Odman, A.M.; Zhang, J.; Keller, E.T.; Kostenuik, P.J.; Dunstan, C.R.; Corey, E. The effect of osteoprotegerin administration on the intra-tibial growth of the osteoblastic LuCaP 23.1 prostate cancer xenograft. Clin. Exp. Metastasis 2004, 21, 381–387. [Google Scholar] [CrossRef]
- Asano, T.; Okamoto, K.; Nakai, Y.; Tsutsumi, M.; Muro, R.; Suematsu, A.; Hashimoto, K.; Okamura, T.; Ehata, S.; Nitta, T.; et al. Soluble RANKL is physiologically dispensable but accelerates tumour metastasis to bone. Nat. Metab. 2019, 1, 868–875. [Google Scholar] [CrossRef]
- Pantano, F.; Rossi, E.; Iuliani, M.; Facchinetti, A.; Simonetti, S.; Ribelli, G.; Zoccoli, A.; Vincenzi, B.; Tonini, G.; Zamarchi, R.; et al. Dynamic changes of Receptor activator of nuclear factor-kappaB expression in Circulating Tumor Cells during Denosumab predict treatment effectiveness in Metastatic Breast Cancer. Sci. Rep. 2020, 10, 1288. [Google Scholar] [CrossRef]
- Min, J.K.; Cho, Y.L.; Choi, J.H.; Kim, Y.; Kim, J.H.; Yu, Y.S.; Rho, J.; Mochizuki, N.; Kim, Y.M.; Oh, G.T.; et al. Receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) increases vascular permeability: Impaired permeability and angiogenesis in eNOS-deficient mice. Blood 2007, 109, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Loberg, R.; Taichman, R.S. The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 2006, 25, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Casimiro, S.; Ferreira, A.R.; Mansinho, A.; Alho, I.; Costa, L. Molecular Mechanisms of Bone Metastasis: Which Targets Came from the Bench to the Bedside? Int. J. Mol. Sci. 2016, 17, 1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Ke, J.; Song, Y. Application of Biomarkers for the Prediction and Diagnosis of Bone Metastasis in Breast Cancer. J. Breast Cancer 2020, 23, 588–598. [Google Scholar] [CrossRef]
- Pavlovic, M.; Arnal-Estape, A.; Rojo, F.; Bellmunt, A.; Tarragona, M.; Guiu, M.; Planet, E.; Garcia-Albeniz, X.; Morales, M.; Urosevic, J.; et al. Enhanced MAF Oncogene Expression and Breast Cancer Bone Metastasis. J. Natl. Cancer Inst. 2015, 107, djv256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westbrook, J.A.; Cairns, D.A.; Peng, J.; Speirs, V.; Hanby, A.M.; Holen, I.; Wood, S.L.; Ottewell, P.D.; Marshall, H.; Banks, R.E.; et al. CAPG and GIPC1: Breast Cancer Biomarkers for Bone Metastasis Development and Treatment. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Itoh, T.; Ito, Y.; Ohtsuki, Y.; Ando, M.; Tsukamasa, Y.; Yamada, N.; Naoe, T.; Akao, Y. Microvesicles released from hormone-refractory prostate cancer cells facilitate mouse pre-osteoblast differentiation. J. Mol. Histol. 2012, 43, 509–515. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.R.; Roudier, M.; Bryant, R.; Morony, S.; Stolina, M.; Kostenuik, P.J.; Dougall, W.C. Inhibition of RANKL blocks skeletal tumor progression and improves survival in a mouse model of breast cancer bone metastasis. Clin. Exp. Metastasis 2008, 25, 119–129. [Google Scholar] [CrossRef]
- George, C.N.; Canuas-Landero, V.; Theodoulou, E.; Muthana, M.; Wilson, C.; Ottewell, P. Oestrogen and zoledronic acid driven changes to the bone and immune environments: Potential mechanisms underlying the differential anti-tumour effects of zoledronic acid in pre- and post-menopausal conditions. J. Bone Oncol. 2020, 25, 100317. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef]
- Cheng, J.N.; Frye, J.B.; Whitman, S.A.; Kunihiro, A.G.; Pandey, R.; Funk, J.L. A Role for TGFbeta Signaling in Preclinical Osteolytic Estrogen Receptor-Positive Breast Cancer Bone Metastases Progression. Int. J. Mol. Sci. 2021, 22, 4463. [Google Scholar] [CrossRef]
- Winding, B.; Misander, H.; Hoegh-Andersen, P.; Brunner, N.; Foged, N.T. Estradiol enhances osteolytic lesions in mice inoculated with human estrogen receptor-negative MDA-231 breast cancer cells in vivo. Breast Cancer Res. Treat. 2003, 78, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Cameron, D.; Dodwell, D.; Bell, R.; Wilson, C.; Rathbone, E.; Keane, M.; Gil, M.; Burkinshaw, R.; Grieve, R.; et al. Adjuvant zoledronic acid in patients with early breast cancer: Final efficacy analysis of the AZURE (BIG 01/04) randomised open-label phase 3 trial. Lancet Oncol. 2014, 15, 997–1006. [Google Scholar] [CrossRef]
- Ottewell, P.D.; Wang, N.; Brown, H.K.; Reeves, K.J.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. Zoledronic acid has differential antitumor activity in the pre- and postmenopausal bone microenvironment in vivo. Clin. Cancer Res. 2014, 20, 2922–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnant, M.; Pfeiler, G.; Steger, G.G.; Egle, D.; Greil, R.; Fitzal, F.; Wette, V.; Balic, M.; Haslbauer, F.; Melbinger-Zeinitzer, E.; et al. Adjuvant denosumab in postmenopausal patients with hormone receptor-positive breast cancer (ABCSG-18): Disease-free survival results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 339–351. [Google Scholar] [CrossRef]
- Raisz, L.G. Pathogenesis of osteoporosis: Concepts, conflicts, and prospects. J. Clin. Investig. 2005, 115, 3318–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofbauer, L.C.; Rachner, T.D.; Coleman, R.E.; Jakob, F. Endocrine aspects of bone metastases. Lancet Diabetes Endocrinol. 2014, 2, 500–512. [Google Scholar] [CrossRef]
- Melani, C.; Sangaletti, S.; Barazzetta, F.M.; Werb, Z.; Colombo, M.P. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res. 2007, 67, 11438–11446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coscia, M.; Quaglino, E.; Iezzi, M.; Curcio, C.; Pantaleoni, F.; Riganti, C.; Holen, I.; Monkkonen, H.; Boccadoro, M.; Forni, G.; et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J. Cell Mol. Med. 2010, 14, 2803–2815. [Google Scholar] [CrossRef] [PubMed]
- Benzaid, I.; Monkkonen, H.; Stresing, V.; Bonnelye, E.; Green, J.; Monkkonen, J.; Touraine, J.L.; Clezardin, P. High phosphoantigen levels in bisphosphonate-treated human breast tumors promote Vgamma9Vdelta2 T-cell chemotaxis and cytotoxicity in vivo. Cancer Res. 2011, 71, 4562–4572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzaid, I.; Monkkonen, H.; Bonnelye, E.; Monkkonen, J.; Clezardin, P. In vivo phosphoantigen levels in bisphosphonate-treated human breast tumors trigger Vgamma9Vdelta2 T-cell antitumor cytotoxicity through ICAM-1 engagement. Clin. Cancer Res. 2012, 18, 6249–6259. [Google Scholar] [CrossRef] [Green Version]
- D’Oronzo, S.; Gregory, W.; Nicholson, S.; Chong, Y.K.; Brown, J.; Coleman, R. Natural history of stage II/III breast cancer, bone metastasis and the impact of adjuvant zoledronate on distribution of recurrences. J. Bone Oncol. 2021, 28, 100367. [Google Scholar] [CrossRef]
- Ferrari-Lacraz, S.; Ferrari, S. Do RANKL inhibitors (denosumab) affect inflammation and immunity? Osteoporos. Int. 2011, 22, 435–446. [Google Scholar] [CrossRef]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Tomoyasu, A.; Goto, M.; Fujise, N.; Mochizuki, S.; Yasuda, H.; Morinaga, T.; Tsuda, E.; Higashio, K. Characterization of monomeric and homodimeric forms of osteoclastogenesis inhibitory factor. Biochem. Biophys. Res. Commun. 1998, 245, 382–387. [Google Scholar] [CrossRef] [PubMed]
- von Moos, R.; Costa, L.; Gonzalez-Suarez, E.; Terpos, E.; Niepel, D.; Body, J.J. Management of bone health in solid tumours: From bisphosphonates to a monoclonal antibody. Cancer Treat. Rev. 2019, 76, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.; Hadji, P.; Body, J.J.; Santini, D.; Chow, E.; Terpos, E.; Oudard, S.; Bruland, O.; Flamen, P.; Kurth, A.; et al. Bone health in cancer: ESMO Clinical Practice Guidelines. Ann. Oncol. 2020, 31, 1650–1663. [Google Scholar] [CrossRef]
- Mhaskar, R.; Kumar, A.; Miladinovic, B.; Djulbegovic, B. Bisphosphonates in multiple myeloma: An updated network meta-analysis. Cochrane Database Syst. Rev. 2017, 12, CD003188. [Google Scholar] [CrossRef]
- Smith, M.R.; Halabi, S.; Ryan, C.J.; Hussain, A.; Vogelzang, N.; Stadler, W.; Hauke, R.J.; Monk, J.P.; Saylor, P.; Bhoopalam, N.; et al. Randomized controlled trial of early zoledronic acid in men with castration-sensitive prostate cancer and bone metastases: Results of CALGB 90202 (alliance). J. Clin. Oncol. 2014, 32, 1143–1150. [Google Scholar] [CrossRef] [Green Version]
- Henry, D.; Vadhan-Raj, S.; Hirsh, V.; von Moos, R.; Hungria, V.; Costa, L.; Woll, P.J.; Scagliotti, G.; Smith, G.; Feng, A.; et al. Delaying skeletal-related events in a randomized phase 3 study of denosumab versus zoledronic acid in patients with advanced cancer: An analysis of data from patients with solid tumors. Support. Care Cancer 2014, 22, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Hirsh, V.; Siena, S.; Henry, D.H.; Woll, P.J.; Manegold, C.; Solal-Celigny, P.; Rodriguez, G.; Krzakowski, M.; Mehta, N.D.; et al. Overall survival improvement in patients with lung cancer and bone metastases treated with denosumab versus zoledronic acid: Subgroup analysis from a randomized phase 3 study. J. Thorac. Oncol. 2012, 7, 1823–1829. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Danson, S.; Hasan, B.; Dafni, U.; Reinmuth, N.; Majem, M.; Tournoy, K.G.; Mark, M.T.; Pless, M.; Cobo, M.; et al. A Randomized Open-Label Phase III Trial Evaluating the Addition of Denosumab to Standard First-Line Treatment in Advanced NSCLC: The European Thoracic Oncology Platform (ETOP) and European Organisation for Research and Treatment of Cancer (EORTC) SPLENDOUR Trial. J. Thorac. Oncol. 2020, 15, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Terpos, E.; Willenbacher, W.; Shimizu, K.; Garcia-Sanz, R.; Durie, B.; Legiec, W.; Krejci, M.; Laribi, K.; Zhu, L.; et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: An international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018, 19, 370–381. [Google Scholar] [CrossRef]
- Morgan, G.J.; Davies, F.E.; Gregory, W.M.; Bell, S.E.; Szubert, A.J.; Cook, G.; Drayson, M.T.; Owen, R.G.; Ross, F.M.; Jackson, G.H.; et al. Long-term follow-up of MRC Myeloma IX trial: Survival outcomes with bisphosphonate and thalidomide treatment. Clin. Cancer Res. 2013, 19, 6030–6038. [Google Scholar] [CrossRef] [Green Version]
- Raje, N.; Vadhan-Raj, S.; Willenbacher, W.; Terpos, E.; Hungria, V.; Spencer, A.; Alexeeva, Y.; Facon, T.; Stewart, A.K.; Feng, A.; et al. Evaluating results from the multiple myeloma patient subset treated with denosumab or zoledronic acid in a randomized phase 3 trial. Blood Cancer J. 2016, 6, e378. [Google Scholar] [CrossRef] [Green Version]
- Tsourdi, E.; Langdahl, B.; Cohen-Solal, M.; Aubry-Rozier, B.; Eriksen, E.F.; Guanabens, N.; Obermayer-Pietsch, B.; Ralston, S.H.; Eastell, R.; Zillikens, M.C. Discontinuation of Denosumab therapy for osteoporosis: A systematic review and position statement by ECTS. Bone 2017, 105, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Cummings, S.R.; Ferrari, S.; Eastell, R.; Gilchrist, N.; Jensen, J.B.; McClung, M.; Roux, C.; Torring, O.; Valter, I.; Wang, A.T.; et al. Vertebral Fractures After Discontinuation of Denosumab: A Post Hoc Analysis of the Randomized Placebo-Controlled FREEDOM Trial and Its Extension. J. Bone Miner. Res. 2018, 33, 190–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, S.; Clezardin, P.; Marquez-Rodas, I.; Niepel, D.; Gedye, C. The RANK-RANKL axis: An opportunity for drug repurposing in cancer? Clin. Transl. Oncol. 2019, 21, 977–991. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.; Finkelstein, D.M.; Barrios, C.; Martin, M.; Iwata, H.; Hegg, R.; Glaspy, J.; Perianez, A.M.; Tonkin, K.; Deleu, I.; et al. Adjuvant denosumab in early breast cancer (D-CARE): An international, multicentre, randomised, controlled, phase 3 trial. Lancet Oncol. 2020, 21, 60–72. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Coleman, R.; Shore, N.; Fizazi, K.; Tombal, B.; Miller, K.; Sieber, P.; Karsh, L.; Damiao, R.; et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: Results of a phase 3, randomised, placebo-controlled trial. Lancet 2012, 379, 39–46. [Google Scholar] [CrossRef] [Green Version]
- The Early Breast Cancer Trialists’ Collaborative Group. Adjuvant bisphosphonate treatment in early breast cancer: Meta-analyses of individual patient data from randomised trials. Lancet 2015, 386, 1353–1361. [Google Scholar] [CrossRef] [Green Version]
- Dhesy-Thind, S.; Fletcher, G.G.; Blanchette, P.S.; Clemons, M.J.; Dillmon, M.S.; Frank, E.S.; Gandhi, S.; Gupta, R.; Mates, M.; Moy, B.; et al. Use of Adjuvant Bisphosphonates and Other Bone-Modifying Agents in Breast Cancer: A Cancer Care Ontario and American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2017, 35, 2062–2081. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E.; Committee, E.G. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1674. [Google Scholar] [CrossRef] [Green Version]
- Gnant, M.; Pfeiler, G.; Dubsky, P.C.; Hubalek, M.; Greil, R.; Jakesz, R.; Wette, V.; Balic, M.; Haslbauer, F.; Melbinger, E.; et al. Adjuvant denosumab in breast cancer (ABCSG-18): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015, 386, 433–443. [Google Scholar] [CrossRef]
- Wirth, M.; Tammela, T.; Cicalese, V.; Gomez Veiga, F.; Delaere, K.; Miller, K.; Tubaro, A.; Schulze, M.; Debruyne, F.; Huland, H.; et al. Prevention of bone metastases in patients with high-risk nonmetastatic prostate cancer treated with zoledronic acid: Efficacy and safety results of the Zometa European Study (ZEUS). Eur. Urol. 2015, 67, 482–491. [Google Scholar] [CrossRef]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Parmar, M.K. STAMPEDE trial and patients with non-metastatic prostate cancer—Authors’ reply. Lancet 2016, 388, 235–236. [Google Scholar] [CrossRef] [Green Version]
- Fata, J.E.; Kong, Y.Y.; Li, J.; Sasaki, T.; Irie-Sasaki, J.; Moorehead, R.A.; Elliott, R.; Scully, S.; Voura, E.B.; Lacey, D.L.; et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell 2000, 103, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Bonizzi, G.; Seagroves, T.N.; Greten, F.R.; Johnson, R.; Schmidt, E.V.; Karin, M. IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell 2001, 107, 763–775. [Google Scholar] [CrossRef] [Green Version]
- Beleut, M.; Rajaram, R.D.; Caikovski, M.; Ayyanan, A.; Germano, D.; Choi, Y.; Schneider, P.; Brisken, C. Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc. Natl. Acad. Sci. USA 2010, 107, 2989–2994. [Google Scholar] [CrossRef] [Green Version]
- Kotsopoulos, J.; Singer, C.; Narod, S.A. Can we prevent BRCA1-associated breast cancer by RANKL inhibition? Breast Cancer Res. Treat. 2017, 161, 11–16. [Google Scholar] [CrossRef]
- Azim, H.A., Jr.; Peccatori, F.A.; Brohee, S.; Branstetter, D.; Loi, S.; Viale, G.; Piccart, M.; Dougall, W.C.; Pruneri, G.; Sotiriou, C. RANK-ligand (RANKL) expression in young breast cancer patients and during pregnancy. Breast Cancer Res. 2015, 17, 24. [Google Scholar] [CrossRef] [Green Version]
- Sigl, V.; Owusu-Boaitey, K.; Joshi, P.A.; Kavirayani, A.; Wirnsberger, G.; Novatchkova, M.; Kozieradzki, I.; Schramek, D.; Edokobi, N.; Hersl, J.; et al. RANKL/RANK control Brca1 mutation-driven mammary tumors. Cell Res. 2016, 26, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Nolan, E.; Vaillant, F.; Branstetter, D.; Pal, B.; Giner, G.; Whitehead, L.; Lok, S.W.; Mann, G.B.; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab); Rohrbach, K.; et al. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat. Med. 2016, 22, 933–939. [Google Scholar] [CrossRef]
- Cuyas, E.; Corominas-Faja, B.; Martin, M.M.; Martin-Castillo, B.; Lupu, R.; Brunet, J.; Bosch-Barrera, J.; Menendez, J.A. BRCA1 haploinsufficiency cell-autonomously activates RANKL expression and generates denosumab-responsive breast cancer-initiating cells. Oncotarget 2017, 8, 35019–35032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widschwendter, M.; Burnell, M.; Fraser, L.; Rosenthal, A.N.; Philpott, S.; Reisel, D.; Dubeau, L.; Cline, M.; Pan, Y.; Yi, P.C.; et al. Osteoprotegerin (OPG), The Endogenous Inhibitor of Receptor Activator of NF-kappaB Ligand (RANKL), is Dysregulated in BRCA Mutation Carriers. EBioMedicine 2015, 2, 1331–1339. [Google Scholar] [CrossRef] [Green Version]
- Renema, N.; Navet, B.; Heymann, M.F.; Lezot, F.; Heymann, D. RANK-RANKL signalling in cancer. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, D.; Perrone, G.; Roato, I.; Godio, L.; Pantano, F.; Grasso, D.; Russo, A.; Vincenzi, B.; Fratto, M.E.; Sabbatini, R.; et al. Expression pattern of receptor activator of NFkappaB (RANK) in a series of primary solid tumors and related bone metastases. J. Cell. Physiol. 2011, 226, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Santini, D.; Schiavon, G.; Vincenzi, B.; Gaeta, L.; Pantano, F.; Russo, A.; Ortega, C.; Porta, C.; Galluzzo, S.; Armento, G.; et al. Receptor activator of NF-kB (RANK) expression in primary tumors associates with bone metastasis occurrence in breast cancer patients. PLoS ONE 2011, 6, e19234. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Teng, Y.; Zhang, Y.; Liu, J.; Xu, L.; Qu, J.; Hou, K.; Yang, X.; Liu, Y.; Qu, X. Receptor activator for nuclear factor kappa B expression predicts poor prognosis in breast cancer patients with bone metastasis but not in patients with visceral metastasis. J. Clin. Pathol. 2012, 65, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Owen, S.; Ye, L.; Sanders, A.J.; Mason, M.D.; Jiang, W.G. Expression profile of receptor activator of nuclear-kappaB (RANK), RANK ligand (RANKL) and osteoprotegerin (OPG) in breast cancer. Anticancer Res. 2013, 33, 199–206. [Google Scholar] [PubMed]
- Pfitzner, B.M.; Branstetter, D.; Loibl, S.; Denkert, C.; Lederer, B.; Schmitt, W.D.; Dombrowski, F.; Werner, M.; Rudiger, T.; Dougall, W.C.; et al. RANK expression as a prognostic and predictive marker in breast cancer. Breast Cancer Res. Treat. 2014, 145, 307–315. [Google Scholar] [CrossRef]
- Park, H.S.; Lee, A.; Chae, B.J.; Bae, J.S.; Song, B.J.; Jung, S.S. Expression of receptor activator of nuclear factor kappa-B as a poor prognostic marker in breast cancer. J. Surg. Oncol. 2014, 110, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Geerts, D.; Chopra, C.; Connelly, L. Osteoprotegerin: Relationship to Breast Cancer Risk and Prognosis. Front. Oncol. 2020, 10, 462. [Google Scholar] [CrossRef] [PubMed]
- Mikami, S.; Katsube, K.; Oya, M.; Ishida, M.; Kosaka, T.; Mizuno, R.; Mochizuki, S.; Ikeda, T.; Mukai, M.; Okada, Y. Increased RANKL expression is related to tumour migration and metastasis of renal cell carcinomas. J. Pathol. 2009, 218, 530–539. [Google Scholar] [CrossRef]
- Palafox, M.; Ferrer, I.; Pellegrini, P.; Vila, S.; Hernandez-Ortega, S.; Urruticoechea, A.; Climent, F.; Soler, M.T.; Munoz, P.; Vinals, F.; et al. RANK induces epithelial-mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res. 2012, 72, 2879–2888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, P.; Cordero, A.; Gallego, M.I.; Dougall, W.C.; Munoz, P.; Pujana, M.A.; Gonzalez-Suarez, E. Constitutive activation of RANK disrupts mammary cell fate leading to tumorigenesis. Stem Cells 2013, 31, 1954–1965. [Google Scholar] [CrossRef]
- Tsubaki, M.; Komai, M.; Fujimoto, S.; Itoh, T.; Imano, M.; Sakamoto, K.; Shimaoka, H.; Takeda, T.; Ogawa, N.; Mashimo, K.; et al. Activation of NF-kappaB by the RANKL/RANK system up-regulates snail and twist expressions and induces epithelial-to-mesenchymal transition in mammary tumor cell lines. J. Exp. Clin. Cancer Res. 2013, 32, 62. [Google Scholar] [CrossRef] [Green Version]
- Benitez, S.; Cordero, A.; Santamaria, P.G.; Redondo-Pedraza, J.; Rocha, A.S.; Collado-Sole, A.; Jimenez, M.; Sanz-Moreno, A.; Yoldi, G.; Santos, J.C.; et al. RANK links senescence to stemness in the mammary epithelia, delaying tumor onset but increasing tumor aggressiveness. Dev. Cell 2021, 56, 1727–1741.e7. [Google Scholar] [CrossRef]
- Zoi, I.; Karamouzis, M.V.; Adamopoulos, C.; Papavassiliou, A.G. RANKL Signaling and ErbB Receptors in Breast Carcinogenesis. Trends Mol. Med. 2016, 22, 839–850. [Google Scholar] [CrossRef]
- Merkhofer, E.C.; Cogswell, P.; Baldwin, A.S. Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha. Oncogene 2010, 29, 1238–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz-Moreno, A.; Palomeras, S.; Pedersen, K.; Morancho, B.; Pascual, T.; Galvan, P.; Benitez, S.; Gomez-Miragaya, J.; Ciscar, M.; Jimenez, M.; et al. RANK signaling increases after anti-HER2 therapy contributing to the emergence of resistance in HER2-positive breast cancer. Breast Cancer Res. 2021, 23, 42. [Google Scholar] [CrossRef]
- van Dam, P.A.; Verhoeven, Y.; Trinh, X.B.; Wouters, A.; Lardon, F.; Prenen, H.; Smits, E.; Baldewijns, M.; Lammens, M. RANK/RANKL signaling inhibition may improve the effectiveness of checkpoint blockade in cancer treatment. Crit. Rev. Oncol. Hematol. 2019, 133, 85–91. [Google Scholar] [CrossRef]
- Luo, J.L.; Tan, W.; Ricono, J.M.; Korchynskyi, O.; Zhang, M.; Gonias, S.L.; Cheresh, D.A.; Karin, M. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature 2007, 446, 690–694. [Google Scholar] [CrossRef]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thallinger, C.; Fureder, T.; Preusser, M.; Heller, G.; Mullauer, L.; Holler, C.; Prosch, H.; Frank, N.; Swierzewski, R.; Berger, W.; et al. Review of cancer treatment with immune checkpoint inhibitors : Current concepts, expectations, limitations and pitfalls. Wien. Klin. Wochenschr. 2018, 130, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Smyth, M.J.; Yagita, H.; McArthur, G.A. Combination Anti-CTLA-4 and Anti-RANKL in Metastatic Melanoma. J. Clin. Oncol. 2016, 34, e104–e106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liede, A.; Hernandez, R.K.; Wade, S.W.; Bo, R.; Nussbaum, N.C.; Ahern, E.; Dougall, W.C.; Smyth, M.J. An observational study of concomitant immunotherapies and denosumab in patients with advanced melanoma or lung cancer. Oncoimmunology 2018, 7, e1480301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.; Gatti-Mays, M.E.; Kalinsky, K.; Korde, L.A.; Sharon, E.; Amiri-Kordestani, L.; Bear, H.; McArthur, H.L.; Frank, E.; Perlmutter, J.; et al. Current Landscape of Immunotherapy in Breast Cancer: A Review. JAMA Oncol. 2019, 5, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.S.; Andre, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.S.; Biganzoli, L.; et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann. Oncol. 2020, 31, 1623–1649. [Google Scholar] [CrossRef] [PubMed]
- Savas, P.; Salgado, R.; Denkert, C.; Sotiriou, C.; Darcy, P.K.; Smyth, M.J.; Loi, S. Clinical relevance of host immunity in breast cancer: From TILs to the clinic. Nat. Rev. Clin. Oncol. 2016, 13, 228–241. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cancer Type(s) | First On-Study SREs (% of Patients; D vs. ZA) | Time to First SRE | Time to First and Subsequent SREs | Ref. |
|---|---|---|---|---|
| Breast (n = 2046) | NE | Denosumab superior (HR 0.82; 95% CI 0.71–0.95; p < 0.001 NI; p = 0.01 S) | Denosumab superior (RR 0.77; 95% CI 0.66–0.89; p = 0.001 S) | [32] |
| CRPC (n = 1901) | 36 vs. 41 | Denosumab superior (HR 0.82; 95% CI 0.71–0.95; p = 0.0002 NI; p = 0.008 S) | Denosumab superior (RR 0.82; 95% CI 0.71–0.94; p = 0.008) | [33] |
| Solid tumors (excluding breast and prostate) and MM (n = 1779) | NE | Denosumab non-inferior, but not statistically superior (HR 0.84; 95% CI, 0.71 to 0.98; p = 0.0007 NI; p = 0.06 S) | Denosumab not statically superior (RR 0.90; 95% CI 0.77–1.04; p = 0.14) | [86] |
| MM (n = 1718) | 44 vs. 45 | Denosumab non-inferior, but not statistically superior (HR 0.98; 95% CI 0.85–1.14; p = 0.01 NI) | Denosumab not statically superior (RR 1.01; 95% CI 0.89–1.15; p = 0.84) | [86] |
| Cancer Type(s) | Number of Patients | Intervention | Disease-Related Outcomes | Trial Identifier/Reference |
|---|---|---|---|---|
| Breast (advanced, all types, pre-and postmenopausal) | 2046 | Denosumab vs. ZA | Similar OS (HR 0.95; 95% CI 0.81–1.11; p = 0.49) and time to disease progression (HR 1.00; 95% CI 0.89–1.11; p = 0.93). | NCT00321464 [32] |
| CRPC | 1901 | Denosumab vs. ZA | Similar OS (HR 1.03; 95% CI 0.91–1.17; p = 0.65) and time to disease progression (HR 1.06; 95% CI 0.95–1.18; p = 0.30). | NCT00321620 [33] |
| Solid tumors (excluding breast and prostate) and MM | 1779 | Denosumab vs. ZA | Similar OS (HR 0.95; 95% CI 0.83–1.08; p = 0.43) and time to disease progression (HR 1.00; 95% CI 0.89–1.12; p = 1.00). Ad hoc analyses favored denosumab for NSCLC patients (HR 0.79; 95% CI 0.65–0.95) and ZA for MM patients (HR 2.26; 95% CI 1.13–4.50). | NCT00330759 [34] |
| NSCLC (stage IV) | 514 | ChT + Denosumab vs. ChT | Similar OS (HR 0.96; 95% CI 0.78–1.19; p = 0.36), PFS (HR 0.99; 95% CI 0.82–1.19; p = 0.46) and ORR (30.5% vs. 29.4%; p = 0.85). | NCT02129699 (SPLENDOUR) [85] |
| MM | 1718 | Denosumab vs. ZA | Denosumab improved PFS by 10.7 months (HR, 0.82; 95% CI 0.68–0.99; p = 0.036). Similar OS (HR, 0.90; 95% CI 0.70–1.16; p = 0.41). | NCT01345019 [86] |
| Cancer Type(s) | Number of Patients | Intervention | Disease-Related Outcomes | Trial Identifier/Reference |
|---|---|---|---|---|
| Breast (adjuvant, early-stage, ER+, posmenopausal, under AIs) | 3425 | Denosumab vs. Placebo | Denosumab increased 5-year DFS by 1.9% and 8-year DFS by 3.1% (HR 0.82; 95% CI 0.69–0.98; p = 0.0260). | NCT00556374 (ABCSG-18) [68] |
| Breast (adjuvant, stage II-III, all types, high-risk, pre-and postmenopausal) | 4509 | Denosumab vs. Placebo | Similar BMFS (HR 0.97; 95% CI 0.82–1.14; p = 0.70), DFS (HR 1.04; 95% CI 0.91–1.19; p = 0.57), DRFS (HR 1.06; 95% CI 0.92–1.21; p = 0.41) and OS (HR 1.03; 95% CI 0.85–1.25; p = 0.76). | NCT01077154 (D-CARE) [92] |
| CRPC (high-risk, non-metastatic) | 1432 | Denosumab vs. Placebo | Denosumab improved BMFS by 4.2 months (HR 0.85; 95% CI 0.73–0.98; p = 0.028) and delayed time to first BM (HR 0.84; 95% CI 0.71–0.98; p = 0.032). Similar OS (HR 1.01; 95% CI 0.85–1.20; p = 0.91). | NCT00286091 [93] |
| Phase | Cancer Type | Intervention | Primary Endpoint | Other Endpoints | Status | Trial Identifier |
|---|---|---|---|---|---|---|
| 1b/2 | Melanoma (unresectable, stage III/IV) | Ipilimumab+ Nivolumab+Denosumab vs. Nivolumab + Denosumab | PFS, grade 3–4 irAEs | OS | Recruiting | NCT03161756 (CHARLI) |
| 1b/2 | NSCLC (neoadjuvant, resectable, stage Ia-IIIa) | Nivolumab + Denosumab vs. Nivolumab | TCR clonality, RNA/transcription profile and genomic changes, markers of interest (IHC) | MPR, rate of R0 resection, radiological response, PFS, OS | Recruiting | ACTRN12618001121257 (POPCORN) |
| 2 | Renal (ccRCC, advanced, refractory to VEGFR-TKIs) | Denosumab + Pembrolizumab (single-arm) | OTR | PFS, time to OTR, DCR, time to first SRE | Recruiting | NCT03280667 (KEYPAD) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casimiro, S.; Vilhais, G.; Gomes, I.; Costa, L. The Roadmap of RANKL/RANK Pathway in Cancer. Cells 2021, 10, 1978. https://doi.org/10.3390/cells10081978
Casimiro S, Vilhais G, Gomes I, Costa L. The Roadmap of RANKL/RANK Pathway in Cancer. Cells. 2021; 10(8):1978. https://doi.org/10.3390/cells10081978
Chicago/Turabian StyleCasimiro, Sandra, Guilherme Vilhais, Inês Gomes, and Luis Costa. 2021. "The Roadmap of RANKL/RANK Pathway in Cancer" Cells 10, no. 8: 1978. https://doi.org/10.3390/cells10081978
APA StyleCasimiro, S., Vilhais, G., Gomes, I., & Costa, L. (2021). The Roadmap of RANKL/RANK Pathway in Cancer. Cells, 10(8), 1978. https://doi.org/10.3390/cells10081978