
H2A-H2B Histone Dimer Plasticity and Its Functional Implications

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
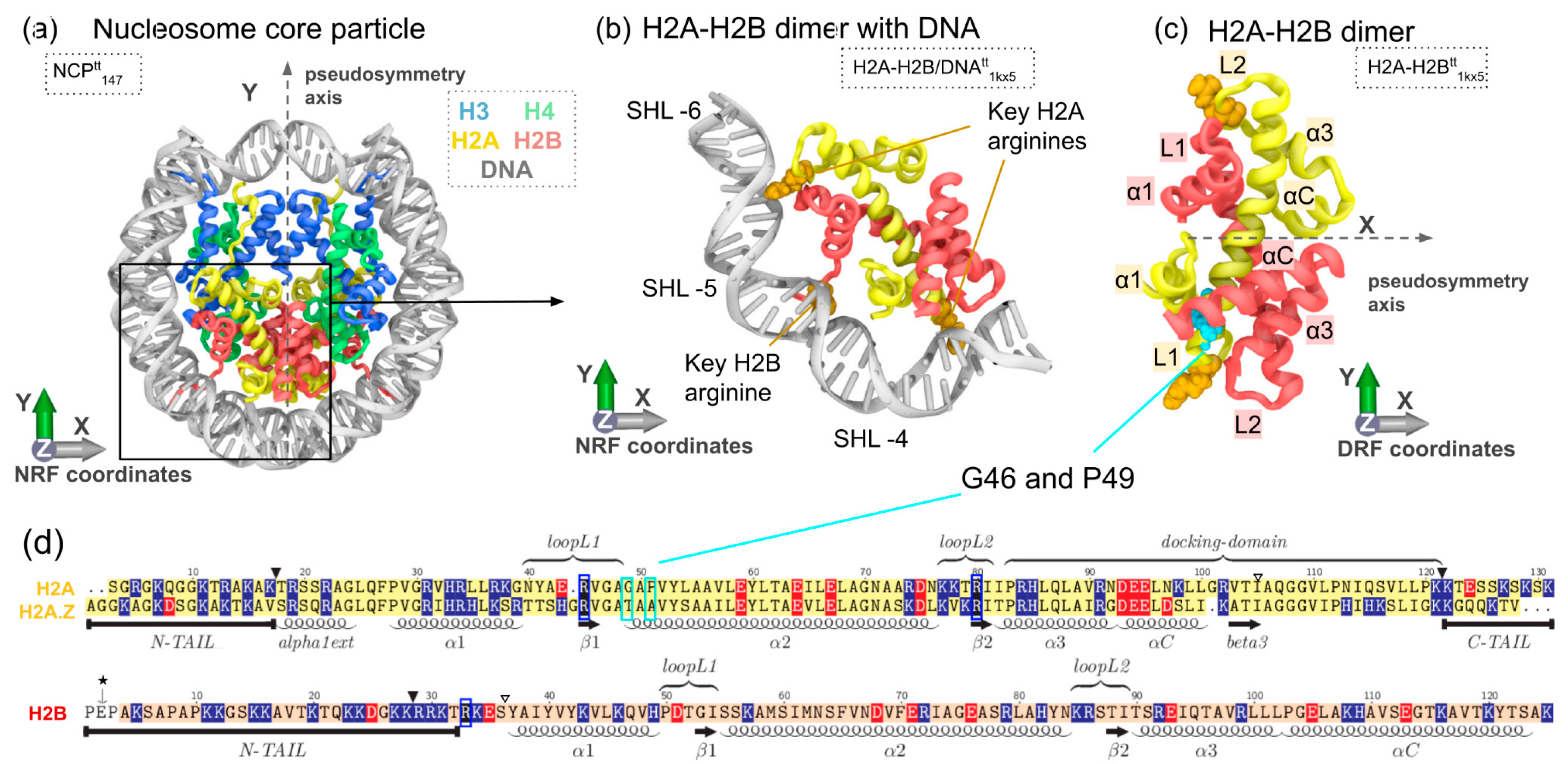
2. Materials and Methods
2.1. Atomistic Models Preparation for Molecular Dynamics (MD) Simulations
2.2. MD Simulations Details
2.3. H2A-H2B Dimer Reference Frame Determination
2.4. Analysis of MD Trajectories
2.5. Principal Component Analysis of H2A-H2B Dynamics
2.6. Structural Analysis of Experimental PDB Structures
2.7. Interactive Materials
3. Results
3.1. Overall Study Design and Research Methodology
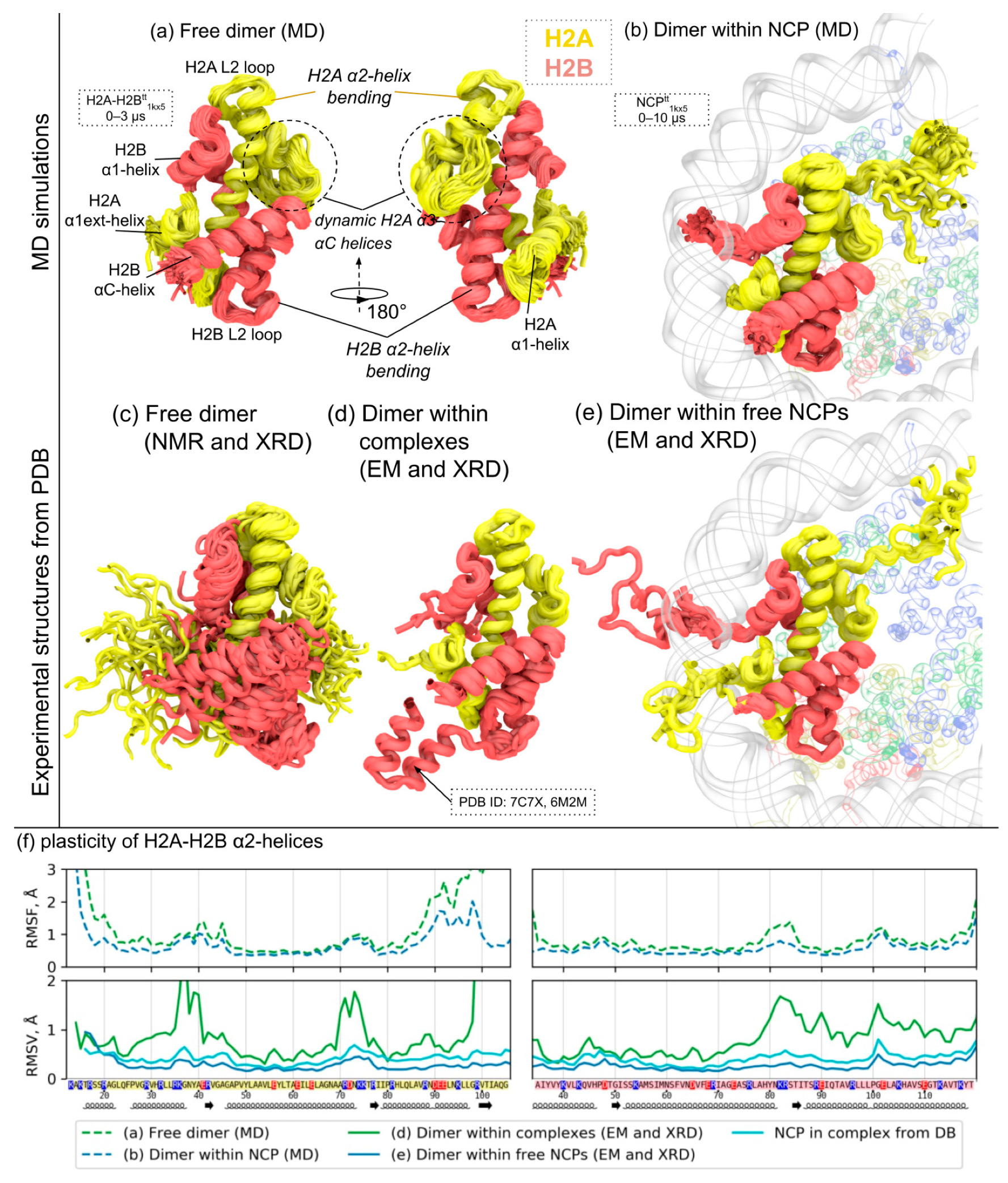
3.2. MD and Experimental Evidence of H2A-H2B Histone Dimer Plasticity
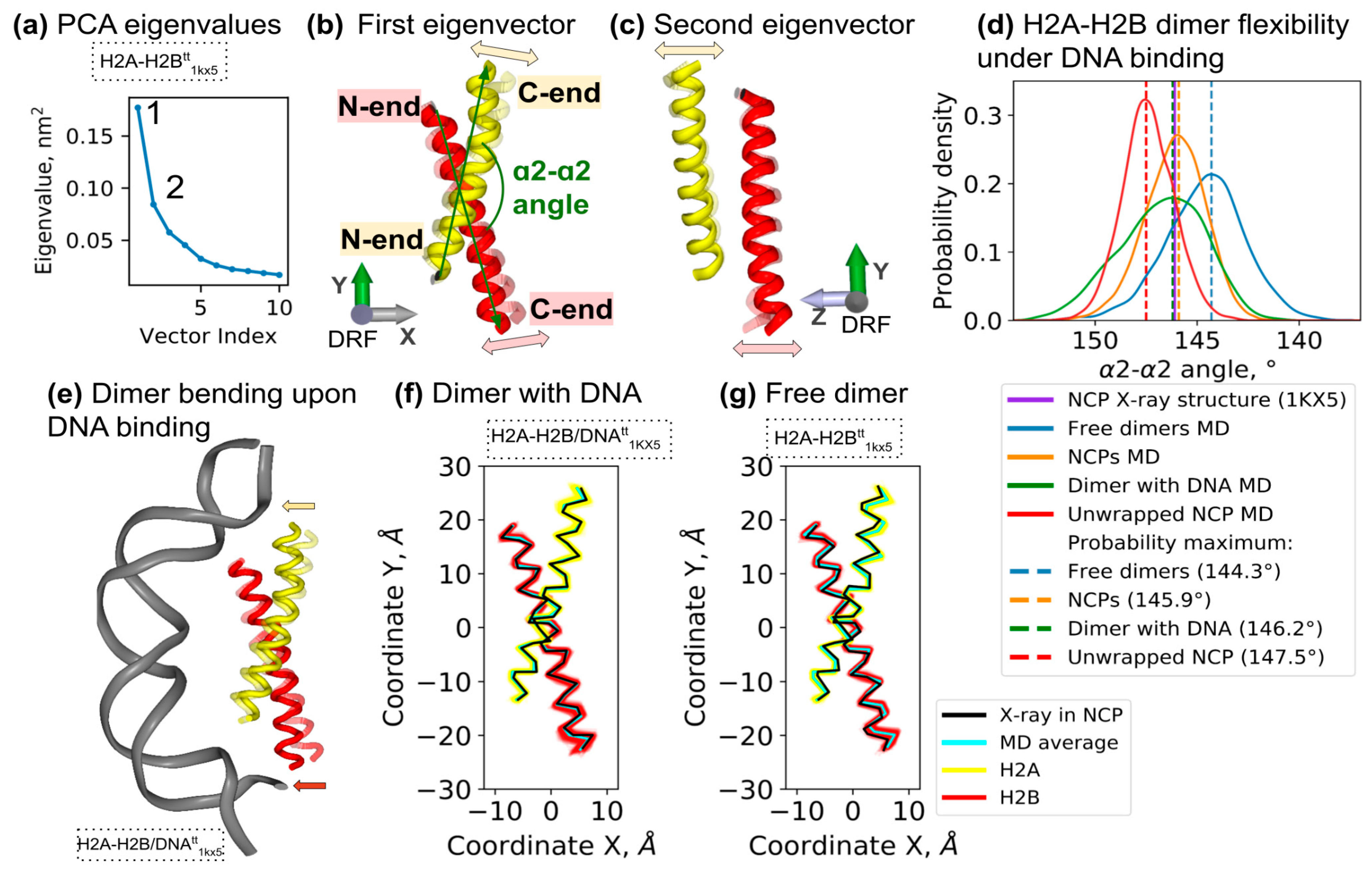
3.3. H2A-H2B Bending Is a Major Dynamical Mode of Free and NCP-Embedded Dimer, Which Is Affected by Interactions with DNA and Other Histones
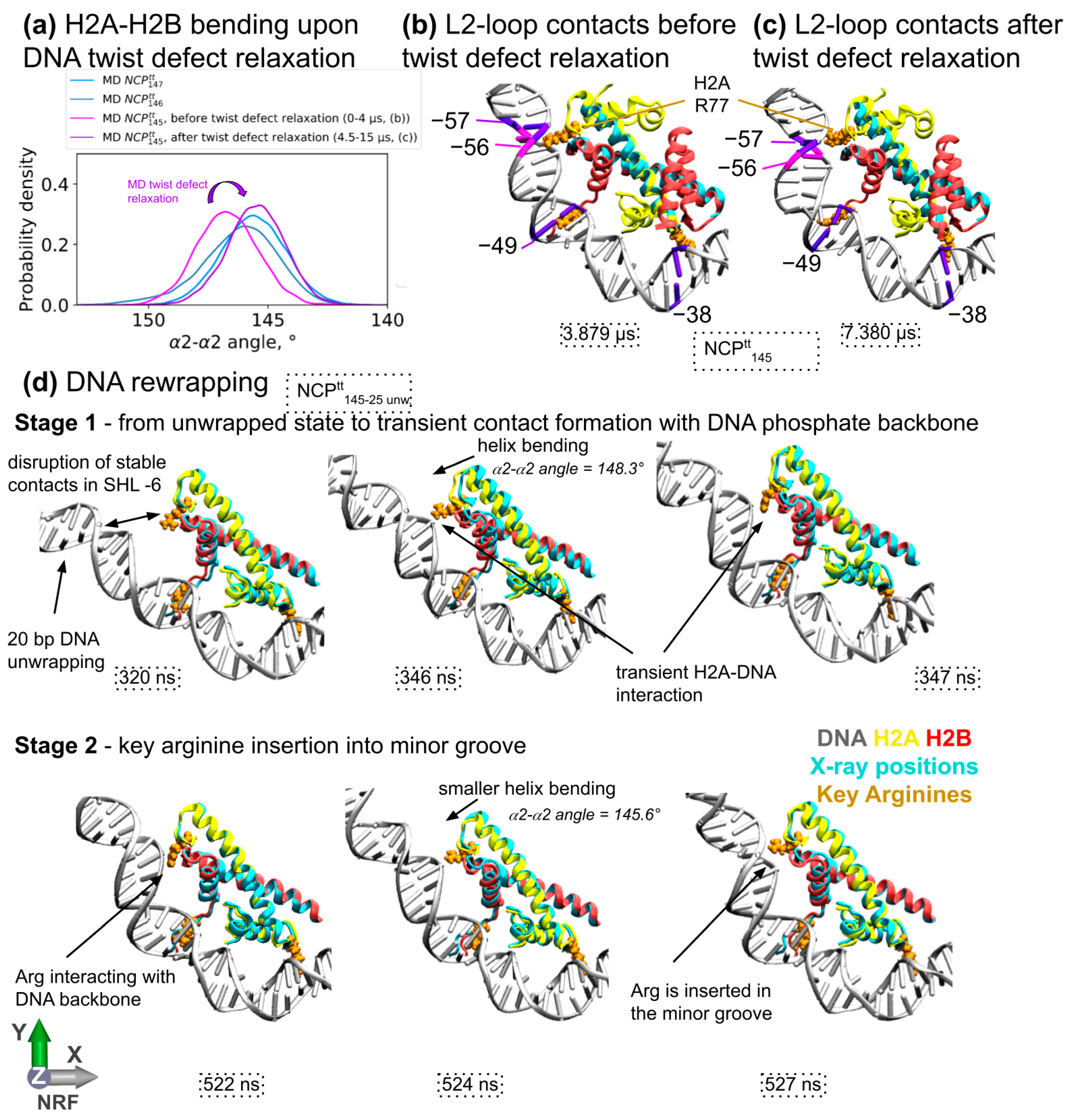
3.4. Influence of DNA Sequence and DNA Unwrapping/Rewrapping on H2A-H2B Dimer Bending
3.5. Histone Sequence Variants May Alter H2A-H2B Dimer Local and Global Dynamics: Example of H2A.Z Histone
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kornberg, R.D. Chromatin Structure: A Repeating Unit of Histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Luger, K. Crystal Structure of the Nucleosome Core Particle at 2.8 A° Resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Armeev, G.A.; Gribkova, A.K.; Pospelova, I.; Komarova, G.A.; Shaytan, A.K. Linking Chromatin Composition and Structural Dynamics at the Nucleosome Level. Curr. Opin. Struct. Biol. 2019, 56, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The Expanding Landscape of “oncohistone” Mutations in Human Cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Mazziotta, C.; Lanzillotti, C.; Gafà, R.; Touzé, A.; Durand, M.-A.; Martini, F.; Rotondo, J.C. The Role of Histone Post-Translational Modifications in Merkel Cell Carcinoma. Front. Oncol. 2022, 12, 832047. [Google Scholar] [CrossRef]
- Tims, H.S.; Gurunathan, K.; Levitus, M.; Widom, J. Dynamics of Nucleosome Invasion by DNA Binding Proteins. J. Mol. Biol. 2011, 411, 430–448. [Google Scholar] [CrossRef]
- Huertas, J.; MacCarthy, C.M.; Schöler, H.R.; Cojocaru, V. Nucleosomal DNA Dynamics Mediate Oct4 Pioneer Factor Binding. Biophys. J. 2020, 118, 2280–2296. [Google Scholar] [CrossRef]
- Kulaeva, O.I.; Hsieh, F.-K.; Chang, H.-W.; Luse, D.S.; Studitsky, V.M. Mechanism of Transcription through a Nucleosome by RNA Polymerase II. Biochim. Biophys. Acta 2013, 1829, 76–83. [Google Scholar] [CrossRef]
- Joseph, J.A.; Reinhardt, A.; Aguirre, A.; Chew, P.Y.; Russell, K.O.; Espinosa, J.R.; Garaizar, A.; Collepardo-Guevara, R. Physics-Driven Coarse-Grained Model for Biomolecular Phase Separation with near-Quantitative Accuracy. Nat. Comput. Sci. 2021, 1, 732–743. [Google Scholar] [CrossRef]
- Bilokapic, S.; Strauss, M.; Halic, M. Structural Rearrangements of the Histone Octamer Translocate DNA. Nat. Commun. 2018, 9, 1330. [Google Scholar] [CrossRef] [Green Version]
- Sinha, K.K.; Gross, J.D.; Narlikar, G.J. Distortion of Histone Octamer Core Promotes Nucleosome Mobilization by a Chromatin Remodeler. Science 2017, 355, eaaa3761. [Google Scholar] [CrossRef] [PubMed]
- Sanulli, S.; Trnka, M.J.; Dharmarajan, V.; Tibble, R.W.; Pascal, B.D.; Burlingame, A.L.; Griffin, P.R.; Gross, J.D.; Narlikar, G.J. HP1 Reshapes Nucleosome Core to Promote Phase Separation of Heterochromatin. Nature 2019, 575, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Armache, J.P.; Gamarra, N.; Johnson, S.L.; Leonard, J.D.; Wu, S.; Narlikar, G.J.; Cheng, Y. Cryo-EM Structures of Remodeler-Nucleosome Intermediates Suggest Allosteric Control through the Nucleosome. eLife 2019, 8, e46057. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Prasanna, C.; Soman, A.; Pervushin, K.; Nordenskiöld, L. Dynamic Networks Observed in the Nucleosome Core Particles Couple the Histone Globular Domains with DNA. Commun. Biol. 2020, 3, 639. [Google Scholar] [CrossRef]
- Peng, Y.; Li, S.; Onufriev, A.; Landsman, D.; Panchenko, A.R. Binding of Regulatory Proteins to Nucleosomes Is Modulated by Dynamic Histone Tails. Nat. Commun. 2021, 12, 5280. [Google Scholar] [CrossRef]
- Rabdano, S.O.; Shannon, M.D.; Izmailov, S.A.; Gonzalez Salguero, N.; Zandian, M.; Purusottam, R.N.; Poirier, M.G.; Skrynnikov, N.R.; Jaroniec, C.P. Histone H4 Tails in Nucleosomes: A Fuzzy Interaction with DNA. Angew. Chem. Int. Ed. 2021, 60, 6480–6487. [Google Scholar] [CrossRef]
- Materese, C.K.; Savelyev, A.; Papoian, G.A. Counterion Atmosphere and Hydration Patterns near a Nucleosome Core Particle. J. Am. Chem. Soc. 2009, 131, 15005–15013. [Google Scholar] [CrossRef]
- Winogradoff, D.; Aksimentiev, A. Molecular Mechanism of Spontaneous Nucleosome Unraveling. J. Mol. Biol. 2019, 431, 323–335. [Google Scholar] [CrossRef]
- Huertas, J.; Cojocaru, V. Breaths, Twists, and Turns of Atomistic Nucleosomes. J. Mol. Biol. 2020, 433, 166744. [Google Scholar] [CrossRef]
- Armeev, G.A.; Kniazeva, A.S.; Komarova, G.A.; Kirpichnikov, M.P.; Shaytan, A.K. Histone Dynamics Mediate DNA Unwrapping and Sliding in Nucleosomes. Nat. Commun. 2021, 12, 2387. [Google Scholar] [CrossRef]
- Rychkov, G.N.; Ilatovskiy, A.V.; Nazarov, I.B.; Shvetsov, A.V.; Lebedev, D.V.; Konev, A.Y.; Isaev-Ivanov, V.V.; Onufriev, A.V. Partially Assembled Nucleosome Structures at Atomic Detail. Biophys. J. 2017, 112, 460–472. [Google Scholar] [CrossRef]
- Bowerman, S.; Wereszczynski, J. Effects of MacroH2A and H2A.Z on Nucleosome Dynamics as Elucidated by Molecular Dynamics Simulations. Biophys. J. 2016, 110, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, S.; Hickok, R.J.; Wereszczynski, J. Unique Dynamics in Asymmetric MacroH2A-H2A Hybrid Nucleosomes Result in Increased Complex Stability. J. Phys. Chem. B 2019, 123, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Yuan, C.; Hua, X.; Zhang, Z. Molecular Mechanism of Histone Variant H2A.B on Stability and Assembly of Nucleosome and Chromatin Structures. Epigenetics Chromatin 2020, 13, 28. [Google Scholar] [CrossRef]
- Kohestani, H.; Wereszczynski, J. Effects of H2A.B Incorporation on Nucleosome Structures and Dynamics. Biophys. J. 2021, 120, 1498–1509. [Google Scholar] [CrossRef]
- Chang, L.; Takada, S. Histone Acetylation Dependent Energy Landscapes in Tri-Nucleosome Revealed by Residue-Resolved Molecular Simulations. Sci. Rep. 2016, 6, 34441. [Google Scholar] [CrossRef]
- Rajagopalan, M.; Balasubramanian, S.; Ioshikhes, I.; Ramaswamy, A. Structural Dynamics of Nucleosome Mediated by Acetylations at H3K56 and H3K115,122. Eur. Biophys. J. 2017, 46, 471–484. [Google Scholar] [CrossRef]
- Fu, I.; Geacintov, N.E.; Broyde, S. Molecular Dynamics Simulations Reveal How H3K56 Acetylation Impacts Nucleosome Structure to Promote DNA Exposure for Lesion Sensing. DNA Repair 2021, 107, 103201. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Park, S.; Maffeo, C.; Ha, T.; Aksimentiev, A. DNA Sequence and Methylation Prescribe the Inside-out Conformational Dynamics and Bending Energetics of DNA Minicircles. Nucleic Acids Res. 2021, 49, 11459–11475. [Google Scholar] [CrossRef]
- Collepardo-Guevara, R.; Portella, G.; Vendruscolo, M.; Frenkel, D.; Schlick, T.; Orozco, M. Chromatin Unfolding by Epigenetic Modifications Explained by Dramatic Impairment of Internucleosome Interactions: A Multiscale Computational Study. J. Am. Chem. Soc. 2015, 137, 10205–10215. [Google Scholar] [CrossRef]
- Shaytan, A.K.; Landsman, D.; Panchenko, A.R. Nucleosome Adaptability Conferred by Sequence and Structural Variations in Histone H2A-H2B Dimers. Curr. Opin. Struct. Biol. 2015, 32, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Gansen, A.; Felekyan, S.; Kühnemuth, R.; Lehmann, K.; Tóth, K.; Seidel, C.A.M.; Langowski, J. High Precision FRET Studies Reveal Reversible Transitions in Nucleosomes between Microseconds and Minutes. Nat. Commun. 2018, 9, 4628. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Dai, L.; Li, C.; Shi, L.; Huang, Y.; Guo, Z.; Wu, F.; Zhu, P.; Zhou, Z. Structural Basis of Nucleosome Dynamics Modulation by Histone Variants H2A.B and H2A.Z.2.2. EMBO J. 2021, 40, e105907. [Google Scholar] [CrossRef] [PubMed]
- Bernier, M.; Luo, Y.; Nwokelo, K.C.; Goodwin, M.; Dreher, S.J.; Zhang, P.; Parthun, M.R.; Fondufe-Mittendorf, Y.; Ottesen, J.J.; Poirier, M.G. Linker Histone H1 and H3K56 Acetylation Are Antagonistic Regulators of Nucleosome Dynamics. Nat. Commun. 2015, 6, 10152. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.; Lee, T.-H. Lysine Acetylation Facilitates Spontaneous DNA Dynamics in the Nucleosome. J. Phys. Chem. B 2015, 119, 15001–15005. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, T.; Hayashi, G.; Okamoto, A. Regulation of the Stability of the Histone H2A–H2B Dimer by H2A Tyr57 Phosphorylation. Biochemistry 2017, 56, 4767–4772. [Google Scholar] [CrossRef]
- Krajewski, W.A.; Li, J.; Dou, Y. Effects of Histone H2B Ubiquitylation on the Nucleosome Structure and Dynamics. Nucleic Acids Res. 2018, 46, 7631–7642. [Google Scholar] [CrossRef]
- Bilokapic, S.; Strauss, M.; Halic, M. Histone Octamer Rearranges to Adapt to DNA Unwrapping. Nat. Struct. Mol. Biol. 2018, 25, 101–108. [Google Scholar] [CrossRef]
- Ranjan, A.; Wang, F.; Mizuguchi, G.; Wei, D.; Huang, Y.; Wu, C. H2A Histone-Fold and DNA Elements in Nucleosome Activate SWR1-Mediated H2A.Z Replacement in Budding Yeast. eLife 2015, 4, e06845. [Google Scholar] [CrossRef]
- Dai, L.; Xiao, X.; Pan, L.; Shi, L.; Xu, N.; Zhang, Z.; Feng, X.; Ma, L.; Dou, S.; Wang, P.; et al. Recognition of the Inherently Unstable H2A Nucleosome by Swc2 Is a Major Determinant for Unidirectional H2A.Z Exchange. Cell Rep. 2021, 35, 109183. [Google Scholar] [CrossRef]
- Suto, R.K.; Clarkson, M.J.; Tremethick, D.J.; Luger, K. Crystal Structure of a Nucleosome Core Particle Containing the Variant Histone H2A.Z. Nat. Struct. Biol. 2000, 7, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. In Protein Structure Prediction; Kihara, D., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; pp. 1–15. ISBN 978-1-4939-0366-5. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-J. 3DNA: A Software Package for the Analysis, Rebuilding and Visualization of Three-Dimensional Nucleic Acid Structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A Refined Force Field for DNA Simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef]
- Yoo, J.; Aksimentiev, A. New Tricks for Old Dogs: Improving the Accuracy of Biomolecular Force Fields by Pair-Specific Corrections to Non-Bonded Interactions. Phys. Chem. Chem. Phys. 2018, 20, 8432–8449. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Voevodin, V.V.; Antonov, A.S.; Nikitenko, D.A.; Shvets, P.A.; Sobolev, S.I.; Sidorov, I.Y.; Stefanov, K.S.; Voevodin, V.V.; Zhumatiy, S.A. Supercomputer Lomonosov-2: Large Scale, Deep Monitoring and Fine Analytics for the User Community. Supercomput. Front. Innov. 2019, 6, 4–11. [Google Scholar] [CrossRef]
- Myers-Turnbull, D.; Bliven, S.E.; Rose, P.W.; Aziz, Z.K.; Youkharibache, P.; Bourne, P.E.; Prlić, A. Systematic Detection of Internal Symmetry in Proteins Using CE-Symm. J. Mol. Biol. 2014, 426, 2255–2268. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array Programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Solving Software Challenges for Exascale; Markidis, S., Laure, E., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2015; Volume 8759, pp. 3–27. ISBN 978-3-319-15975-1. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 27–28, 33–38. [Google Scholar] [CrossRef]
- Nguyen, H.; Case, D.A.; Rose, A.S. NGLview–Interactive Molecular Graphics for Jupyter Notebooks. Bioinformatics 2018, 34, 1241–1242. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Lowary, P.T.; Widom, J. New DNA Sequence Rules for High Affinity Binding to Histone Octamer and Sequence-Directed Nucleosome Positioning. J. Mol. Biol. 1998, 276, 19–42. [Google Scholar] [CrossRef]
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent Mediated Interactions in the Structure of the Nucleosome Core Particle at 1.9 a Resolution. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef]
- Shaytan, A.K.; Armeev, G.A.; Goncearenco, A.; Zhurkin, V.B.; Landsman, D.; Panchenko, A.R. Coupling between Histone Conformations and DNA Geometry in Nucleosomes on a Microsecond Timescale: Atomistic Insights into Nucleosome Functions. J. Mol. Biol. 2016, 428, 221–237. [Google Scholar] [CrossRef] [Green Version]
- Valieva, M.E.; Armeev, G.A.; Kudryashova, K.S.; Gerasimova, N.S.; Shaytan, A.K.; Kulaeva, O.I.; McCullough, L.L.; Formosa, T.; Georgiev, P.G.; Kirpichnikov, M.P.; et al. Large-Scale ATP-Independent Nucleosome Unfolding by a Histone Chaperone. Nat. Struct. Mol. Biol. 2016, 23, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Eirín-López, J.M.; González-Romero, R.; Dryhurst, D.; Ishibashi, T.; Ausió, J. The Evolutionary Differentiation of Two Histone H2A.Z Variants in Chordates (H2A.Z-1 and H2A.Z-2) Is Mediated by a Stepwise Mutation Process That Affects Three Amino Acid Residues. BMC Evol. Biol. 2009, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Giaimo, B.D.; Ferrante, F.; Herchenröther, A.; Hake, S.B.; Borggrefe, T. The Histone Variant H2A.Z in Gene Regulation. Epigenetics Chromatin 2019, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Draizen, E.J.; Shaytan, A.K.; Mariño-Ramírez, L.; Talbert, P.B.; Landsman, D.; Panchenko, A.R. HistoneDB 2.0: A Histone Database with Variants—an Integrated Resource to Explore Histones and Their Variants. Database 2016, 2016, baw014. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Xu, N.; Zhou, Z. NMR Investigations on H2A-H2B Heterodimer Dynamics Conferred by Histone Variant H2A.Z. Biochem. Biophys. Res. Commun. 2019, 518, 752–758. [Google Scholar] [CrossRef]
- Moriwaki, Y.; Yamane, T.; Ohtomo, H.; Ikeguchi, M.; Kurita, J.I.; Sato, M.; Nagadoi, A.; Shimojo, H.; Nishimura, Y. Solution Structure of the Isolated Histone H2A-H2B Heterodimer. Sci. Rep. 2016, 6, 24999. [Google Scholar] [CrossRef]
- Sato, S.; Takizawa, Y.; Hoshikawa, F.; Dacher, M.; Tanaka, H.; Tachiwana, H.; Kujirai, T.; Iikura, Y.; Ho, C.-H.; Adachi, N.; et al. Cryo-EM Structure of the Nucleosome Core Particle Containing Giardia Lamblia Histones. Nucleic Acids Res. 2021, 49, 8934–8946. [Google Scholar] [CrossRef]
- Bowman, G.D.; Poirier, M.G. Post-Translational Modifications of Histones That Influence Nucleosome Dynamics. Chem. Rev. 2015, 115, 2274–2295. [Google Scholar] [CrossRef]
- Fenley, A.T.; Anandakrishnan, R.; Kidane, Y.H.; Onufriev, A.V. Modulation of Nucleosomal DNA Accessibility via Charge-Altering Post-Translational Modifications in Histone Core. Epigenetics Chromatin 2018, 11, 11. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kniazeva, A.S.; Armeev, G.A.; Shaytan, A.K. H2A-H2B Histone Dimer Plasticity and Its Functional Implications. Cells 2022, 11, 2837. https://doi.org/10.3390/cells11182837
Kniazeva AS, Armeev GA, Shaytan AK. H2A-H2B Histone Dimer Plasticity and Its Functional Implications. Cells. 2022; 11(18):2837. https://doi.org/10.3390/cells11182837
Chicago/Turabian StyleKniazeva, Anastasiia S., Grigoriy A. Armeev, and Alexey K. Shaytan. 2022. "H2A-H2B Histone Dimer Plasticity and Its Functional Implications" Cells 11, no. 18: 2837. https://doi.org/10.3390/cells11182837
APA StyleKniazeva, A. S., Armeev, G. A., & Shaytan, A. K. (2022). H2A-H2B Histone Dimer Plasticity and Its Functional Implications. Cells, 11(18), 2837. https://doi.org/10.3390/cells11182837