
In Vitro Characterization of a Tissue Renin-Angiotensin System in Human Nucleus Pulposus Cells

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human NP Cells Isolation and Expansion
2.2. Experimental Design
2.3. Cell Metabolic Activity
2.4. Immunofluorescent Staining
2.5. Gene Expression Analysis
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Western Blot
2.8. Statistical Analysis
3. Results
3.1. Evaluation of tRAS Protein Translation in Human NP Cells via Immunofluorescence
3.2. Impact of Angiotensin II Addition on Human Nucleus Pulposus Cells
3.2.1. Cell Metabolic Activity Analysis
3.2.2. Gene Expression Analysis
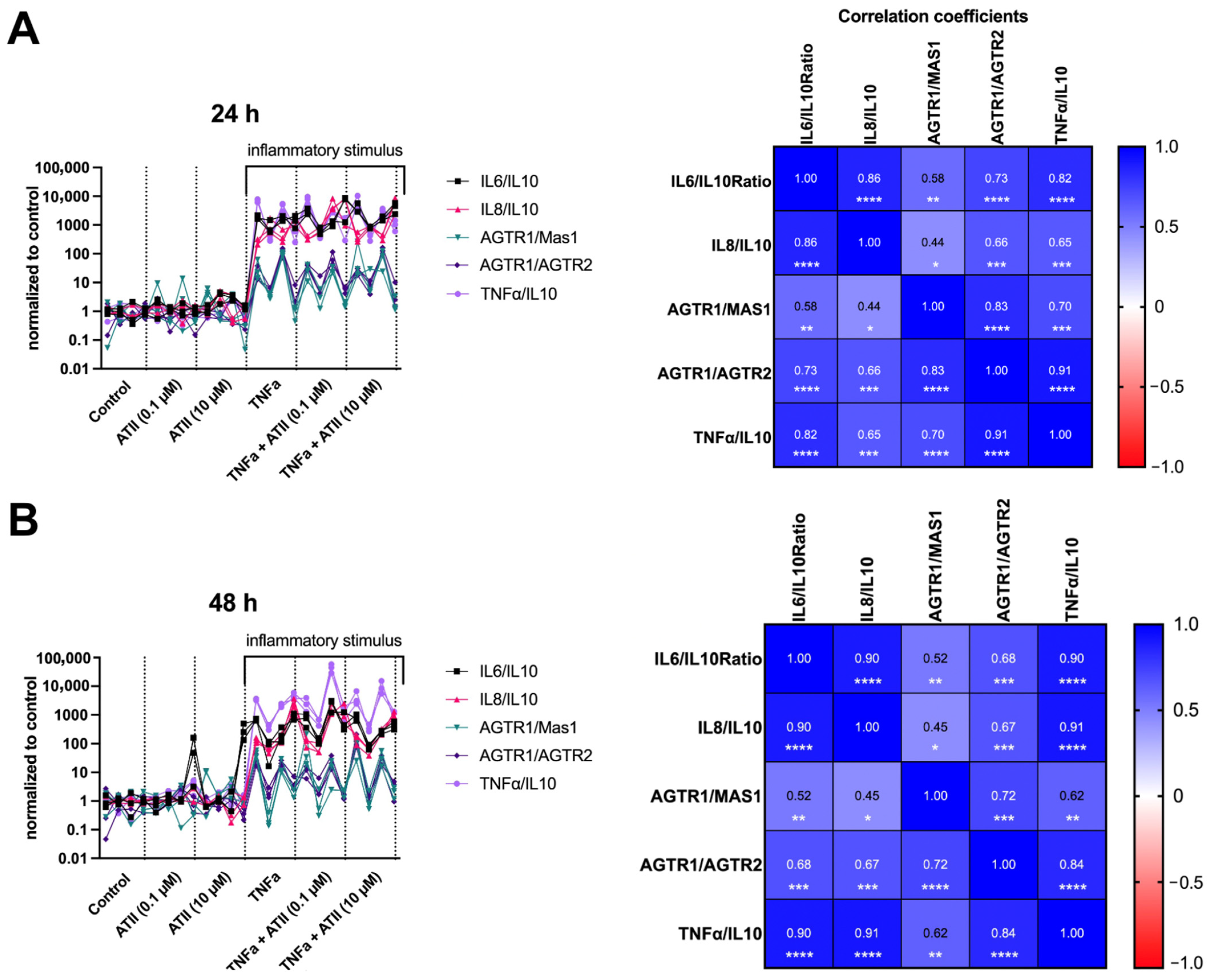
3.2.3. Nitrite Release from the Metabolism of Nitric Oxide
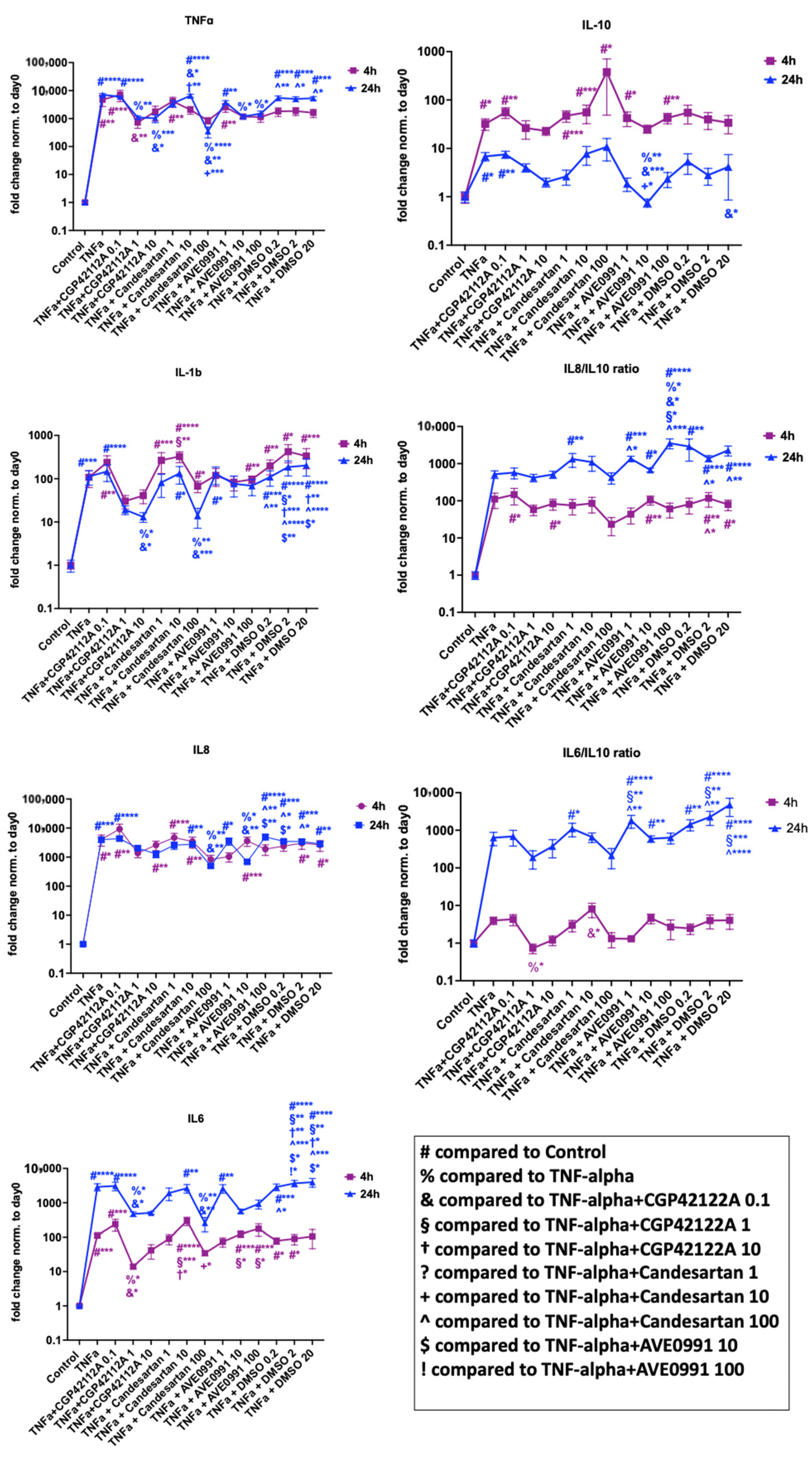
3.3. Impact of Pathological Axis (AGTR1) Inhibition versus Protective Axis (AGTR2, MAS1) Stimulation on Human Nucleus Pulposus Cells
3.3.1. Cell Metabolic Activity Analysis
3.3.2. Gene Expression Analysis
Inflammation
Tissue Degeneration and Phenotype Modulation
tRAS Markers
3.3.3. Protein Validation
4. Discussion
4.1. Main Findings
4.2. Role of Angiotensin II in Human NP Cells
4.3. ACE2 Is Expressed in Human NP Cells
4.4. tRAS Expression Is Correlated with the Inflammatory State of NP Cells
4.5. Modulation of tRAS as a Potential Target in Intervertebral Disc Disease
4.6. Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koes, B.W.; van Tulder, M.W.; Thomas, S. Diagnosis and Treatment of Low Back Pain. BMJ 2006, 332, 1430–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; March, L.; Zheng, X.; Huang, J.; Wang, X.; Zhao, J.; Blyth, F.M.; Smith, E.; Buchbinder, R.; Hoy, D. Global Low Back Pain Prevalence and Years Lived with Disability from 1990 to 2017: Estimates from the Global Burden of Disease Study 2017. Ann. Transl. Med. 2020, 8, 299. [Google Scholar] [CrossRef] [PubMed]
- Lyu, F.-J.; Cui, H.; Pan, H.; MC Cheung, K.; Cao, X.; Iatridis, J.C.; Zheng, Z. Painful Intervertebral Disc Degeneration and Inflammation: From Laboratory Evidence to Clinical Interventions. Bone Res. 2021, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.; Evans, H.; Trivedi, J.; Menage, J. Histology and Pathology of the Human Intervertebral Disc. J. Bone Joint Surg. Am. 2006, 88 (Suppl. S2), 10–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binch, A.L.A.; Cole, A.A.; Breakwell, L.M.; Michael, A.L.R.; Chiverton, N.; Creemers, L.B.; Cross, A.K.; Le Maitre, C.L. Nerves Are More Abundant than Blood Vessels in the Degenerate Human Intervertebral Disc. Arthritis Res. Ther. 2015, 17, 370. [Google Scholar] [CrossRef] [Green Version]
- Fournier, D.E.; Kiser, P.K.; Shoemaker, J.K.; Battié, M.C.; Séguin, C.A. Vascularization of the Human Intervertebral Disc: A Scoping Review. JOR Spine 2020, 3, e1123. [Google Scholar] [CrossRef]
- He, M.; Pang, J.; Sun, H.; Zheng, G.; Lin, Y.; Ge, W. P14ARF Inhibits Regional Inflammation and Vascularization in Intervertebral Disc Degeneration by Upregulating TIMP3. Am. J. Physiol. Cell Physiol. 2020, 318, C751–C761. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, B.; Liu, W.; Wang, P.; Lv, X.; Chen, S.; Shao, Z. The Role of Structure and Function Changes of Sensory Nervous System in Intervertebral Disc-Related Low Back Pain. Osteoarthr. Cartil. 2021, 29, 17–27. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, B.; Luo, Z.-J. The Immune Privilege of the Intervertebral Disc: Implications for Intervertebral Disc Degeneration Treatment. Int. J. Med. Sci. 2020, 17, 685–692. [Google Scholar] [CrossRef] [Green Version]
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-Regulatory Renin–Angiotensin System in Cardiovascular Disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef]
- Saravi, B.; Li, Z.; Lang, C.N.; Schmid, B.; Lang, F.K.; Grad, S.; Alini, M.; Richards, R.G.; Schmal, H.; Südkamp, N.; et al. The Tissue Renin-Angiotensin System and Its Role in the Pathogenesis of Major Human Diseases: Quo Vadis? Cells 2021, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Skov, J.; Persson, F.; Frøkiær, J.; Christiansen, J.S. Tissue Renin-Angiotensin Systems: A Unifying Hypothesis of Metabolic Disease. Front. Endocrinol 2014, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, T.; Steckelings, U.M.; dos Santos, R.A.S. The Protective Arm of the Renin Angiotensin System (RAS): Functional Aspects and Therapeutic Implications; Academic Press: Cambridge, MA, USA, 2015; ISBN 0-12-801485-7. [Google Scholar]
- Sriram, K.; Loomba, R.; Insel, P.A. Targeting the Renin−angiotensin Signaling Pathway in COVID-19: Unanswered Questions, Opportunities, and Challenges. Proc. Natl. Acad. Sci. USA 2020, 117, 29274–29282. [Google Scholar] [CrossRef] [PubMed]
- Hasan, L.K.; Deadwiler, B.; Haratian, A.; Bolia, I.K.; Weber, A.E.; Petrigliano, F.A. Effects of COVID-19 on the Musculoskeletal System: Clinician’s Guide. Orthop. Res. Rev. 2021, 13, 141–150. [Google Scholar] [CrossRef]
- dos Santos, P.K.; Sigoli, E.; Bragança, L.J.G.; Cornachione, A.S. The Musculoskeletal Involvement After Mild to Moderate COVID-19 Infection. Front. Physiol. 2022, 13, 813924. [Google Scholar] [CrossRef]
- Džubera, A.; Chochol, J.; Illéš, R.; Chocholová, A.; Zemková, E. Vertebral Algic Syndrome Treatment in Long COVID-Cases Reports. Int. J. Environ. Res. Public Health 2021, 18, 11457. [Google Scholar] [CrossRef]
- Bakılan, F.; Gökmen, İ.G.; Ortanca, B.; Uçan, A.; Eker Güvenç, Ş.; Şahin Mutlu, F.; Gökmen, H.M.; Ekim, A. Musculoskeletal Symptoms and Related Factors in Postacute COVID-19 Patients. Int. J. Clin. Pract. 2021, 75, e14734. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takeshita, H.; Rakugi, H. ACE2, Angiotensin 1-7 and Skeletal Muscle: Review in the Era of COVID-19. Clin. Sci. 2020, 134, 3047–3062. [Google Scholar] [CrossRef]
- Semprun-Prieto, L.C.; Sukhanov, S.; Yoshida, T.; Rezk, B.M.; Gonzalez-Villalobos, R.A.; Vaughn, C.; Michael Tabony, A.; Delafontaine, P. Angiotensin II Induced Catabolic Effect and Muscle Atrophy Are Redox Dependent. Biochem. Biophys. Res. Commun. 2011, 409, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Saravi, B.; Lang, G.; Ülkümen, S.; Burchard, T.; Weihrauch, V.; Patzelt, S.; Boeker, M.; Li, Z.; Woelber, J.P. The Tissue Renin-Angiotensin System (TRAS) and the Impact of Its Inhibition on Inflammation and Bone Loss in the Periodontal Tissue. Eur. Cell Mater. 2020, 40, 203–226. [Google Scholar] [CrossRef]
- Saravi, B.; Vollmer, A.; Lang, G.; Adolphs, N.; Li, Z.; Giers, V.; Stoll, P. Impact of Renin-Angiotensin System Inhibitors and Beta-Blockers on Dental Implant Stability. Int. J. Implant. Dent. 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, A.; Scavone, C.; Rafaniello, C.; Ferrajolo, C.; Racagni, G.; Berrino, L.; Paolisso, G.; Rossi, F.; Capuano, A. Renin-Angiotensin System and Coronavirus Disease 2019: A Narrative Review. Front. Cardiovasc. Med. 2020, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.H. COVID-19-Does This Disease Kill Due to Imbalance of the Renin Angiotensin System (RAS) Caused by Genetic and Gender Differences in the Response to Viral ACE 2 Attack? Heart Lung Circ. 2020, 29, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Wiese, O.J.; Allwood, B.W.; Zemlin, A.E. COVID-19 and the Renin-Angiotensin System (RAS): A Spark That Sets the Forest Alight? Med. Hypotheses 2020, 144, 110231. [Google Scholar] [CrossRef]
- Seltzer, S. Linking ACE2 and Angiotensin II to Pulmonary Immunovascular Dysregulation in SARS-CoV-2 Infection. Int. J. Infect. Dis. 2020, 101, 42–45. [Google Scholar] [CrossRef]
- Pucci, F.; Bogaerts, P.; Rooman, M. Modeling the Molecular Impact of SARS-CoV-2 Infection on the Renin-Angiotensin System. Viruses 2020, 12, 1367. [Google Scholar] [CrossRef]
- Pucci, F.; Annoni, F.; Dos Santos, R.A.S.; Taccone, F.S.; Rooman, M. Quantifying Renin-Angiotensin-System Alterations in COVID-19. Cells 2021, 10, 2755. [Google Scholar] [CrossRef]
- Morimoto, R.; Akeda, K.; Iida, R.; Nishimura, A.; Tsujii, M.; Obata, S.; Kasai, Y.; Uchida, A.; Sudo, A. Tissue Renin-Angiotensin System in the Intervertebral Disc. Spine (Phila Pa 1976) 2013, 38, E129–E136. [Google Scholar] [CrossRef]
- Li, Z.; Wystrach, L.; Bernstein, A.; Grad, S.; Alini, M.; Richards, R.G.; Kubosch, D.; Südkamp, N.; Izadpanah, K.; Kubosch, E.J.; et al. The Tissue-Renin-Angiotensin-System of the Human Intervertebral Disc. Eur. Cell Mater. 2020, 40, 115–132. [Google Scholar] [CrossRef]
- Saravi, B.; Li, Z.; Pfannkuche, J.; Wystrach, L.; Häckel, S.; Albers, C.E.; Grad, S.; Alini, M.; Richards, R.G.; Lang, C.; et al. Angiotensin II Type 1 Receptor Antagonist Losartan Inhibits TNF-α-Induced Inflammation and Degeneration Processes in Human Nucleus Pulposus Cells. Appl. Sci. 2021, 11, 417. [Google Scholar] [CrossRef]
- Gromotowicz-Poplawska, A.; Szoka, P.; Kolodziejczyk, P.; Kramkowski, K.; Wojewodzka-Zelezniakowicz, M.; Chabielska, E. New Agents Modulating the Renin-Angiotensin-Aldosterone System-Will There Be a New Therapeutic Option? Exp. Biol. Med. 2016, 241, 1888–1899. [Google Scholar] [CrossRef] [Green Version]
- Koh, K.K.; Quon, M.J.; Han, S.H.; Chung, W.-J.; Lee, Y.; Shin, E.K. Anti-Inflammatory and Metabolic Effects of Candesartan in Hypertensive Patients. Int. J. Cardiol. 2006, 108, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, S.; Merino-Serrais, P.; Di Grande, A.; Dussmann, H.; Prehn, J.H.M.; Ní Chonghaile, T.; Henshall, D.C.; Jimenez-Mateos, E.M. The Anti-Inflammatory Compound Candesartan Cilexetil Improves Neurological Outcomes in a Mouse Model of Neonatal Hypoxia. Front. Immunol. 2019, 10, 1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T. Targeting AT2 Receptors in Renal Disease. Am. J. Physiol. Ren. Physiol. 2021, 320, F1025–F1027. [Google Scholar] [CrossRef]
- Skiba, D.S.; Nosalski, R.; Mikolajczyk, T.P.; Siedlinski, M.; Rios, F.J.; Montezano, A.C.; Jawien, J.; Olszanecki, R.; Korbut, R.; Czesnikiewicz-Guzik, M.; et al. Anti-Atherosclerotic Effect of the Angiotensin 1-7 Mimetic AVE0991 Is Mediated by Inhibition of Perivascular and Plaque Inflammation in Early Atherosclerosis. Br. J. Pharmacol. 2017, 174, 4055–4069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Rueden, C.T.; Hiner, M.C.; Eliceiri, K.W. The ImageJ Ecosystem: An Open Platform for Biomedical Image Analysis. Mol. Reprod. Dev. 2015, 82, 518–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslova, I.; Slobodin, T.; Mykhailovska, N.; Lisyanyi, N. Dynamics of Proinflammatory and Anti-Inflammatory Cytokines in Blood Plasma in the Treatment of Back Pain with Non-Steroidal Anti-Inflammatory Drugs. Sci. Med. Sci. 2019, 6, 32–36. [Google Scholar] [CrossRef]
- Teodorczyk-Injeyan, J.A.; Triano, J.J.; Injeyan, H.S. Nonspecific Low Back Pain: Inflammatory Profiles of Patients With Acute and Chronic Pain. Clin. J. Pain 2019, 35, 818–825. [Google Scholar] [CrossRef]
- Sun, K.; Sun, X.; Sun, J.; Jiang, Y.; Lin, F.; Kong, F.; Li, F.; Zhu, J.; Huan, L.; Zheng, B.; et al. Tissue Renin-Angiotensin System (TRAS) Induce Intervertebral Disc Degeneration by Activating Oxidative Stress and Inflammatory Reaction. Oxidative Med. Cell. Longev. 2021, 2021, 3225439. [Google Scholar] [CrossRef]
- Lee, Y.H.; Mungunsukh, O.; Tutino, R.L.; Marquez, A.P.; Day, R.M. Angiotensin-II-Induced Apoptosis Requires Regulation of Nucleolin and Bcl-XL by SHP-2 in Primary Lung Endothelial Cells. J. Cell Sci. 2010, 123, 1634–1643. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The Protein Expression Profile of ACE2 in Human Tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Zamorano Cuervo, N.; Grandvaux, N. ACE2: Evidence of Role as Entry Receptor for SARS-CoV-2 and Implications in Comorbidities. eLife 2020, 9, e61390. [Google Scholar] [CrossRef]
- Rodrigues-Machado, M.G.; Magalhães, G.S.; Cardoso, J.A.; Kangussu, L.M.; Murari, A.; Caliari, M.V.; Oliveira, M.L.; Cara, D.C.; Noviello, M.L.M.; Marques, F.D.; et al. AVE 0991, a Non-Peptide Mimic of Angiotensin-(1-7) Effects, Attenuates Pulmonary Remodelling in a Model of Chronic Asthma. Br. J. Pharmacol. 2013, 170, 835–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiemer, G.; Dobrucki, L.W.; Louka, F.R.; Malinski, T.; Heitsch, H. AVE 0991, a Nonpeptide Mimic of the Effects of Angiotensin-(1–7) on the Endothelium. Hypertension 2002, 40, 847–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1–7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [Green Version]
- Rivas-Santisteban, R.; Lillo, J.; Muñoz, A.; Rodríguez-Pérez, A.I.; Labandeira-García, J.L.; Navarro, G.; Franco, R. Novel Interactions Involving the Mas Receptor Show Potential of the Renin–Angiotensin System in the Regulation of Microglia Activation: Altered Expression in Parkinsonism and Dyskinesia. Neurotherapeutics 2021, 18, 998–1016. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.; Lu, Q.; Ren, D.; Sun, X.; Rousselle, T.; Li, J.; Leng, J. AMPK: A Balancer of the Renin–Angiotensin System. Biosci. Rep. 2019, 39, BSR20181994. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Alfonso, M.S.; González, C. Nitric Oxide and the Renin-Angiotensin System. Is There a Physiological Interplay between the Systems? J. Hypertens. 1999, 17, 1355–1361. [Google Scholar] [CrossRef]
- Olson, S.C.; Dowds, T.A.; Pino, P.A.; Barry, M.T.; Burke-Wolin, T. ANG II Stimulates Endothelial Nitric Oxide Synthase Expression in Bovine Pulmonary Artery Endothelium. Am. J. Physiol. 1997, 273, L315–L321. [Google Scholar] [CrossRef]
- Saito, S.; Hirata, Y.; Emori, T.; Imai, T.; Marumo, F. Angiotensin II Activates Endothelial Constitutive Nitric Oxide Synthase via AT1 Receptors. Hypertens. Res. 1996, 19, 201–206. [Google Scholar] [CrossRef]
- Hennington, B.S.; Zhang, H.; Miller, M.T.; Granger, J.P.; Reckelhoff, J.F. Angiotensin II Stimulates Synthesis of Endothelial Nitric Oxide Synthase. Hypertension 1998, 31, 283–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, I.; Kawahara, Y.; Tsuda, T.; Okuda, M.; Yokoyama, M. Angiotensin II Inhibits Cytokine-Stimulated Inducible Nitric Oxide Synthase Expression in Vascular Smooth Muscle Cells. J. Biol. Chem. 1994, 269, 11628–11633. [Google Scholar] [CrossRef]
- Chandler, L.J.; Kopnisky, K.; Richards, E.; Crews, F.T.; Sumners, C. Angiotensin II Decreases Inducible Nitric Oxide Synthase Expression in Rat Astroglial Cultures. Am. J. Physiol. 1995, 268, C700–C707. [Google Scholar] [CrossRef] [PubMed]
- Montón, M.; López-Farré, A.; Mosquera, J.R.; Sánchez de Miguel, L.; García-Durán, M.; Sierra, M.P.; Bellver, T.; Rico, L.; Casado, S. Endogenous Angiotensin II Produced by Endothelium Regulates Interleukin-1beta-Stimulated Nitric Oxide Generation in Rat Isolated Vessels. Hypertension 1997, 30, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Labandeira-Garcia, J.L.; Labandeira, C.M.; Valenzuela, R.; Pedrosa, M.A.; Quijano, A.; Rodriguez-Perez, A.I. Drugs Modulating Renin-Angiotensin System in COVID-19 Treatment. Biomedicines 2022, 10, 502. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Group | Medium Composition | Supplier Reference Number | Manufacturer |
|---|---|---|---|
| Control | Basal Medium | - | - |
| Angiotensin II | Basal Medium + Angiotensin II: 0.1 µM, 10 µM | A9525-5MG | Sigma Aldrich |
| TNF-α | Basal Medium + 10 ng/mL TNF-α | 210-TA-020 | R&D Systems Europe, Ltd. |
| TNF-α + Angiotensin II | Basal Medium + TNF-α: 10 ng/mL + Angiotensin II: 0.1 µM, 10 µM | TNF-α: 210-TA-020 Angiotensin II: A9525-5MG | TNF-α: R&D Systems Europe, Ltd. Angiotensin II: Sigma Aldrich |
| TNF-α + CGP42112A | Basal Medium + TNF-α: 10 ng/mL + CGP42112A: 0.1 µM, 1 µM, 10 µM | TNF-α: 210-TA-020 CGP42112A: C160-1MG | TNF-α: R&D Systems Europe, Ltd. CGP42112A: Sigma Aldrich |
| TNF-α + AVE 0991 | Basal Medium + TNF-α: 10 ng/mL + AVE 0991: 1 µM, 10 µM, 100 µM | TNF-α: 210-TA-020 AVE 0991: HY-15778-5MG | TNF-α: R&D Systems Europe, Ltd. AVE 0991: Lucerna-Chem |
| TNF-α + Candesartan | Basal Medium + TNF-α: 10 ng/mL + Candesartan: 1 µM, 10 µM, 100 µM | TNF-α: 210-TA-020 Candesartan: SML0245-10MG | TNF-α: R&D Systems Europe, Ltd. CGP42112A: Sigma Aldrich |
| TNF-α + DMSO | Basal Medium + Solvent control: DMSO (0.1 µL/mL, 1 µL/mL, 10 µL/mL) | - | - |
| Primary Antibody | Dilution | Supplier Reference Number | Manufacturer |
|---|---|---|---|
| AGTR1 Polyclonal Antibody | 1:125 | PA5-20812 | Invitrogen |
| AGTR2 Polyclonal Antibody | 1:125 | PA3-210 | Invitrogen |
| ACE2 Polyclonal Antibody | 1:100 | PA5-20040 | Invitrogen |
| MAS1 Polyclonal Antibody | 1:100 | PA5-77282 | Invitrogen |
| Gene Acronym | Gene Full Name | Primer Sequence or Catalogue Number | Reporter/Quencher |
|---|---|---|---|
| hRPLP0 | Human 60S acidic ribosomal protein P0 | Forward seq.: 5′-TGG GCA AGA ACA CCA TGA TG-3′ Reverse primer seq.: 5′-CGG ATA TGA GGC AGC AGT TTC-3′ | FAM/TAMRA |
| hACAN | Human aggrecan | Forward seq.: 5′-AGT CTT CAA GCC TCC TGT ACT CA-3′ Reverse primer seq.: 5′-CGG GAA GTG GCG GTA ACA-3′ | FAM/TAMRA |
| Human angiotensinogen | Hs01586213_m1 | ||
| hACE | Human angiotensin-converting enzyme | Hs00174179_m1 | FAM/NFQ-MGB |
| hAGTR1a | Human angiotensin-II receptor type 1 | Hs00258938_m1 | FAM/NFQ-MGB |
| hAGTR2 | Human angiotensin-II receptor type 2 | Hs02621316_s1 | FAM/NFQ-MGB |
| hMAS1 | Human MAS1 proto-oncogene, G protein-coupled receptor | Hs00267157_s1 | FAM/NFQ-MGB |
| hCTSD | Human cathepsin D | Hs00157205_m1 | FAM/NFQ-MGB |
| hCOL1A1 | Human collagen type 1 alpha 1 chain | Forward seq.: 5′-CCC TGG AAA GAA TGG AGA TGA T-3′ Reverse primer seq.: 5′-ACT GAA ACC TCT GTG TCC CTT CA-’3 | FAM/TAMRA |
| hCOL2A1 | Human collagen type 2 alpha 1 chain | Forward seq.: 5′-GGC AAT AGC AGG TTC ACG TAC A-3′ Reverse primer seq.: 5′-GAT AAC AGT CTT GCC CCA CTT ACC-3′ | FAM/TAMRA |
| hIL6 | Human interleukin 6 | Hs00985639_m1 | FAM/NFQ-MGB |
| hIL8 | Human interleukin 8 | Hs00174103_m1 | FAM/NFQ-MGB |
| hMMP1 | Human matrix-metalloproteinase 1 | Hs00899568_m1 | FAM/NFQ-MGB |
| hMMP3 | Human matrix-metalloproteinase 3 | Hs00968305_m1 | FAM/NFQ-MGB |
| hIL1-b | Human interleukin 1b | Hs00174097_m1 | FAM/NFQ-MGB |
| hTLR4 | Human toll-like receptor 4 | Hs00152939_m1 | FAM/NFQ-MGB |
| hTNFα | Human tumor necrosis factor α | Hs00174128_m1 | FAM/NFQ-MGB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saravi, B.; Li, Z.; Basoli, V.; Grad, S.; Häckel, S.; Albers, C.E.; Alini, M.; Schmal, H.; Obid, P.; Lang, G. In Vitro Characterization of a Tissue Renin-Angiotensin System in Human Nucleus Pulposus Cells. Cells 2022, 11, 3418. https://doi.org/10.3390/cells11213418
Saravi B, Li Z, Basoli V, Grad S, Häckel S, Albers CE, Alini M, Schmal H, Obid P, Lang G. In Vitro Characterization of a Tissue Renin-Angiotensin System in Human Nucleus Pulposus Cells. Cells. 2022; 11(21):3418. https://doi.org/10.3390/cells11213418
Chicago/Turabian StyleSaravi, Babak, Zhen Li, Valentina Basoli, Sibylle Grad, Sonja Häckel, Christoph E. Albers, Mauro Alini, Hagen Schmal, Peter Obid, and Gernot Lang. 2022. "In Vitro Characterization of a Tissue Renin-Angiotensin System in Human Nucleus Pulposus Cells" Cells 11, no. 21: 3418. https://doi.org/10.3390/cells11213418