

Levodopa-Induced Dyskinesia in Parkinson’s Disease: Pathogenesis and Emerging Treatment Strategies



Abstract
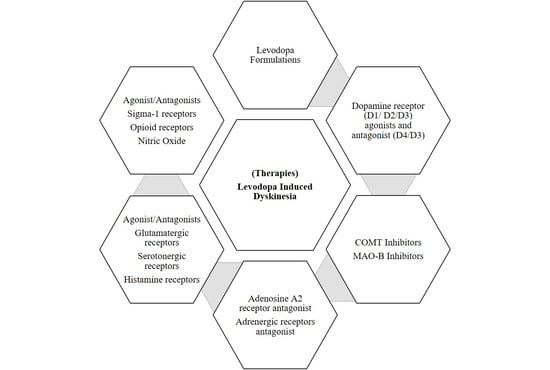
1. Introduction
2. Epidemiology and Risk Factors
3. Overview of LID in PD
4. Causes and Mechanisms
5. Dopaminergic Management and Treatment
6. Non-Dopaminergic Management and Treatment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeted System | Mechanism of Action | Side Effects | Name of Drug | Route of Administration | References |
|---|---|---|---|---|---|
| Adenosine | Adenosine A2A receptor antagonist | Falls in systolic and diastolic blood pressure, nausea, dizziness, insomnia, stiffness, vomiting, headache, hallucinations | ST1535 | Oral | [121,122] |
| Istradefylline (KW-6002) | Oral | [123] | |||
| Preladenant (SCH-420814) | Oral | [121,124] | |||
| Tozadenant (SYN115) | Oral | [121,124] | |||
| Vipadenant (V2006) | Oral | [121,124] | |||
| Ciforadenant (CPI-444, V81444) | Oral | [121,124] | |||
| PBF-509 | Oral | [121,124] | |||
| ST4206 | Oral | [121,124] | |||
| Nonspecific adenosine receptor antagonist | N/A | Caffeine | Oral | [125] | |
| Adrenergic | Alpha-2 adrenergic receptor antagonist | Nausea, vomiting, dysgeusia, headache, oral hypoesthesia, flushing, and increases in systolic and diastolic blood pressure; potentially severe adverse effects affecting the cardiovascular system | Idazoxan | Oral | [20] |
| Yohimbine | Oral | [126] | |||
| Fipamezole (JP-1730) | Oral | [127] | |||
| Beta-2 adrenergic receptor antagonist | Risk of bronchospasm, masked symptoms of hypoglycemia, bradycardia, hypotension, dizziness, lowered heart rate and blood pressure | Propranolol | Oral | [128] | |
| Salbutamol | Oral | [129] | |||
| Glutamatergic | NMDA receptor antagonist | confusion, worsening of hallucinations, peripheral oedema, skin rash, re-emergence of palpitations, nausea, dry mouth, swelling of feet and constipation | Amantadine & Amantadine extended-release capsules (Gocovri®) | Oral | [113,130,131] |
| Amnesia, dissociation | Traxoprodil | Oral | [132] | ||
| tiredness, vertigo, increased off time | Memantine | Oral | [133] | ||
| Neurotoxicity, hyperactivity | Dizocilpine (MK-801) | Oral | [134] | ||
| Perceptual distortions, detachment, anxiety, nausea, dissociation, confusion | Ketamine (FDA Approves IND for Ketamine in Parkinson Disease Dyskinesia, 2021) | Intravenous infusion, oral | [135,136] | ||
| GluN2B antagonist | Amnesia, dissociation | Ifenprodil | Oral | [132] | |
| Traxoprodil (CP-101606) | Oral, intravenous infusion | [132] | |||
| AMPA receptor antagonist | Dry eyes and mouth, hallucinations, worsening dyskinesia, anxiety/depression, breathing problems | Topiramate | Oral | [137] | |
| Somnolence, dizziness, worsening of dyskinesia | Perampanel | Oral | [138] | ||
| N/A (ongoing clinical trial) | Talampanel (LY-300164) | N/A (ongoing clinical trial) | [132] | ||
| Tezampanel (LY-293558) | N/A (ongoing clinical trial) | [132] | |||
| mGlu4R positive allosteric modulator | N/A (preclinical) | ADX88178 | N/A (preclinical) | [132] | |
| Lu AF21934 | N/A (preclinical) | [132] | |||
| on and off phenomenon, dyskinesia, headache | Foliglurax | Oral | [139] | ||
| mGluR5 receptor antagonist | Dizziness, hallucination, fatigue, nasopharyngitis, diarrhea, insomnia | Mavoglurant | Oral | [140] | |
| mGlu5R negative allosteric modulator | N/A (preclinical) | MPEP [2-Methyl-6-(phenylethynyl)-pyridine] | N/A (preclinical) | [141] | |
| MTEP (3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine) | N/A (preclinical) | [141] | |||
| memory loss, dizziness, hallucinations | Fenobam | Oral | [142] | ||
| Dizziness, nausea | Dipraglurant (ADX48621) | Oral | [141] | ||
| mGlu7R allosteric agonist | N/A (preclinical) | AMN082 (N,N′-dibenzhydrylethane-1,2-diamine) | N/A (preclinical) | [143] | |
| Serotonergic | 5-HT1AR agonist | Dizziness, increase in “off” time | Buspirone | Oral | [144] |
| N/A (ongoing clinical trial) | Befiradol (F-13,640; NLX-112) | N/A (ongoing clinical trial) | [145] | ||
| N/A (preclinical) | F13714 | N/A (preclinical) | [146] | ||
| 5-HT1BR agonist | N/A (preclinical) | CP-94253 | N/A (preclinical) | [147] | |
| 5-HT1AR/5-HT1BR agonist, 5-HT2CR antagonist | Fatigue, nausea, dizziness, mild somnolence/sedation | Eltoprazine | Oral | [88] | |
| 5-HT1AR agonist, partial D2 agonist | Worsening of parkinsonism, falls, aggravated tremor, somnolence, fatigue, headache, and arthralgia | Sarizotan | Oral | [148] | |
| Partial 5-HT1AR agonist | Sedation, muscle relaxation | Tandospirone | Oral | [149] | |
| 5-HT2AR inverse agonist and antagonist | Urinary tract infections, falls | Pimavanserin (ACP-103) | Oral | [150] | |
| 5-HT2AR, 5-HT2CR antagonist, D1R agonist, D2R, D4R antagonist | Diurnal drowsiness, somnolence, excessive sweating, hypereosinophilia, worsening of parkinsonism | Clozapine | Oral | [151] | |
| 5-HT2AR agonist and adrenergic, muscarinic, histaminergic, dopaminergic receptor action | Sedation, dizziness | Quetiapine | Oral | [152] | |
| 5-HT2AR antagonist, partial 5-HT1AR agonist, partial D2 agonist | headache, insomnia, agitation, anxiety, dyspepsia, nausea, lightheadedness, somnolence, akathisia | Aripiprazole | Intramuscular injection, oral | [153] | |
| 5-HT2R agonist/5-HT3R antagonist | Hallucinations, confusion | Mirtazapine | Oral | [154] | |
| Worsening of parkinsonism | Mianserin | Oral | [132] | ||
| Histamine | H3 antagonist | N/A (preclinical) | Thioperamide | N/A (preclinical) | [155] |
| H3 receptor agonist | N/A (preclinical) | Immepip | N/A (preclinical) | [156,157] | |
| N/A (preclinical) | Imetit | N/A (preclinical) | [157] | ||
| Cholinergic | M1 receptor antagonist | dry mouth, blurred vision, constipation, confusion, hallucination, memory disturbance, urinary retention | Trihexyphenidyl | Oral | [158] |
| Benzatropine | Oral | [159] | |||
| M4 positive allosteric moderator | N/A (preclinical) | VU0467154 | N/A (preclinical) | [132] | |
| M4 positive allosteric moderator | N/A (preclinical) | VU0476406 | N/A (preclinical) | [132] | |
| Nicotinic agonist | nausea, vomiting, lung damage, dizziness, | Nicotine | Oral, inhaled | [160,161] | |
| Non-selective nicotinic receptor antagonist | constipation, blurred vision, dry mouth, orthostatic hypotension | Mecamylamine | Oral | [162] | |
| ɑ7 nicotinic receptor agonist | N/A (preclinical) | ABT-107 | N/A (preclinical) | [163] | |
| agitation, constipation, diarrhea, fall, and headache | ABT-126 | Oral | [163,164] | ||
| ꞵ2 nicotinic receptor agonist | Worsening of parkinsonism | ABT-089 | Oral | [163] | |
| ABT-894 | Oral | [163] | |||
| Opioid | к agonist, μ antagonist | Sedation (Non-human primate models) | Nalbuphine | Injection (Non-human primate models) | [165] |
| Mu-delta opioid receptor agonist | N/A (preclinical) | Lactomorphin (MMP-2200) | N/A (preclinical) | [166] | |
| δ antagonist | N/A (preclinical) | Naltrindole | N/A (preclinical) | [167] | |
| Kappa (к) opioid receptor agonist | Worsening of parkinsonism | U50,488 | N/A (preclinical) | [168] | |
| Sigma-1 | σ1 receptor antagonist | N/A (preclinical) | BMY-14802 | N/A (preclinical) | [169] |
| σ1 receptor agonist | Fatigue, somnolence, dizziness, constipation | Dextromethorphan | Oral | [170] | |
| Nitric Oxide | NO Donor | Worsening of parkinsonism | Molsidomine | Oral | [171] |
7. Surgical Options
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat. Rev. Neurosci. 2008, 9, 665–677. [Google Scholar] [CrossRef]
- Bezard, E. Experimental reappraisal of continuous dopaminergic stimulation against L-dopa-induced dyskinesia. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 1021–1022. [Google Scholar] [CrossRef]
- Cerri, S.; Blandini, F. An update on the use of non-ergot dopamine agonists for the treatment of Parkinson’s disease. Expert Opin. Pharmacother. 2020, 21, 2279–2291. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.E.; Muenter, M.D. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov. Disord. 2001, 16, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet. Neurol. 2018, 17, 939–953. [CrossRef] [PubMed]
- Ou, Z.; Pan, J.; Tang, S.; Duan, D.; Yu, D.; Nong, H.; Wang, Z. Global Trends in the Incidence, Prevalence, and Years Lived with Disability of Parkinson’s Disease in 204 Countries/Territories from 1990 to 2019. Front. Public Health 2021, 9, 776847. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Srivanitchapoom, P. Levodopa-induced Dyskinesia: Clinical Features, Pathophysiology, and Medical Management. Ann. Indian Acad. Neurol. 2017, 20, 190–198. [Google Scholar] [CrossRef]
- Sharma, J.C.; Macnamara, L.; Hasoon, M.; Vassallo, M.; Ross, I. Cascade of levodopa dose and weight-related dyskinesia in Parkinson’s disease (LD-WD-PD cascade). Park. Relat. Disord. 2006, 12, 499–505. [Google Scholar] [CrossRef]
- Cilia, R.; Akpalu, A.; Sarfo, F.S.; Cham, M.; Amboni, M.; Cereda, E.; Fabbri, M.; Adjei, P.; Akassi, J.; Bonetti, A.; et al. The modern pre-levodopa era of Parkinson’s disease: Insights into motor complications from sub-Saharan Africa. Brain 2014, 137, 2731–2742. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.; Glass, G.A. Age of Parkinson’s disease onset as a predictor for the development of dyskinesia. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, S.; Mash, D.C. Motor fluctuations and dyskinesias in advanced/end stage Parkinson’s disease: A study from a population of brain donors. J. Neural Transm. 2007, 114, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Warren Olanow, C.; Kieburtz, K.; Rascol, O.; Poewe, W.; Schapira, A.H.; Emre, M.; Nissinen, H.; Leinonen, M.; Stocchi, F. Factors predictive of the development of Levodopa-induced dyskinesia and wearing-off in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 1064–1071. [Google Scholar] [CrossRef]
- Zappia, M.; Annesi, G.; Nicoletti, G.; Arabia, G.; Annesi, F.; Messina, D.; Pugliese, P.; Spadafora, P.; Tarantino, P.; Carrideo, S.; et al. Sex differences in clinical and genetic determinants of levodopa peak-dose dyskinesias in Parkinson disease: An exploratory study. Arch. Neurol. 2005, 62, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.A.; Sullivan, K.L.; Hauser, R.A. Levodopa-induced dyskinesia in Parkinson’s disease: Epidemiology, etiology, and treatment. Curr. Neurol. Neurosci. Rep. 2007, 7, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Di Filippo, M.; Ghiglieri, V.; Tambasco, N.; Picconi, B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: Filling the bench-to-bedside gap. Lancet. Neurol. 2010, 9, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Caviness, J.N.; Brown, P. Myoclonus: Current concepts and recent advances. Lancet. Neurol. 2004, 3, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, D.; Jankovic, J. Drug-Induced Dyskinesia, Part 1: Treatment of Levodopa-Induced Dyskinesia. Drugs 2016, 76, 759–777. [Google Scholar] [CrossRef] [PubMed]
- Rascol, O.; Arnulf, I.; Peyro-Saint Paul, H.; Brefel-Courbon, C.; Vidailhet, M.; Thalamas, C.; Bonnet, A.M.; Descombes, S.; Bejjani, B.; Fabre, N.; et al. Idazoxan, an alpha-2 antagonist, and L-DOPA-induced dyskinesias in patients with Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2001, 16, 708–713. [Google Scholar] [CrossRef]
- Nutt, J.G. Levodopa-induced dyskinesia: Review, observations, and speculations. Neurology 1990, 40, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Picconi, B.; Tozzi, A.; Ghiglieri, V.; Di Filippo, M. Direct and indirect pathways of basal ganglia: A critical reappraisal. Nat. Neurosci. 2014, 17, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- McGregor, M.M.; Nelson, A.B. Circuit Mechanisms of Parkinson’s Disease. Neuron 2019, 101, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Yousaf, T.; Politis, M. PET Molecular Imaging Research of Levodopa-Induced Dyskinesias in Parkinson’s Disease. Curr. Neurol. Neurosci. Rep. 2017, 17, 90. [Google Scholar] [CrossRef]
- Lindgren, H.S.; Andersson, D.R.; Lagerkvist, S.; Nissbrandt, H.; Cenci, M.A. L-DOPA-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: Temporal and quantitative relationship to the expression of dyskinesia. J. Neurochem. 2010, 112, 1465–1476. [Google Scholar] [CrossRef]
- Yahr, M.D.; Duvoisin, R.C.; Hoehn, M.M.; Schear, M.J.; Barrett, R.E. L-Dopa (L-3,4-dihydroxyphenylanine)—Its clinical effects in parkinsonism. Trans. Am. Neurol. Assoc. 1968, 93, 56–63. [Google Scholar]
- Muriel, M.P.; Bernard, V.; Levey, A.I.; Laribi, O.; Abrous, D.N.; Agid, Y.; Bloch, B.; Hirsch, E.C. Levodopa induces a cytoplasmic localization of D1 dopamine receptors in striatal neurons in Parkinson’s disease. Ann. Neurol. 1999, 46, 103–111. [Google Scholar] [CrossRef]
- Bezard, E.; Brotchie, J.M.; Gross, C.E. Pathophysiology of levodopa-induced dyskinesia: Potential for new therapies. Nat. Rev. Neurosci. 2001, 2, 577–588. [Google Scholar] [CrossRef]
- Contin, M.; Martinelli, P. Pharmacokinetics of levodopa. J. Neurol. 2010, 257, S253–S261. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Keefe, K.A.; Gauda, E.B. D1 and D2 dopamine receptor function in the striatum: Coactivation of D1- and D2-dopamine receptors on separate populations of neurons results in potentiated immediate early gene response in D1-containing neurons. J. Neurosci. Off. J. Soc. Neurosci. 1995, 15, 8167–8176. [Google Scholar] [CrossRef]
- Guigoni, C.; Aubert, I.; Li, Q.; Gurevich, V.V.; Benovic, J.L.; Ferry, S.; Mach, U.; Stark, H.; Leriche, L.; Håkansson, K.; et al. Pathogenesis of levodopa-induced dyskinesia: Focus on D1 and D3 dopamine receptors. Park. Relat. Disord. 2005, 11 (Suppl. S1), S25–S29. [Google Scholar] [CrossRef] [PubMed]
- Bézard, E.; Ferry, S.; Mach, U.; Stark, H.; Leriche, L.; Boraud, T.; Gross, C.; Sokoloff, P. Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat. Med. 2003, 9, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Rodríguez-Oroz, M.C.; Rodríguez, M.; Lanciego, J.L.; Artieda, J.; Gonzalo, N.; Olanow, C.W. Pathophysiology of the basal ganglia in Parkinson’s disease. Trends Neurosci. 2000, 23, S8–S19. [Google Scholar] [CrossRef]
- Olanow, C.W.; Obeso, J.A.; Stocchi, F. Drug insight: Continuous dopaminergic stimulation in the treatment of Parkinson’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Obeso, J.A.; Stocchi, F. Continuous dopamine-receptor treatment of Parkinson’s disease: Scientific rationale and clinical implications. Lancet. Neurol. 2006, 5, 677–687. [Google Scholar] [CrossRef]
- Nutt, J.G.; Obeso, J.A.; Stocchi, F. Continuous dopamine-receptor stimulation in advanced Parkinson’s disease. Trends Neurosci. 2000, 23, S109–S115. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.V.; Borgkvist, A.; Sulzer, D. Presynaptic effects of levodopa and their possible role in dyskinesia. Mov. Disord. 2015, 30, 45–53. [Google Scholar] [CrossRef]
- Nutt, J.G. Pharmacokinetics and pharmacodynamics of levodopa. Mov. Disord. 2008, 23 (Suppl. S3), S580–S584. [Google Scholar] [CrossRef]
- Corsi, S.; Stancampiano, R.; Carta, M. Serotonin/dopamine interaction in the induction and maintenance of L-DOPA-induced dyskinesia: An update. Prog. Brain Res. 2021, 261, 287–302. [Google Scholar] [CrossRef]
- Navailles, S.; De Deurwaerdère, P. Imbalanced Dopaminergic Transmission Mediated by Serotonergic Neurons in L-DOPA-Induced Dyskinesia. Park. Dis. 2012, 2012, 323686. [Google Scholar] [CrossRef]
- Bandopadhyay, R.; Mishra, N.; Rana, R.; Kaur, G.; Ghoneim, M.M.; Alshehri, S.; Mustafa, G.; Ahmad, J.; Alhakamy, N.A.; Mishra, A. Molecular Mechanisms and Therapeutic Strategies for Levodopa-Induced Dyskinesia in Parkinson’s Disease: A Perspective Through Preclinical and Clinical Evidence. Front. Pharmacol. 2022, 13, 805388. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Oertel, W.H.; Wu, K.; Quinn, N.P.; Pogarell, O.; Brooks, D.J.; Bjorklund, A.; Lindvall, O.; Piccini, P. Graft-induced dyskinesias in Parkinson’s disease: High striatal serotonin/dopamine transporter ratio. Mov. Disord. 2011, 26, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Wu, K.; Loane, C.; Quinn, N.P.; Brooks, D.J.; Rehncrona, S.; Bjorklund, A.; Lindvall, O.; Piccini, P. Serotonergic neurons mediate dyskinesia side effects in Parkinson’s patients with neural transplants. Sci. Transl. Med. 2010, 2, 38ra46. [Google Scholar] [CrossRef] [PubMed]
- Carta, M.; Carlsson, T.; Kirik, D.; Björklund, A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain 2007, 130, 1819–1833. [Google Scholar] [CrossRef]
- Kempadoo, K.A.; Mosharov, E.V.; Choi, S.J.; Sulzer, D.; Kandel, E.R. Dopamine release from the locus coeruleus to the dorsal hippocampus promotes spatial learning and memory. Proc. Natl. Acad. Sci. USA 2016, 113, 14835–14840. [Google Scholar] [CrossRef]
- Bishop, C.; George, J.A.; Buchta, W.; Goldenberg, A.A.; Mohamed, M.; Dickinson, S.O.; Eissa, S.; Eskow Jaunarajs, K.L. Serotonin transporter inhibition attenuates l-DOPA-induced dyskinesia without compromising l-DOPA efficacy in hemi-parkinsonian rats. Eur. J. Neurosci. 2012, 36, 2839–2848. [Google Scholar] [CrossRef]
- Calabresi, P.; Centonze, D.; Gubellini, P.; Marfia, G.A.; Pisani, A.; Sancesario, G.; Bernardi, G. Synaptic transmission in the striatum: From plasticity to neurodegeneration. Prog. Neurobiol. 2000, 61, 231–265. [Google Scholar] [CrossRef]
- Ahmed, I.; Bose, S.K.; Pavese, N.; Ramlackhansingh, A.; Turkheimer, F.; Hotton, G.; Hammers, A.; Brooks, D.J. Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain 2011, 134, 979–986. [Google Scholar] [CrossRef]
- Sgambato-Faure, V.; Cenci, M.A. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog. Neurobiol. 2012, 96, 69–86. [Google Scholar] [CrossRef]
- Menegoz, M.; Lau, L.F.; Hervé, D.; Huganir, R.L.; Girault, J.A. Tyrosine phosphorylation of NMDA receptor in rat striatum: Effects of 6-OH-dopamine lesions. Neuroreport 1995, 7, 125–128. [Google Scholar] [CrossRef]
- Nash, J.E.; Brotchie, J.M. Characterisation of striatal NMDA receptors involved in the generation of parkinsonian symptoms: Intrastriatal microinjection studies in the 6-OHDA-lesioned rat. Mov. Disord. 2002, 17, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, D.; Conti, M.M.; Ostock, C.Y.; George, J.A.; Goldenberg, A.A.; Melikhov-Sosin, M.; Nuss, E.E.; Bishop, C. The Role of Primary Motor Cortex (M1) Glutamate and GABA Signaling in l-DOPA-Induced Dyskinesia in Parkinsonian Rats. J. Neurosci. 2016, 36, 9873–9887. [Google Scholar] [CrossRef] [PubMed]
- Konradi, C.; Westin, J.E.; Carta, M.; Eaton, M.E.; Kuter, K.; Dekundy, A.; Lundblad, M.; Cenci, M.A. Transcriptome analysis in a rat model of L-DOPA-induced dyskinesia. Neurobiol. Dis. 2004, 17, 219–236. [Google Scholar] [CrossRef] [PubMed]
- Klawans, H.L.; Goetz, C.; Nausieda, P.A.; Weiner, W.J. Levodopa-induced dopamine receptor hypersensitivity. Trans. Am. Neurol. Assoc. 1977, 102, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Nadjar, A.; Gerfen, C.R.; Bezard, E. Priming for l-dopa-induced dyskinesia in Parkinson’s disease: A feature inherent to the treatment or the disease? Prog. Neurobiol. 2009, 87, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wachtel, S.R.; Abercrombie, E.D. L-3,4-dihydroxyphenylalanine-induced dopamine release in the striatum of intact and 6-hydroxydopamine-treated rats: Differential effects of monoamine oxidase A and B inhibitors. J. Neurochem. 1994, 63, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Niccolini, F.; Foltynie, T.; Reis Marques, T.; Muhlert, N.; Tziortzi, A.C.; Searle, G.E.; Natesan, S.; Kapur, S.; Rabiner, E.A.; Gunn, R.N.; et al. Loss of phosphodiesterase 10A expression is associated with progression and severity in Parkinson’s disease. Brain 2015, 138, 3003–3015. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Levy, R.; Herrero, M.T.; Ruberg, M.; Faucheux, B.; Obeso, J.A.; Agid, Y.; Hirsch, E.C. Consequences of nigrostriatal denervation on the functioning of the basal ganglia in human and nonhuman primates: An in situ hybridization study of cytochrome oxidase subunit I mRNA. J. Neurosci. 1997, 17, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.E.; Boraud, T.; Guehl, D.; Bioulac, B.; Bezard, E. From experimentation to the surgical treatment of Parkinson’s disease: Prelude or suite in basal ganglia research? Prog. Neurobiol. 1999, 59, 509–532. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, G.; Brotchie, J.M.; Grandas, F.; Nomoto, M.; Goetz, C.G. Levodopa-induced dyskinesias. Mov. Disord. 2007, 22, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Konitsiotis, S.; Blanchet, P.J.; Verhagen, L.; Lamers, E.; Chase, T.N. AMPA receptor blockade improves levodopa-induced dyskinesia in MPTP monkeys. Neurology 2000, 54, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Leblois, A.; Boraud, T.; Meissner, W.; Bergman, H.; Hansel, D. Competition between feedback loops underlies normal and pathological dynamics in the basal ganglia. J. Neurosci. 2006, 26, 3567–3583. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchi, S.; Frosini, D.; Bonuccelli, U.; Ceravolo, R. Current treatment and future prospects of dopa-induced dyskinesias. Drugs Today 2015, 51, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Timpka, J.; Fox, T.; Fox, K.; Honig, H.; Odin, P.; Martinez-Martin, P.; Antonini, A.; Chaudhuri, K.R. Improvement of dyskinesias with L-dopa infusion in advanced Parkinson’s disease. Acta Neurol. Scand. 2016, 133, 451–458. [Google Scholar] [CrossRef]
- Freire-Alvarez, E.; Kurča, E.; Lopez Manzanares, L.; Pekkonen, E.; Spanaki, C.; Vanni, P.; Liu, Y.; Sánchez-Soliño, O.; Barbato, L.M. Levodopa-Carbidopa Intestinal Gel Reduces Dyskinesia in Parkinson’s Disease in a Randomized Trial. Mov. Disord. 2021, 36, 2615–2623. [Google Scholar] [CrossRef]
- Udd, M.; Lyytinen, J.; Eerola-Rautio, J.; Kenttämies, A.; Lindström, O.; Kylänpää, L.; Pekkonen, E. Problems related to levodopa-carbidopa intestinal gel treatment in advanced Parkinson’s disease. Brain Behav. 2017, 7, e00737. [Google Scholar] [CrossRef]
- Rascol, O.; Perez-Lloret, S.; Ferreira, J.J. New treatments for levodopa-induced motor complications. Mov. Disord. 2015, 30, 1451–1460. [Google Scholar] [CrossRef]
- Hauser, R.A.; Rascol, O.; Korczyn, A.D.; Jon Stoessl, A.; Watts, R.L.; Poewe, W.; De Deyn, P.P.; Lang, A.E. Ten-year follow-up of Parkinson’s disease patients randomized to initial therapy with ropinirole or levodopa. Mov. Disord. 2007, 22, 2409–2417. [Google Scholar] [CrossRef]
- Choi, J.; Horner, K.A. Dopamine Agonists. In StatPearls; StatPearls Publishing Copyright © 2022; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Voon, V.; Fernagut, P.O.; Wickens, J.; Baunez, C.; Rodriguez, M.; Pavon, N.; Juncos, J.L.; Obeso, J.A.; Bezard, E. Chronic dopaminergic stimulation in Parkinson’s disease: From dyskinesias to impulse control disorders. Lancet. Neurol. 2009, 8, 1140–1149. [Google Scholar] [CrossRef]
- Clarke, C.E.; Deane, K.H. Cabergoline for levodopa-induced complications in Parkinson’s disease. Cochrane Database Syst. Rev. 2001, Cd001518. [Google Scholar] [CrossRef]
- Mailland, E.; Magnani, P.; Ottillinger, B. Alpha-dihydroergocryptine in the long-term therapy of Parkinson’s disease. Arzneim. -Forsch. 2004, 54, 647–654. [Google Scholar] [CrossRef]
- Clarke, C.E.; Deane, K.H. Ropinirole versus bromocriptine for levodopa-induced complications in Parkinson’s disease. Cochrane Database Syst. Rev. 2001, Cd001517. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.E.; Speller, J.M. Pergolide for levodopa-induced complications in Parkinson’s disease. Cochrane Database Syst. Rev. 2000, Cd000235. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Barone, P.; Ceravolo, R.; Fabbrini, G.; Tinazzi, M.; Abbruzzese, G. Role of pramipexole in the management of Parkinson’s disease. CNS Drugs 2010, 24, 829–841. [Google Scholar] [CrossRef]
- Frampton, J.E. Rotigotine Transdermal Patch: A Review in Parkinson’s Disease. CNS Drugs 2019, 33, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.A.; Jackson, M.J.; Johnston, L.; Kuoppamaki, M.; Rose, S.; Al-Barghouthy, G.; Del Signore, S.; Jenner, P. Switching from levodopa to the long-acting dopamine D2/D3 agonist piribedil reduces the expression of dyskinesia while maintaining effective motor activity in MPTP-treated primates. Clin. Neuropharmacol. 2006, 29, 112–125. [Google Scholar] [CrossRef]
- Bhidayasiri, R.; Garcia Ruiz, P.J.; Henriksen, T. Practical management of adverse events related to apomorphine therapy. Park. Relat. Disord. 2016, 33 (Suppl. S1), S42–S48. [Google Scholar] [CrossRef]
- Jenner, P.; Katzenschlager, R. Apomorphine—Pharmacological properties and clinical trials in Parkinson’s disease. Park. Relat. Disord. 2016, 33 (Suppl. S1), S13–S21. [Google Scholar] [CrossRef]
- Katzenschlager, R.; Hughes, A.; Evans, A.; Manson, A.J.; Hoffman, M.; Swinn, L.; Watt, H.; Bhatia, K.; Quinn, N.; Lees, A.J. Continuous subcutaneous apomorphine therapy improves dyskinesias in Parkinson’s disease: A prospective study using single-dose challenges. Mov. Disord. 2005, 20, 151–157. [Google Scholar] [CrossRef]
- Sohur, U.S.; Gray, D.L.; Duvvuri, S.; Zhang, Y.; Thayer, K.; Feng, G. Phase 1 Parkinson’s Disease Studies Show the Dopamine D1/D5 Agonist PF-06649751 is Safe and Well Tolerated. Neurol. Ther. 2018, 7, 307–319. [Google Scholar] [CrossRef]
- Bruns, R.F.; Mitchell, S.N.; Wafford, K.A.; Harper, A.J.; Shanks, E.A.; Carter, G.; O’Neill, M.J.; Murray, T.K.; Eastwood, B.J.; Schaus, J.M.; et al. Preclinical profile of a dopamine D1 potentiator suggests therapeutic utility in neurological and psychiatric disorders. Neuropharmacology 2018, 128, 351–365. [Google Scholar] [CrossRef]
- Simms, S.L.; Huettner, D.P.; Kortagere, S. In vivo characterization of a novel dopamine D3 receptor agonist to treat motor symptoms of Parkinson’s disease. Neuropharmacology 2016, 100, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, D.; Das, B.; Conti, M.M.; Meadows, S.M.; Dutta, A.K.; Bishop, C. D-512, a novel dopamine D(2/3) receptor agonist, demonstrates greater anti-Parkinsonian efficacy than ropinirole in Parkinsonian rats. Br. J. Pharmacol. 2017, 174, 3058–3071. [Google Scholar] [CrossRef] [PubMed]
- Elmabruk, A.; Das, B.; Yedlapudi, D.; Xu, L.; Antonio, T.; Reith, M.E.A.; Dutta, A.K. Design, Synthesis, and Pharmacological Characterization of Carbazole Based Dopamine Agonists as Potential Symptomatic and Neuroprotective Therapeutic Agents for Parkinson’s Disease. ACS Chem. Neurosci. 2019, 10, 396–411. [Google Scholar] [CrossRef]
- Rascol, O.; Bronzova, J.; Hauser, R.A.; Lang, A.E.; Sampaio, C.; Theeuwes, A.; van de Witte, S.V. Pardoprunox as adjunct therapy to levodopa in patients with Parkinson’s disease experiencing motor fluctuations: Results of a double-blind, randomized, placebo-controlled, trial. Park. Relat. Disord. 2012, 18, 370–376. [Google Scholar] [CrossRef]
- Sebastianutto, I.; Maslava, N.; Hopkins, C.R.; Cenci, M.A. Validation of an improved scale for rating l-DOPA-induced dyskinesia in the mouse and effects of specific dopamine receptor antagonists. Neurobiol. Dis. 2016, 96, 156–170. [Google Scholar] [CrossRef]
- Svenningsson, P.; Johansson, A.; Nyholm, D.; Tsitsi, P.; Hansson, F.; Sonesson, C.; Tedroff, J. Safety and tolerability of IRL790 in Parkinson’s disease with levodopa-induced dyskinesia-a phase 1b trial. NPJ Park. Dis. 2018, 4, 35. [Google Scholar] [CrossRef]
- Müller, T. Entacapone. Expert Opin. Drug Metab. Toxicol. 2010, 6, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.H.; Deuschl, G.; Gordin, A.; Kultalahti, E.R.; Leinonen, M. Efficacy and safety of entacapone in Parkinson’s disease patients with suboptimal levodopa response: A 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol. Scand. 2002, 105, 245–255. [Google Scholar] [CrossRef]
- Micek, S.T.; Ernst, M.E. Tolcapone: A novel approach to Parkinson’s disease. Am. J. Health-Syst. Pharm. 1999, 56, 2195–2205. [Google Scholar] [CrossRef]
- Reichmann, H.; Lees, A.; Rocha, J.F.; Magalhães, D.; Soares-da-Silva, P. Effectiveness and safety of opicapone in Parkinson’s disease patients with motor fluctuations: The OPTIPARK open-label study. Transl. Neurodegener. 2020, 9, 9. [Google Scholar] [CrossRef]
- Blair, H.A.; Dhillon, S. Safinamide: A Review in Parkinson’s Disease. CNS Drugs 2017, 31, 169–176. [Google Scholar] [CrossRef]
- Schaeffer, E.; Pilotto, A.; Berg, D. Pharmacological strategies for the management of levodopa-induced dyskinesia in patients with Parkinson’s disease. CNS Drugs 2014, 28, 1155–1184. [Google Scholar] [CrossRef] [PubMed]
- Tsunekawa, H.; Takahata, K.; Okano, M.; Ishikawa, T.; Satoyoshi, H.; Nishimura, T.; Hoshino, N.; Muraoka, S. Selegiline increases on time without exacerbation of dyskinesia in 6-hydroxydopamine-lesioned rats displaying l-Dopa-induced wearing-off and abnormal involuntary movements. Behav. Brain Res. 2018, 347, 350–359. [Google Scholar] [CrossRef]
- Oldfield, V.; Keating, G.M.; Perry, C.M. Rasagiline: A review of its use in the management of Parkinson’s disease. Drugs 2007, 67, 1725–1747. [Google Scholar] [CrossRef]
- Perez-Lloret, S.; Rascol, O. Safety of rasagiline for the treatment of Parkinson’s disease. Expert Opin. Drug Saf. 2011, 10, 633–643. [Google Scholar] [CrossRef]
- Woitalla, D.; Krüger, R.; Lorenzl, S.; Müller, T.; Oelwein, G.; Storch, A.; Wolz, M.; Wüllner, U. The role of inhibitors of COMT and MAO-B in the therapy of Parkinson’s disease. Fortschr. Der Neurol. -Psychiatr. 2020, 88, 620–633. [Google Scholar] [CrossRef]
- Borgohain, R.; Szasz, J.; Stanzione, P.; Meshram, C.; Bhatt, M.H.; Chirilineau, D.; Stocchi, F.; Lucini, V.; Giuliani, R.; Forrest, E.; et al. Two-year, randomized, controlled study of safinamide as add-on to levodopa in mid to late Parkinson’s disease. Mov. Disord. 2014, 29, 1273–1280. [Google Scholar] [CrossRef]
- Schapira, A.H.; Fox, S.H.; Hauser, R.A.; Jankovic, J.; Jost, W.H.; Kenney, C.; Kulisevsky, J.; Pahwa, R.; Poewe, W.; Anand, R. Assessment of Safety and Efficacy of Safinamide as a Levodopa Adjunct in Patients with Parkinson Disease and Motor Fluctuations: A Randomized Clinical Trial. JAMA Neurol. 2017, 74, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.E.; Ruiz-Lopez, M.; Fox, S.H. Novel Levodopa Formulations for Parkinson’s Disease. CNS Drugs 2016, 30, 1079–1095. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Huff, F.J.; Hauser, R.A.; Chen, D.; Lissin, D.; Zomorodi, K.; Cundy, K.C. Double-blind study of the actively transported levodopa prodrug XP21279 in Parkinson’s disease. Mov. Disord. 2014, 29, 75–82. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Hauser, R.A.; Grosset, D.G.; Stocchi, F.; Saint-Hilaire, M.H.; Ellenbogen, A.; Leinonen, M.; Hampson, N.B.; DeFeo-Fraulini, T.; Freed, M.I.; et al. A randomized trial of inhaled levodopa (CVT-301) for motor fluctuations in Parkinson’s disease. Mov. Disord. 2016, 31, 1356–1365. [Google Scholar] [CrossRef]
- Wang, W.W.; Zhang, X.R.; Lin, J.Y.; Zhang, Z.R.; Wang, Z.; Chen, S.Y.; Xie, C.L. Levodopa/Benserazide PLGA Microsphere Prevents L-Dopa-Induced Dyskinesia via Lower β-Arrestin2 in 6-Hydroxydopamine Parkinson’s Rats. Front. Pharmacol. 2019, 10, 660. [Google Scholar] [CrossRef]
- Xie, C.L.; Wang, W.W.; Zhang, S.F.; Yuan, M.L.; Che, J.Y.; Gan, J.; Song, L.; Yuan, W.E.; Liu, Z.G. Levodopa/benserazide microsphere (LBM) prevents L-dopa induced dyskinesia by inactivation of the DR1/PKA/P-tau pathway in 6-OHDA-lesioned Parkinson’s rats. Sci. Rep. 2014, 4, 7506. [Google Scholar] [CrossRef]
- Cao, X.; Hou, D.; Wang, L.; Li, S.; Sun, S.; Ping, Q.; Xu, Y. Effects and molecular mechanism of chitosan-coated levodopa nanoliposomes on behavior of dyskinesia rats. Biol. Res. 2016, 49, 32. [Google Scholar] [CrossRef]
- Poewe, W.; Antonini, A. Novel formulations and modes of delivery of levodopa. Mov. Disord. 2015, 30, 114–120. [Google Scholar] [CrossRef]
- Trenkwalder, C.; Kuoppamäki, M.; Vahteristo, M.; Müller, T.; Ellmén, J. Increased dose of carbidopa with levodopa and entacapone improves “off” time in a randomized trial. Neurology 2019, 92, e1487–e1496. [Google Scholar] [CrossRef]
- Olanow, C.W.; Espay, A.J.; Stocchi, F.; Ellenbogen, A.L.; Leinonen, M.; Adar, L.; Case, R.J.; Orenbach, S.F.; Yardeni, T.; Oren, S.; et al. Continuous Subcutaneous Levodopa Delivery for Parkinson’s Disease: A Randomized Study. J. Park. Dis. 2021, 11, 177–186. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Chen, J. Levodopa-Carbidopa Intestinal Gel in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2018, 9, 620. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Giladi, N.; Navon, N. Pharmacokinetics and efficacy of a novel formulation of carbidopa-levodopa (Accordion Pill(®)) in Parkinson’s disease. Park. Relat. Disord. 2019, 65, 131–138. [Google Scholar] [CrossRef]
- Navon, N. The Accordion Pill(®): Unique oral delivery to enhance pharmacokinetics and therapeutic benefit of challenging drugs. Ther. Deliv. 2019, 10, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Snow, B.J.; Macdonald, L.; McAuley, D.; Wallis, W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: A double-blind, placebo-controlled study. Clin. Neuropharmacol. 2000, 23, 82–85. [Google Scholar] [CrossRef]
- Wolf, E.; Seppi, K.; Katzenschlager, R.; Hochschorner, G.; Ransmayr, G.; Schwingenschuh, P.; Ott, E.; Kloiber, I.; Haubenberger, D.; Auff, E.; et al. Long-term antidyskinetic efficacy of amantadine in Parkinson’s disease. Mov. Disord. 2010, 25, 1357–1363. [Google Scholar] [CrossRef]
- Stanic, J.; Mellone, M.; Napolitano, F.; Racca, C.; Zianni, E.; Minocci, D.; Ghiglieri, V.; Thiolat, M.L.; Li, Q.; Longhi, A.; et al. Rabphilin 3A: A novel target for the treatment of levodopa-induced dyskinesias. Neurobiol. Dis. 2017, 108, 54–64. [Google Scholar] [CrossRef]
- Fabbrini, A.; Guerra, A. Pathophysiological Mechanisms and Experimental Pharmacotherapy for L-Dopa-Induced Dyskinesia. J. Exp. Pharmacol. 2021, 13, 469–485. [Google Scholar] [CrossRef]
- Gomez-Mancilla, B.; Bédard, P.J. Effect of nondopaminergic drugs on L-dopa-induced dyskinesias in MPTP-treated monkeys. Clin. Neuropharmacol. 1993, 16, 418–427. [Google Scholar] [CrossRef]
- Henry, B.; Fox, S.H.; Peggs, D.; Crossman, A.R.; Brotchie, J.M. The alpha2-adrenergic receptor antagonist idazoxan reduces dyskinesia and enhances anti-parkinsonian actions of L-dopa in the MPTP-lesioned primate model of Parkinson’s disease. Mov. Disord. 1999, 14, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Savola, J.M.; Hill, M.; Engstrom, M.; Merivuori, H.; Wurster, S.; McGuire, S.G.; Fox, S.H.; Crossman, A.R.; Brotchie, J.M. Fipamezole (JP-1730) is a potent alpha2 adrenergic receptor antagonist that reduces levodopa-induced dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Mov. Disord. 2003, 18, 872–883. [Google Scholar] [CrossRef]
- Pilleri, M.; Antonini, A. Therapeutic strategies to prevent and manage dyskinesias in Parkinson’s disease. Expert Opin. Drug Saf. 2015, 14, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Pinna, A. Adenosine A2A receptor antagonists in Parkinson’s disease: Progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs 2014, 28, 455–474. [Google Scholar] [CrossRef]
- Stasi, M.A.; Borsini, F.; Varani, K.; Vincenzi, F.; Di Cesare, M.A.; Minetti, P.; Ghirardi, O.; Carminati, P. ST 1535: A preferential A2A adenosine receptor antagonist. Int. J. Neuropsychopharmacol. 2006, 9, 575–584. [Google Scholar] [CrossRef][Green Version]
- Müller, T. Suitability of the adenosine antagonist istradefylline for the treatment of Parkinson’s disease: Pharmacokinetic and clinical considerations. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1015–1024. [Google Scholar] [CrossRef]
- Bara-Jimenez, W.; Sherzai, A.; Dimitrova, T.; Favit, A.; Bibbiani, F.; Gillespie, M.; Morris, M.J.; Mouradian, M.M.; Chase, T.N. Adenosine A(2A) receptor antagonist treatment of Parkinson’s disease. Neurology 2003, 61, 293–296. [Google Scholar] [CrossRef]
- Wills, A.M.; Eberly, S.; Tennis, M.; Lang, A.E.; Messing, S.; Togasaki, D.; Tanner, C.M.; Kamp, C.; Chen, J.F.; Oakes, D.; et al. Caffeine consumption and risk of dyskinesia in CALM-PD. Mov. Disord. 2013, 28, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Swann, A.C.; Lijffijt, M.; Lane, S.D.; Cox, B.; Steinberg, J.L.; Moeller, F.G. Norepinephrine and impulsivity: Effects of acute yohimbine. Psychopharmacology 2013, 229, 83–94. [Google Scholar] [CrossRef]
- Lewitt, P.A.; Hauser, R.A.; Lu, M.; Nicholas, A.P.; Weiner, W.; Coppard, N.; Leinonen, M.; Savola, J.M. Randomized clinical trial of fipamezole for dyskinesia in Parkinson disease (FJORD study). Neurology 2012, 79, 163–169. [Google Scholar] [CrossRef]
- Julius, A.; Longfellow, K. Movement Disorders: A Brief Guide in Medication Management. Med. Clin. North Am. 2016, 100, 733–761. [Google Scholar] [CrossRef]
- Hopfner, F.; Höglinger, G.U.; Kuhlenbäumer, G.; Pottegård, A.; Wod, M.; Christensen, K.; Tanner, C.M.; Deuschl, G. β-adrenoreceptors and the risk of Parkinson’s disease. Lancet. Neurol. 2020, 19, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.D.; Lyons, K.E.; Pahwa, R. Amantadine extended-release capsules for levodopa-induced dyskinesia in patients with Parkinson’s disease. Ther. Clin. Risk Manag. 2018, 14, 665–673. [Google Scholar] [CrossRef]
- Hauser, R.A.; Walsh, R.R.; Pahwa, R.; Chernick, D.; Formella, A.E. Amantadine ER (Gocovri(®)) Significantly Increases ON Time without Any Dyskinesia: Pooled Analyses from Pivotal Trials in Parkinson’s Disease. Front. Neurol. 2021, 12, 645706. [Google Scholar] [CrossRef]
- Del Bello, F.; Giannella, M.; Giorgioni, G.; Piergentili, A.; Quaglia, W. Receptor Ligands as Helping Hands to L-DOPA in the Treatment of Parkinson’s Disease. Biomolecules 2019, 9, 142. [Google Scholar] [CrossRef] [PubMed]
- Wictorin, K.; Widner, H. Memantine and reduced time with dyskinesia in Parkinson’s Disease. Acta Neurol. Scand. 2016, 133, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.J.; Bartlett, M.J.; So, L.Y.; Laude, N.D.; Parent, K.L.; Heien, M.L.; Sherman, S.J.; Falk, T. Differential effects of the NMDA receptor antagonist MK-801 on dopamine receptor D1- and D2-induced abnormal involuntary movements in a preclinical model. Neurosci. Lett. 2014, 564, 48–52. [Google Scholar] [CrossRef]
- Bartlett, M.J.; Joseph, R.M.; LePoidevin, L.M.; Parent, K.L.; Laude, N.D.; Lazarus, L.B.; Heien, M.L.; Estevez, M.; Sherman, S.J.; Falk, T. Long-term effect of sub-anesthetic ketamine in reducing L-DOPA-induced dyskinesias in a preclinical model. Neurosci. Lett. 2016, 612, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.J.; Estevez, M.; Magill, A.B.; Falk, T. Case Reports Showing a Long-Term Effect of Subanesthetic Ketamine Infusion in Reducing l-DOPA-Induced Dyskinesias. Case Rep. Neurol. 2016, 8, 53–58. [Google Scholar] [CrossRef]
- Kobylecki, C.; Burn, D.J.; Kass-Iliyya, L.; Kellett, M.W.; Crossman, A.R.; Silverdale, M.A. Randomized clinical trial of topiramate for levodopa-induced dyskinesia in Parkinson’s disease. Park. Relat. Disord. 2014, 20, 452–455. [Google Scholar] [CrossRef]
- Lattanzi, S.; Grillo, E.; Brigo, F.; Silvestrini, M. Efficacy and safety of perampanel in Parkinson’s disease. A systematic review with meta-analysis. J. Neurol. 2018, 265, 733–740. [Google Scholar] [CrossRef]
- Rascol, O.; Medori, R.; Baayen, C.; Such, P.; Meulien, D. A Randomized, Double-Blind, Controlled Phase II Study of Foliglurax in Parkinson’s Disease. Mov. Disord. 2022, 37, 1088–1093. [Google Scholar] [CrossRef]
- Negida, A.; Ghaith, H.S.; Fala, S.Y.; Ahmed, H.; Bahbah, E.I.; Ebada, M.A.; Aziz, M.A.E. Mavoglurant (AFQ056) for the treatment of levodopa-induced dyskinesia in patients with Parkinson’s disease: A meta-analysis. Neurol. Sci. 2021, 42, 3135–3143. [Google Scholar] [CrossRef]
- Rascol, O.; Fox, S.; Gasparini, F.; Kenney, C.; Di Paolo, T.; Gomez-Mancilla, B. Use of metabotropic glutamate 5-receptor antagonists for treatment of levodopa-induced dyskinesias. Park. Relat. Disord. 2014, 20, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Ko, W.K.; Pioli, E.; Li, Q.; McGuire, S.; Dufour, A.; Sherer, T.B.; Bezard, E.; Facheris, M.F. Combined fenobam and amantadine treatment promotes robust antidyskinetic effects in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned primate model of Parkinson’s disease. Mov. Disord. 2014, 29, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Sebastianutto, I.; Cenci, M.A. mGlu receptors in the treatment of Parkinson’s disease and L-DOPA-induced dyskinesia. Curr. Opin. Pharmacol. 2018, 38, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Fabrizio, E.; Cipriani, R.; Vanacore, N.; Meco, G. Buspirone in levodopa-induced dyskinesias. Clin. Neuropharmacol. 1994, 17, 73–82. [Google Scholar] [CrossRef]
- Huot, P.; Kang, W.; Kim, E.; Bédard, D.; Belliveau, S.; Frouni, I.; Kwan, C. Levodopa-induced dyskinesia: A brief review of the ongoing clinical trials. Neurodegener. Dis. Manag. 2022, 12, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Newman-Tancredi, A.; Varney, M.A.; McCreary, A.C. Effects of the Serotonin 5-HT1A Receptor Biased Agonists, F13714 and F15599, on Striatal Neurotransmitter Levels Following l-DOPA Administration in Hemi-Parkinsonian Rats. Neurochem. Res. 2018, 43, 1035–1046. [Google Scholar] [CrossRef]
- Iderberg, H.; Rylander, D.; Bimpisidis, Z.; Cenci, M.A. Modulating mGluR5 and 5-HT1A/1B receptors to treat l-DOPA-induced dyskinesia: Effects of combined treatment and possible mechanisms of action. Exp. Neurol. 2013, 250, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.G.; Damier, P.; Hicking, C.; Laska, E.; Müller, T.; Olanow, C.W.; Rascol, O.; Russ, H. Sarizotan as a treatment for dyskinesias in Parkinson’s disease: A double-blind placebo-controlled trial. Mov. Disord. 2007, 22, 179–186. [Google Scholar] [CrossRef]
- Huang, X.; Yang, J.; Yang, S.; Cao, S.; Qin, D.; Zhou, Y.; Li, X.; Ye, Y.; Wu, J. Role of tandospirone, a 5-HT1A receptor partial agonist, in the treatment of central nervous system disorders and the underlying mechanisms. Oncotarget 2017, 8, 102705–102720. [Google Scholar] [CrossRef]
- Cummings, J.; Isaacson, S.; Mills, R.; Williams, H.; Chi-Burris, K.; Corbett, A.; Dhall, R.; Ballard, C. Pimavanserin for patients with Parkinson’s disease psychosis: A randomised, placebo-controlled phase 3 trial. Lancet 2014, 383, 533–540. [Google Scholar] [CrossRef]
- Durif, F.; Debilly, B.; Galitzky, M.; Morand, D.; Viallet, F.; Borg, M.; Thobois, S.; Broussolle, E.; Rascol, O. Clozapine improves dyskinesias in Parkinson disease: A double-blind, placebo-controlled study. Neurology 2004, 62, 381–388. [Google Scholar] [CrossRef]
- Morgante, L.; Epifanio, A.; Spina, E.; Zappia, M.; Di Rosa, A.E.; Marconi, R.; Basile, G.; Di Raimondo, G.; La Spina, P.; Quattrone, A. Quetiapine and clozapine in parkinsonian patients with dopaminergic psychosis. Clin. Neuropharmacol. 2004, 27, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Kohen, I.; Lester, P.E.; Lam, S. Antipsychotic treatments for the elderly: Efficacy and safety of aripiprazole. Neuropsychiatr. Dis. Treat. 2010, 6, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Meco, G.; Fabrizio, E.; Di Rezze, S.; Alessandri, A.; Pratesi, L. Mirtazapine in L-dopa-induced dyskinesias. Clin. Neuropharmacol. 2003, 26, 179–181. [Google Scholar] [CrossRef]
- Nowak, P.; Bortel, A.; Dabrowska, J.; Biedka, I.; Slomian, G.; Roczniak, W.; Kostrzewa, R.M.; Brus, R. Histamine H(3) receptor ligands modulate L-dopa-evoked behavioral responses and L-dopa derived extracellular dopamine in dopamine-denervated rat striatum. Neurotox. Res. 2008, 13, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Avila-Luna, A.; Ríos, C.; Gálvez-Rosas, A.; Montes, S.; Arias-Montaño, J.A.; Bueno-Nava, A. Chronic administration of the histamine H(3) receptor agonist immepip decreases L-Dopa-induced dyskinesias in 6-hydroxydopamine-lesioned rats. Psychopharmacology 2019, 236, 1937–1948. [Google Scholar] [CrossRef]
- Gomez-Ramirez, J.; Johnston, T.H.; Visanji, N.P.; Fox, S.H.; Brotchie, J.M. Histamine H3 receptor agonists reduce L-dopa-induced chorea, but not dystonia, in the MPTP-lesioned nonhuman primate model of Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2006, 21, 839–846. [Google Scholar] [CrossRef]
- Chambers, N.E.; Meadows, S.M.; Taylor, A.; Sheena, E.; Lanza, K.; Conti, M.M.; Bishop, C. Effects of Muscarinic Acetylcholine m1 and m4 Receptor Blockade on Dyskinesia in the Hemi-Parkinsonian Rat. Neuroscience 2019, 409, 180–194. [Google Scholar] [CrossRef]
- Brocks, D.R. Anticholinergic drugs used in Parkinson’s disease: An overlooked class of drugs from a pharmacokinetic perspective. J. Pharm. Pharm. Sci. 1999, 2, 39–46. [Google Scholar]
- Ma, C.; Liu, Y.; Neumann, S.; Gao, X. Nicotine from cigarette smoking and diet and Parkinson disease: A review. Transl. Neurodegener. 2017, 6, 18. [Google Scholar] [CrossRef]
- Xie, C.L.; Pan, J.L.; Zhang, S.F.; Gan, J.; Liu, Z.G. Effect of nicotine on L-dopa-induced dyskinesia in animal models of Parkinson’s disease: A systematic review and meta-analysis. Neurol. Sci. 2014, 35, 653–662. [Google Scholar] [CrossRef]
- Young, J.M.; Shytle, R.D.; Sanberg, P.R.; George, T.P. Mecamylamine: New therapeutic uses and toxicity/risk profile. Clin. Ther. 2001, 23, 532–565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Bordia, T.; McGregor, M.; McIntosh, J.M.; Decker, M.W.; Quik, M. ABT-089 and ABT-894 reduce levodopa-induced dyskinesias in a monkey model of Parkinson’s disease. Mov. Disord. 2014, 29, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Florian, H.; Meier, A.; Gauthier, S.; Lipschitz, S.; Lin, Y.; Tang, Q.; Othman, A.A.; Robieson, W.Z.; Gault, L.M. Efficacy and Safety of ABT-126 in Subjects with Mild-to-Moderate Alzheimer’s Disease on Stable Doses of Acetylcholinesterase Inhibitors: A Randomized, Double-Blind, Placebo-Controlled Study. J. Alzheimer’s Dis. JAD 2016, 51, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Potts, L.F.; Park, E.S.; Woo, J.M.; Dyavar Shetty, B.L.; Singh, A.; Braithwaite, S.P.; Voronkov, M.; Papa, S.M.; Mouradian, M.M. Dual κ-agonist/μ-antagonist opioid receptor modulation reduces levodopa-induced dyskinesia and corrects dysregulated striatal changes in the nonhuman primate model of Parkinson disease. Ann. Neurol. 2015, 77, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.J.; Bartlett, M.J.; Root, B.K.; Parent, K.L.; Heien, M.L.; Porreca, F.; Polt, R.; Sherman, S.J.; Falk, T. The combination of the opioid glycopeptide MMP-2200 and a NMDA receptor antagonist reduced l-DOPA-induced dyskinesia and MMP-2200 by itself reduced dopamine receptor 2-like agonist-induced dyskinesia. Neuropharmacology 2018, 141, 260–271. [Google Scholar] [CrossRef]
- Henry, B.; Fox, S.H.; Crossman, A.R.; Brotchie, J.M. Mu- and delta-opioid receptor antagonists reduce levodopa-induced dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Exp. Neurol. 2001, 171, 139–146. [Google Scholar] [CrossRef]
- Cox, H.; Togasaki, D.M.; Chen, L.; Langston, J.W.; Di Monte, D.A.; Quik, M. The selective kappa-opioid receptor agonist U50,488 reduces L-dopa-induced dyskinesias but worsens parkinsonism in MPTP-treated primates. Exp. Neurol. 2007, 205, 101–107. [Google Scholar] [CrossRef][Green Version]
- Paquette, M.A.; Brudney, E.G.; Putterman, D.B.; Meshul, C.K.; Johnson, S.W.; Berger, S.P. Sigma ligands, but not N-methyl-D-aspartate antagonists, reduce levodopa-induced dyskinesias. Neuroreport 2008, 19, 111–115. [Google Scholar] [CrossRef]
- Fox, S.H.; Metman, L.V.; Nutt, J.G.; Brodsky, M.; Factor, S.A.; Lang, A.E.; Pope, L.E.; Knowles, N.; Siffert, J. Trial of dextromethorphan/quinidine to treat levodopa-induced dyskinesia in Parkinson’s disease. Mov. Disord. 2017, 32, 893–903. [Google Scholar] [CrossRef]
- Solís, O.; Espadas, I.; Del-Bel, E.A.; Moratalla, R. Nitric oxide synthase inhibition decreases l-DOPA-induced dyskinesia and the expression of striatal molecular markers in Pitx3(-/-) aphakia mice. Neurobiol. Dis. 2015, 73, 49–59. [Google Scholar] [CrossRef]
- Ferré, S.; Fredholm, B.B.; Morelli, M.; Popoli, P.; Fuxe, K. Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997, 20, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Adenosine and its receptors: Multiple modulatory functions and potential therapeutic targets for neurologic disease. Neurology 2008, 70, 231–236. [Google Scholar] [CrossRef]
- Iravani, M.M.; Jenner, P. Mechanisms underlying the onset and expression of levodopa-induced dyskinesia and their pharmacological manipulation. J. Neural Transm. 2011, 118, 1661–1690. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.Y.; Pearce, R.K.; MacKenzie, G.M.; Jenner, P. Alterations in preproenkephalin and adenosine-2a receptor mRNA, but not preprotachykinin mRNA correlate with occurrence of dyskinesia in normal monkeys chronically treated with L-DOPA. Eur. J. Neurosci. 2000, 12, 1096–1104. [Google Scholar] [CrossRef]
- Morissette, M.; Dridi, M.; Calon, F.; Hadj Tahar, A.; Meltzer, L.T.; Bédard, P.J.; Di Paolo, T. Prevention of dyskinesia by an NMDA receptor antagonist in MPTP monkeys: Effect on adenosine A2A receptors. Synapse 2006, 60, 239–250. [Google Scholar] [CrossRef]
- Shook, B.C.; Jackson, P.F. Adenosine A(2A) Receptor Antagonists and Parkinson’s Disease. ACS Chem. Neurosci. 2011, 2, 555–567. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A.; Guttman, M.; Tetrud, J.W.; Tuite, P.J.; Mori, A.; Chaikin, P.; Sussman, N.M. Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: A double-blind, randomized, multicenter clinical trial (6002-US-005). Ann. Neurol. 2008, 63, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Cassin, J.J.; Healy, B.; Burdett, T.C.; Chen, J.F.; Fredholm, B.B.; Schwarzschild, M.A. Deletion of adenosine A₁ or A(₂A) receptors reduces L-3,4-dihydroxyphenylalanine-induced dyskinesia in a model of Parkinson’s disease. Brain Res. 2011, 1367, 310–318. [Google Scholar] [CrossRef]
- Schlicker, E.; Fink, K.; Detzner, M.; Göthert, M. Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. J. Neural Transm. Gen. Sect. 1993, 93, 1–10. [Google Scholar] [CrossRef]
- Yanovsky, Y.; Li, S.; Klyuch, B.P.; Yao, Q.; Blandina, P.; Passani, M.B.; Lin, J.S.; Haas, H.L.; Sergeeva, O.A. L-Dopa activates histaminergic neurons. J. Physiol. 2011, 589, 1349–1366. [Google Scholar] [CrossRef]
- Conn, P.J.; Jones, C.K.; Lindsley, C.W. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol. Sci. 2009, 30, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Langmead, C.J.; Watson, J.; Reavill, C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 2008, 117, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bai, L.; He, J.; Zhong, L.; Duan, X.; Ouyang, L.; Zhu, Y.; Wang, T.; Zhang, Y.; Shi, J. Recent advances in discovery and development of natural products as source for anti-Parkinson’s disease lead compounds. Eur. J. Med. Chem. 2017, 141, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Bordia, T.; Perez, X.A. Cholinergic control of striatal neurons to modulate L-dopa-induced dyskinesias. Eur. J. Neurosci. 2019, 49, 859–868. [Google Scholar] [CrossRef]
- Kosillo, P.; Zhang, Y.F.; Threlfell, S.; Cragg, S.J. Cortical Control of Striatal Dopamine Transmission via Striatal Cholinergic Interneurons. Cereb. Cortex 2016, 26, 4160–4169. [Google Scholar] [CrossRef]
- Johansson, P.A.; Andersson, M.; Andersson, K.E.; Cenci, M.A. Alterations in cortical and basal ganglia levels of opioid receptor binding in a rat model of l-DOPA-induced dyskinesia. Neurobiol. Dis. 2001, 8, 220–239. [Google Scholar] [CrossRef][Green Version]
- Sgroi, S.; Tonini, R. Opioidergic Modulation of Striatal Circuits, Implications in Parkinson’s Disease and Levodopa Induced Dyskinesia. Front. Neurol. 2018, 9, 524. [Google Scholar] [CrossRef]
- Eve, D.J.; Nisbet, A.P.; Kingsbury, A.E.; Hewson, E.L.; Daniel, S.E.; Lees, A.J.; Marsden, C.D.; Foster, O.J. Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Brain Res. Mol. Brain Res. 1998, 63, 62–71. [Google Scholar] [CrossRef]
- Meurers, B.H.; Dziewczapolski, G.; Shi, T.; Bittner, A.; Kamme, F.; Shults, C.W. Dopamine depletion induces distinct compensatory gene expression changes in DARPP-32 signal transduction cascades of striatonigral and striatopallidal neurons. J. Neurosci. 2009, 29, 6828–6839. [Google Scholar] [CrossRef]
- Garthwaite, J.; Charles, S.L.; Chess-Williams, R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 1988, 336, 385–388. [Google Scholar] [CrossRef]
- Lorenc-Koci, E.; Czarnecka, A.; Lenda, T.; Kamińska, K.; Konieczny, J. Molsidomine, a nitric oxide donor, modulates rotational behavior and monoamine metabolism in 6-OHDA lesioned rats treated chronically with L-DOPA. Neurochem. Int. 2013, 63, 790–804. [Google Scholar] [CrossRef] [PubMed]
- Mishina, M.; Ishiwata, K.; Ishii, K.; Kitamura, S.; Kimura, Y.; Kawamura, K.; Oda, K.; Sasaki, T.; Sakayori, O.; Hamamoto, M.; et al. Function of sigma1 receptors in Parkinson’s disease. Acta Neurol. Scand. 2005, 112, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Hasegawa, K.; Kanazawa, I.; Fukasaka, J.; Kochi, K.; Shimazu, R. Zonisamide improves wearing-off in Parkinson’s disease: A randomized, double-blind study. Mov. Disord. 2015, 30, 1343–1350. [Google Scholar] [CrossRef]
- Katzenschlager, R.; Manson, A.J.; Evans, A.; Watt, H.; Lees, A.J. Low dose quetiapine for drug induced dyskinesias in Parkinson’s disease: A double blind cross over study. J. Neurol. Neurosurg. Psychiatry 2004, 75, 295–297. [Google Scholar]
- Kleiner-Fisman, G.; Fisman, D.N.; Zamir, O.; Dostrovsky, J.O.; Sime, E.; Saint-Cyr, J.A.; Lozano, A.M.; Lang, A.E. Subthalamic nucleus deep brain stimulation for parkinson’s disease after successful pallidotomy: Clinical and electrophysiological observations. Mov. Disord. 2004, 19, 1209–1214. [Google Scholar] [CrossRef]
- Oyama, G.; Foote, K.D.; Jacobson, C.E.t.; Velez-Lago, F.; Go, C.; Limotai, N.; Zeilman, P.R.; Romrell, J.; Wu, S.S.; Neal, D.; et al. GPi and STN deep brain stimulation can suppress dyskinesia in Parkinson’s disease. Park. Relat. Disord. 2012, 18, 814–818. [Google Scholar] [CrossRef]
- Espay, A.J.; Morgante, F.; Merola, A.; Fasano, A.; Marsili, L.; Fox, S.H.; Bezard, E.; Picconi, B.; Calabresi, P.; Lang, A.E. Levodopa-induced dyskinesia in Parkinson disease: Current and evolving concepts. Ann. Neurol. 2018, 84, 797–811. [Google Scholar] [CrossRef]
- Mansouri, A.; Taslimi, S.; Badhiwala, J.H.; Witiw, C.D.; Nassiri, F.; Odekerken, V.J.J.; De Bie, R.M.A.; Kalia, S.K.; Hodaie, M.; Munhoz, R.P.; et al. Deep brain stimulation for Parkinson’s disease: Meta-analysis of results of randomized trials at varying lengths of follow-up. J. Neurosurg. 2018, 128, 1199–1213. [Google Scholar] [CrossRef]
- Marion, M.H.; Stocchi, F.; Quinn, N.P.; Jenner, P.; Marsden, C.D. Repeated levodopa infusions in fluctuating Parkinson’s disease: Clinical and pharmacokinetic data. Clin. Neuropharmacol. 1986, 9, 165–181. [Google Scholar] [CrossRef]
- Sayın, S.; Cakmur, R.; Yener, G.G.; Yaka, E.; Uğurel, B.; Uzunel, F. Low-frequency repetitive transcranial magnetic stimulation for dyskinesia and motor performance in Parkinson’s disease. J. Clin. Neurosci. 2014, 21, 1373–1376. [Google Scholar] [CrossRef]
- Jankovic, J.; Lai, E.; Ben-Arie, L.; Krauss, J.K.; Grossman, R. Levodopa-induced dyskinesias treated by pallidotomy. J. Neurol. Sci. 1999, 167, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, H.M.; Krishna, V.; Elias, W.J.; Cosgrove, G.R.; Gandhi, D.; Aldrich, C.E.; Fishman, P.S. MR-guided focused ultrasound pallidotomy for Parkinson’s disease: Safety and feasibility. J. Neurosurg. 2020, 135, 792–798. [Google Scholar] [CrossRef] [PubMed]





| Dyskinesia Types | Movement | Body Part | Occurrence |
|---|---|---|---|
| Peak dose (IDI) | Primarily choreiform movement; may also be characterized by dystonia, myoclonus, or ballism | Primarily neck, axial, and proximal limbs | High plasma levels, coincidence with antiparkinsonian benefit |
| Diphasic (DID) | Repetitive dystonia, ballism | Lower limbs | Plasma levels rise or fall at the start of the levodopa effect and as levodopa wears off |
| Off-period Dystonia (Early Morning Dystonia) | Dystonia | Usually, one foot | Low plasma levels, “off” period |
| Mechanism of Action | Name of Drug | Route of Administration | Side Effects | References |
|---|---|---|---|---|
| Dopamine receptor agonists | Cabergoline (ergot D2 agonist) | Oral | Nausea, postural hypotension, somnolence, sleep disorder, hallucination | [71] |
| Dihydroergocryptine (ergot D2 agonist, partial D1 agonist) | Oral | Nausea, vomiting, dry mouth, diarrhea, abdominal pain | [72] | |
| Bromocriptine (ergot partial D2 agonist) | Oral | Nausea, postural hypotension, hallucination, vomiting, dizziness, | [73] | |
| Pergolide (ergot D2/D3 agonist) | Oral | Nausea, hallucination | [74] | |
| Pramipexole (non-ergot) | Oral | nausea, vomiting, postural hypotension, oedema, hallucinations, somnolence, sudden-onset sleep disorder | [75] | |
| Ropinirole (non-ergot D2 agonist) | Oral | Nausea, hallucination, postural hypotension | [73] | |
| Rotigotine (non-ergot D3/D2/D1 agonist) | Transdermal patch | nausea, vomiting, somnolence, dizziness and application-site reactions | [76] | |
| Piribedil (non-ergot D2/D3 agonist) | Oral | nausea, postural hypotension, vomiting, confusion, hallucinations, agitation, dizziness, drowsiness | [77] | |
| Apomorphine (non-ergot D2 agonist) | Intermittent subcutaneous injection or continuous subcutaneous infusion | Administration site reactions, nausea, vomiting, transient sedation, somnolence, dizziness, confusion, hallucinations | [78,79,80] | |
| Tavapdon (D1/D5 agonist) | Oral | headache, nausea, and vomiting | [81] | |
| DETQ (D1 agonist) | N/A (preclinical) | N/A (preclinical) | [82] | |
| SK609 (D3 agonist) | N/A (preclinical) | N/A (preclinical) | [83] | |
| D-512 (D2/D3 agonist) | N/A (preclinical) | N/A (preclinical) | [84] | |
| D-636 (D2/D3 agonist) | N/A (preclinical) | N/A (preclinical) | [85] | |
| D-536 (D2/D3 agonist) | N/A (preclinical) | N/A (preclinical) | [85] | |
| D-656 (D2/D3 agonist) | N/A (preclinical) | N/A (preclinical) | [85] | |
| Pardoprunox (D2/D3 partial agonist, 5-HT1A receptor agonist) | Oral | Nausea, vomiting, dizziness, somnolence, headache, insomnia, hallucination | [86] | |
| D4 antagonist | VU6004461 | N/A (preclinical | N/A (preclinical) | [87] |
| D3 antagonist | IRL790 | Oral | Asthenia, dissociation, headache, worsening of parkinsonism | [88] |
| COMT inhibitor | Entacapone | Oral | Diarrhea, nausea, constipation, abdominal pain | [89,90] |
| COMT inhibitor | Tolcapone | Oral | dyskinesia, nausea, sleep disorders, dystonia, orthostatic hypotension, diarrhea, dizziness, hallucinations, potential elevated liver transaminase concentrations, possible fulminant hepatic failure | [91] |
| COMT inhibitor | Opicapone | Oral | Dry mouth, dizziness, nausea, constipation, insomnia, hallucination, fall | [92] |
| MAO-B inhibitor | Safinamide | Oral | Headache, hypertension, cataract, back pain | [93,94] |
| MAO-B inhibitor | Selegiline | Oral | insomnia, nausea, benign cardiac arrhythmias, dizziness and headache | [95] |
| MAO-B inhibitor | Rasagiline | Oral | Infection, headache, musculoskeletal pain | [96,97] |
| Name of Drug | Mechanism of Action | Route of Administration | Side Effects | References |
|---|---|---|---|---|
| XP21279 | L-DOPA prodrug, activated by carboxylesterase, creates more stable plasma concentration | Oral | Headache, dizziness, anorexia, insomnia, gastroesophageal reflux disease, and somnolence | [101,102] |
| CVT-301 | L-DOPA powder | Inhaled | Dizziness, cough, and nausea | [101,103] |
| L-DOPA/benserazide microspheres | Inactivity of D1R/Shp-2/ERK1/2 pathway, inhibition of tau protein phosphorylation and PKA signaling | N/A (preclinical) | N/A (preclinical) | [104,105] |
| Chitosan-coated nanoliposomes | Reduced expression of ERK ½, DARPP-32, and FosB/ΔFosB | N/A (preclinical) | N/A (preclinical) | [106] |
| ODM-101 | LD/CD/ENT formulation | Oral | nausea, dizziness, headache, diarrhea, and insomnia | [107,108] |
| ND0612 | Liquid formulation of LD/CD, continuous administration | Transcutaneous, patch-pump device | Small transient papules at infusion sites, infusion site bruising, and erythema | [101,109] |
| LCIG | Gel formulation of LD/CD, finer and more continuous titrations of levodopa | Intestinal, injected through PEG-J tube | peristomal complications, problems flushing the tube, accidental removal of the tube, tube occlusion, weight loss, nausea, and hallucinations | [66,110] |
| Accordion Pill | Immediate-release CD and both immediate and extended-release LD | Oral | Nausea, vomiting, mild somnolence, and fatigue | [111,112] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, D.K.; Kwatra, M.; Wang, J.; Ko, H.S. Levodopa-Induced Dyskinesia in Parkinson’s Disease: Pathogenesis and Emerging Treatment Strategies. Cells 2022, 11, 3736. https://doi.org/10.3390/cells11233736
Kwon DK, Kwatra M, Wang J, Ko HS. Levodopa-Induced Dyskinesia in Parkinson’s Disease: Pathogenesis and Emerging Treatment Strategies. Cells. 2022; 11(23):3736. https://doi.org/10.3390/cells11233736
Chicago/Turabian StyleKwon, Destany K., Mohit Kwatra, Jing Wang, and Han Seok Ko. 2022. "Levodopa-Induced Dyskinesia in Parkinson’s Disease: Pathogenesis and Emerging Treatment Strategies" Cells 11, no. 23: 3736. https://doi.org/10.3390/cells11233736
APA StyleKwon, D. K., Kwatra, M., Wang, J., & Ko, H. S. (2022). Levodopa-Induced Dyskinesia in Parkinson’s Disease: Pathogenesis and Emerging Treatment Strategies. Cells, 11(23), 3736. https://doi.org/10.3390/cells11233736