
Recent Advances of Studies on Cell-Penetrating Peptides Based on Molecular Dynamics Simulations
Abstract
:1. Introduction
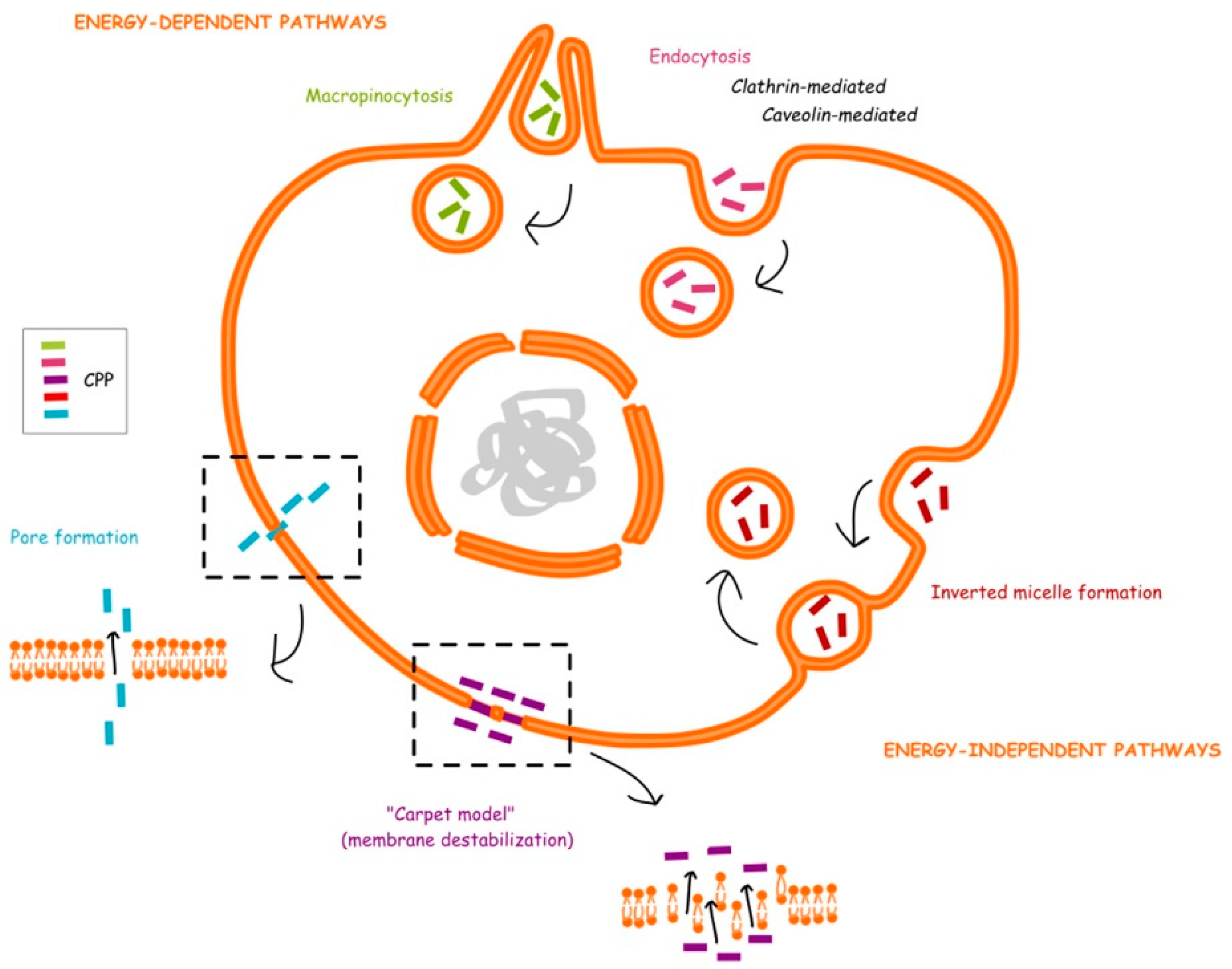
2. Categories and Cellular Uptake Mechanisms of CPPs
3. Techniques for CPP Simulations
3.1. All-Atom MD Simulations, Coarse-Grained Simulations and Implicit Membrane Models
3.2. Enhanced Sampling Approaches
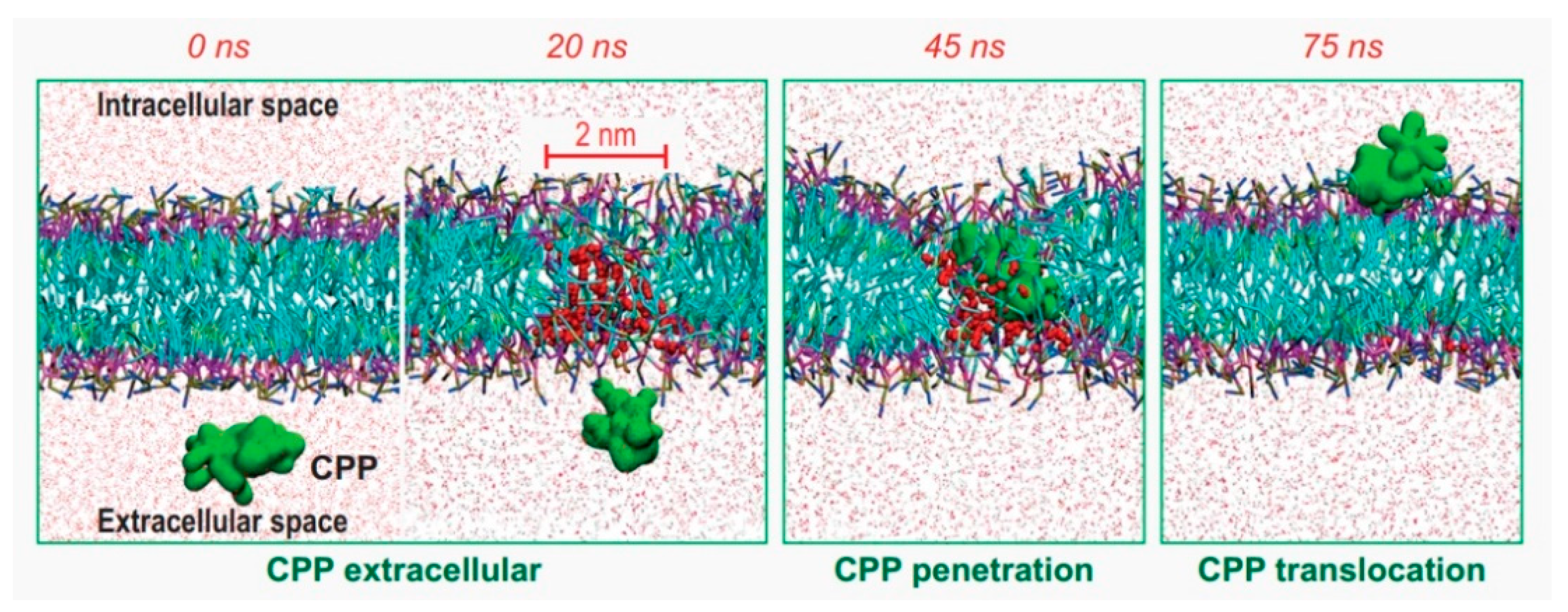
4. Simulations on CPP Internalization Mechanisms
4.1. Role of Arginine Residues
4.2. Role of the Secondary Structures of CPPs
4.3. Role of Hydrophobic Aromatic Residues
4.4. Other Factors Affecting the Uptake Mechanism
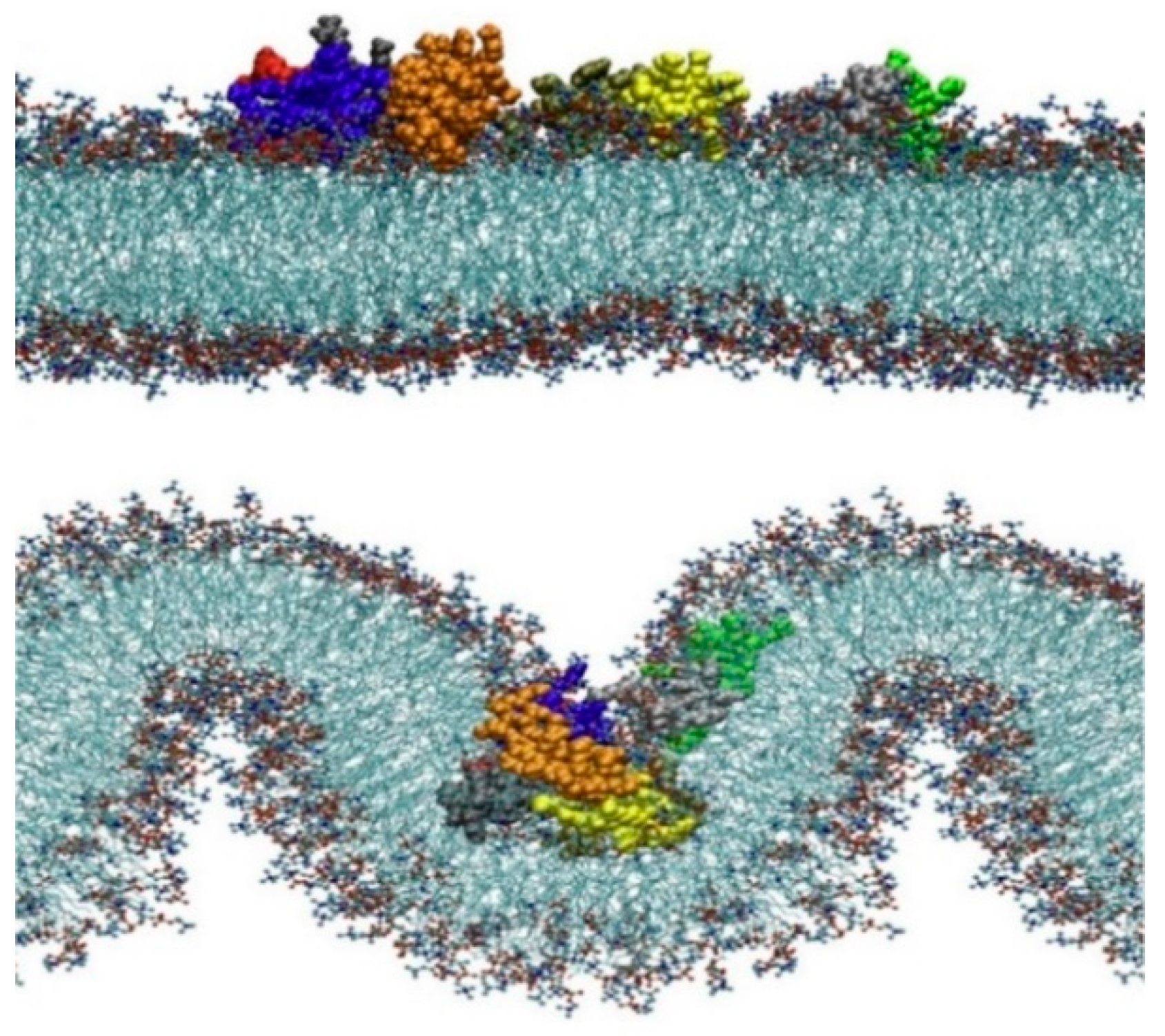
4.5. Comparison and Evaluation of the Characteristics of Different CPPs
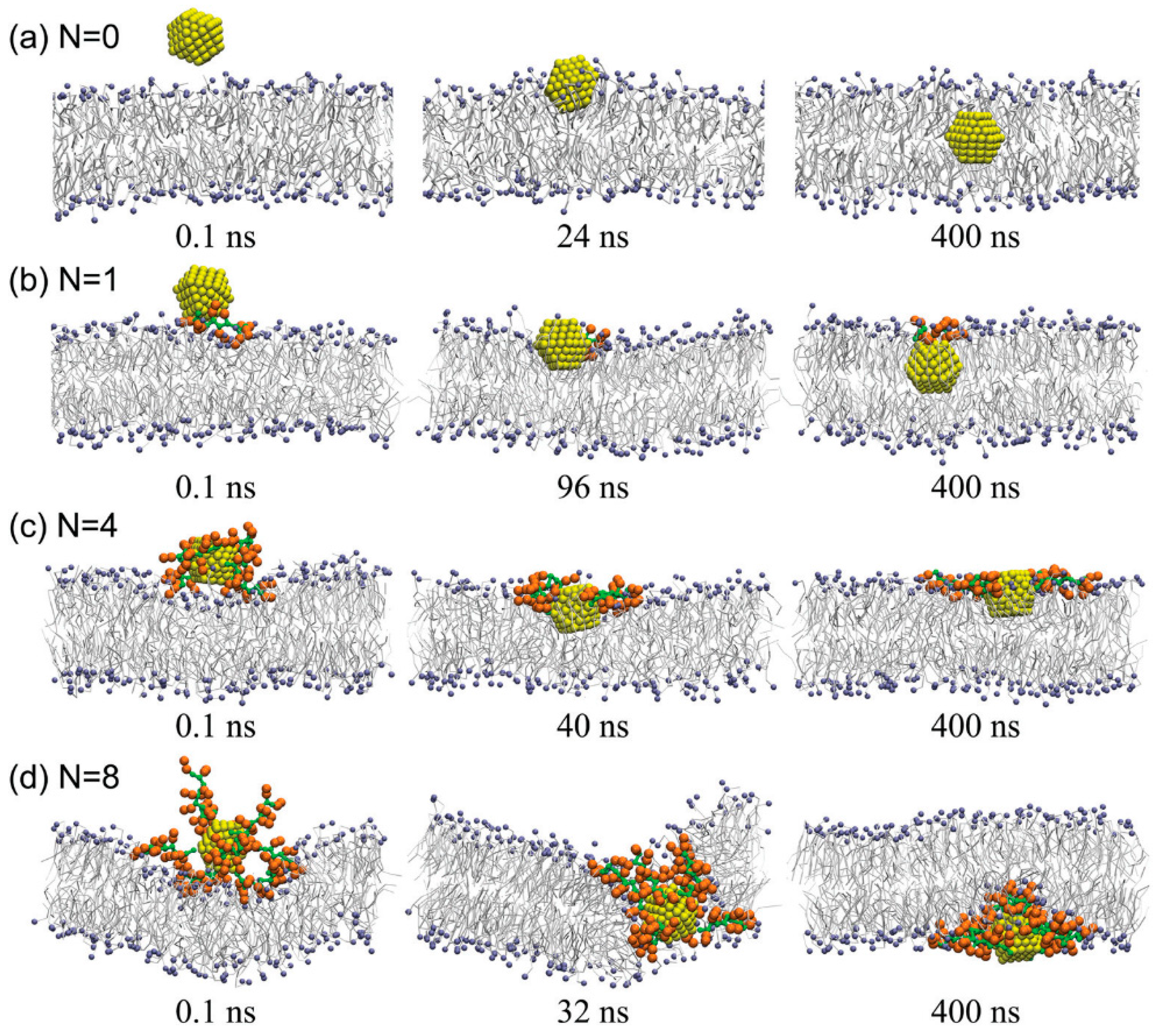
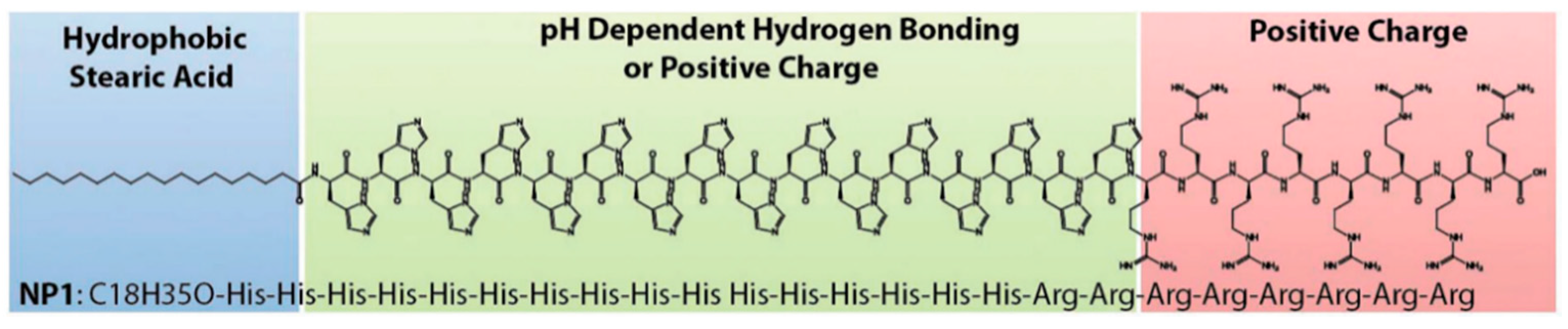
4.6. Larger Systems
4.7. Design Strategies of CPPs
5. Simulations of CPP Decoration and Coupling
5.1. Chemical Modifications of CPPs
5.2. CPP Coupling with Cargoes
5.3. The Design of Self-Complementary Peptides
6. Simulations on Membrane Modification and Simulations of Multi-Component Membranes
6.1. The Effect of the Membrane Compositions on CPP Uptake Process
6.2. The Effect of Membrane Tension and Transmembrane Potential
6.3. Peptide-Induced Membrane Response
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Eiriksdottir, E.; Konate, K.; Langel, U.; Divita, G.; Deshayes, S. Secondary structure of cell-penetrating peptides controls membrane interaction and insertion. Biochim. Biophys. Acta-Biomembr. 2010, 1798, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, E.L.; Dowdy, S.F. Cell penetrating peptides in drug delivery. Pharm. Res. 2004, 21, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Niesner, U.; Halin, C.; Lozzi, L.; Gunthert, M.; Neri, P.; Wunderli-Allenspach, H.; Zardi, L.; Neri, D. Quantitation of the tumor-targeting properties of antibody fragments conjugated to cell-permeating HIV-1 TAT peptides. Bioconjug. Chem. 2002, 13, 729–736. [Google Scholar] [CrossRef]
- Silva, S.; Almeida, A.J.; Vale, N. Combination of Cell-Penetrating Peptides with Nanoparticles for Therapeutic Application: A Review. Biomolecules 2019, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Tseng, Y.L.; Liu, J.J.; Hong, R.L. Translocation of liposomes into cancer cells by cell-penetrating peptides penetratin and TAT: A kinetic and efficacy study. Mol. Pharmacol. 2002, 62, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Torchilin, V.P.; Levchenko, T.S.; Rammohan, R.; Volodina, N.; Papahadjopoulos-Sternberg, B.; D’Souza, G.G.M. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 1972–1977. [Google Scholar] [CrossRef] [Green Version]
- Astriab-Fisher, A.; Sergueev, D.; Fisher, M.; Shaw, B.R.; Juliano, R.L. Conjugates of antisense oligonucleotides with the Tat and antennapedia cell-penetrating peptides: Effects on cellular uptake, binding to target sequences, and biologic actions. Pharm. Res. 2002, 19, 744–754. [Google Scholar] [CrossRef]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Borrelli, A.; Tornesello, A.L.; Tornesello, M.L.; Buonaguro, F.M. Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents. Molecules 2018, 23, 295. [Google Scholar] [CrossRef] [Green Version]
- Gessner, I.; Neundorf, I. Nanoparticles Modified with Cell-Penetrating Peptides: Conjugation Mechanisms, Physicochemical Properties, and Application in Cancer Diagnosis and Therapy. Int. J. Mol. Sci. 2020, 21, 2536. [Google Scholar] [CrossRef]
- Grau, M.; Walker, P.R.; Derouazi, M. Mechanistic insights into the efficacy of cell penetrating peptide-based cancer vaccines. Cell. Mol. Life Sci. 2018, 75, 2887–2896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habault, J.; Poyet, J.L. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, S.H.; Jang, J.; Cheon, D.H.; Chong, S.E.; Ahn, J.H.; Hyun, S.; Yu, J.; Lee, Y. pH-Activatable cell penetrating peptide dimers for potent delivery of anticancer drug to triple-negative breast cancer. J. Control. Release 2021, 330, 898–906. [Google Scholar] [CrossRef]
- Wu, J.; Li, J.S.; Wang, H.; Liu, C.B. Mitochondrial-targeted penetrating peptide delivery for cancer therapy. Expert Opin. Drug Deliv. 2018, 15, 951–964. [Google Scholar] [CrossRef]
- Fukuoka, Y.; Khafagy, E.; Goto, T.; Kamei, N.; Takayama, K.; Peppas, N.A.; Takeda-Morishita, M. Combination Strategy with Complexation Hydrogels and Cell-Penetrating Peptides for Oral Delivery of Insulin. Biol. Pharm. Bull. 2018, 41, 811–814. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.S.; Chen, Z.T.; Liao, T.Y.; Lin, C.; Shen, H.C.H.; Wang, Y.H.; Chang, C.W.; Liu, R.S.; Chen, R.P.Y.; Tu, P.H. An intranasally delivered peptide drug ameliorates cognitive decline in Alzheimer transgenic mice. EMBO Mol. Med. 2017, 9, 703–715. [Google Scholar] [CrossRef]
- Tansi, F.L.; Filatova, M.P.; Koroev, D.O.; Volpina, O.M.; Lange, S.; Schumann, C.; Teichgraber, U.K.; Reissmann, S.; Hilger, I. New generation CPPs show distinct selectivity for cancer and noncancer cells. J. Cell. Biochem. 2019, 120, 6528–6541. [Google Scholar] [CrossRef] [PubMed]
- Movafegh, B.; Jalal, R.; Mohanunadi, Z.; Aldaghi, S.A. Poly-L-arginine: Enhancing Cytotoxicity and Cellular Uptake of Doxorubicin and Necrotic Cell Death. Anti-Cancer Agents Med. Chem. 2018, 18, 1448–1456. [Google Scholar] [CrossRef]
- Du, Y.Q.; Wang, L.; Wang, W.Y.; Guo, T.; Zhang, M.Z.; Zhang, P.; Zhang, Y.N.; Wu, K.J.; Li, A.X.; Wang, X.Y.; et al. Novel Application of Cell Penetrating R11 Peptide for Diagnosis of Bladder Cancer. J. Biomed. Nanotechnol. 2018, 14, 161–167. [Google Scholar] [CrossRef]
- Berneche, S.; Nina, M.; Roux, B. Molecular dynamics simulation of melittin in a dimyristoylphosphatidylcholine bilayer membrane. Biophys. J. 1998, 75, 1603–1618. [Google Scholar] [CrossRef]
- Biggin, P.C.; Sansom, M.S.P. Interactions of alpha-helices with lipid bilayers: A review of simulation studies. Biophys. Chem. 1999, 76, 161–183. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, P.; Biggin, P.C.; Tieleman, D.P.; Sansom, M.S.P. Simulation studies of the interaction of antimicrobial peptides and lipid bilayers. Biochim. Biophys. Acta-Biomembr. 1999, 1462, 185–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial Peptides in Action. J. Am. Chem. Soc. 2006, 128, 12156–12161. [Google Scholar] [CrossRef] [Green Version]
- Trofimenko, E.; Grasso, G.; Heulot, M.; Chevalier, N.; Deriu, M.A.; Dubuis, G.; Arribat, Y.; Serulla, M.; Michel, S.; Vantomme, G.; et al. Genetic, cellular, and structural characterization of the membrane potential-dependent cell-penetrating peptide translocation pore. Elife 2021, 10, e69832. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Barsoum, P.J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar] [CrossRef] [Green Version]
- Torchilin, V.P. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv. Drug Deliv. Rev. 2008, 60, 548–558. [Google Scholar] [CrossRef]
- Tao, F.; Johns, R.A. Tat-Mediated Peptide Intervention in Analgesia and Anesthesia. Drug Dev. Res. 2010, 71, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The 3rd helix of the Antennapedia homeodomain translocates through biological-membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Derossi, D.; Chassaing, G.; Prochiantz, A. Trojan peptides: The penetratin system for intracellular delivery. Trends Cell Biol. 1998, 8, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, M.; Biverstahl, H.; Graslund, A.; Maler, L. Structure and positioning comparison of two variants of penetratin in two different membrane mimicking systems by NMR. Eur. J. Biochem. 2003, 270, 3055–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoren, P.E.G.; Persson, D.; Karlsson, M.; Norden, B. The Antennapedia peptide penetratin translocates across lipid bilayers—The first direct observation. FEBS Lett. 2000, 482, 265–268. [Google Scholar] [CrossRef] [Green Version]
- Futaki, S.; Ohashi, W.; Suzuki, T.; Niwa, M.; Tanaka, S.; Ueda, K.; Harashima, H.; Sugiura, Y. Stearylated arginine-rich peptides: A new class of transfection systems. Bioconj. Chem. 2001, 12, 1005–1011. [Google Scholar] [CrossRef]
- Pooga, M.; Hallbrink, M.; Zorko, M.; Langel, U. Cell penetration by transportan. FASEB J. 1998, 12, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Soomets, U.; Lindgren, M.; Gallet, X.; Hallbrink, M.; Elmquist, A.; Balaspiri, L.; Zorko, M.; Pooga, M.; Brasseur, R.; Langel, U. Deletion analogues of transportan. Biochim. Biophys. Acta-Biomembr. 2000, 1467, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Pooga, M.; Langel, U. Classes of Cell-Penetrating Peptides. In Cell-Penetrating Peptides: Methods and Protocols, 2nd ed.; Langel, U., Ed.; Humana: Atlanta, GA, USA, 2015; Volume 1324, pp. 3–28. [Google Scholar]
- Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q.N.; McMaster, G.K.; Brasseur, R.; Heitz, F.; Divita, G. A New Potent Secondary Amphipathic Cell-penetrating Peptide for siRNA Delivery Into Mammalian Cells. Mol. Ther. 2009, 17, 95–103. [Google Scholar] [CrossRef]
- Oehlke, J.; Scheller, A.; Wiesner, B.; Krause, E.; Beyermann, M.; Klauschenz, E.; Melzig, M.; Bienert, M. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta-Biomembr. 1998, 1414, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [Green Version]
- Elmquist, A.; Lindgren, M.; Bartfai, T.; Langel, U. VE-cadherin-derived cell-penetrating peptide, pVEC, with carrier functions. Exp. Cell Res. 2001, 269, 237–244. [Google Scholar] [CrossRef]
- Ziegler, A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv. Drug Deliv. Rev. 2008, 60, 580–597. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Depollier, J.; Mery, J.; Heitz, F.; Divita, G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 2001, 19, 1173–1176. [Google Scholar] [CrossRef]
- Lin, Y.Z.; Yao, S.Y.; Veach, R.A.; Torgerson, T.R.; Hawiger, J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J. Biol. Chem. 1995, 270, 14255–14258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Di Pisa, M.; Chassaing, G.; Swiecicki, J.M. Translocation Mechanism(s) of Cell-Penetrating Peptides: Biophysical Studies Using Artificial Membrane Bilayers. Biochemistry 2015, 54, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, W.B.; Fuselier, T.; He, J.; Wimley, W.C. Mechanism Maters: A Taxonomy of Cell Penetrating Peptices. Trends Biochem. Sci. 2015, 40, 749–764. [Google Scholar] [CrossRef] [Green Version]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef]
- Ter-Avetisyan, G.; Tuennemann, G.; Nowak, D.; Nitschke, M.; Herrmann, A.; Drab, M.; Cardoso, M.C. Cell Entry of Arginine-rich Peptides Is Independent of Endocytosis. J. Biol. Chem. 2009, 284, 3370–3378. [Google Scholar] [CrossRef] [Green Version]
- Gazit, E.; Lee, W.J.; Brey, P.T.; Shai, Y.C. Mode of Action of the Antibacterial Cecropin B2: A Spectrofluorometric Study. Biochemistry 1994, 33, 10681–10692. [Google Scholar] [CrossRef]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell internalization of the third helix of the antennapedia homeodomain is receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef] [Green Version]
- Prochiantz, A. Getting hydrophilic compounds into cells: Lessons from homeopeptides—Commentary. Curr. Opin. Neurobiol. 1996, 6, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of Antimicrobial Dermaseptin and Its Fluorescently Labeled Analogues with Phospholipid Membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Furuta, G.T.; Synnestvedt, K.; Colgan, S.P. Phosphorylation-dependent targeting of cAMP response element binding protein to the ubiquitin/proteasome pathway in hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 12091–12096. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.T.; Hung, W.C.; Chen, F.Y.; Huang, H.W. Many-body effect of antimicrobial peptides: On the correlation between lipid’s spontaneous curvature and pore formation. Biophys. J. 2005, 89, 4006–4016. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Wang, Y.; Zhang, X.; Zhang, W.; Guo, S.; Jin, F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J. Control. Release 2014, 174, 126–136. [Google Scholar] [CrossRef]
- Fretz, M.M.; Penning, N.A.; Al-Taei, S.; Futaki, S.; Takeuchi, T.; Nakase, I.; Storm, G.; Jones, A.T. Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34(+) leukaemia cells. Biochem. J. 2007, 403, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Mueller, J.; Kretzschmar, I.; Volkmer, R.; Boisguerin, P. Comparison of Cellular Uptake Using 22 CPPs in 4 Different Cell Lines. Bioconj. Chem. 2008, 19, 2363–2374. [Google Scholar] [CrossRef]
- Crosio, M.A.; Via, M.A.; Camara, C.I.; Mangiarotti, A.; Del Popolo, M.G.; Wilke, N. Interaction of a Polyarginine Peptide with Membranes of Different Mechanical Properties. Biomolecules 2019, 9, 625. [Google Scholar] [CrossRef]
- Tunnemann, G.; Martin, R.M.; Haupt, S.; Patsch, C.; Edenhofer, F.; Cardoso, M.C. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 2006, 20, 1775–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.T.; Sayers, E.J. Cell entry of cell penetrating peptides: Tales of tails wagging dogs. J. Control. Release 2012, 161, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Alder, B.J.; Wainwright, T.E. Phase Transition for a Hard Sphere System. J. Chem. Phys. 1957, 27, 1208–1209. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A. Correlations in the Motion of Atoms in Liquid Argon. Phys. Rev. 1964, 136, A405–A411. [Google Scholar] [CrossRef]
- Lensink, M.F. Membrane-Associated Proteins and Peptides. In Molecular Modeling of Proteins, 2nd ed.; Kukol, A., Ed.; Humana: New York, NY, USA, 2015; pp. 109–124. [Google Scholar]
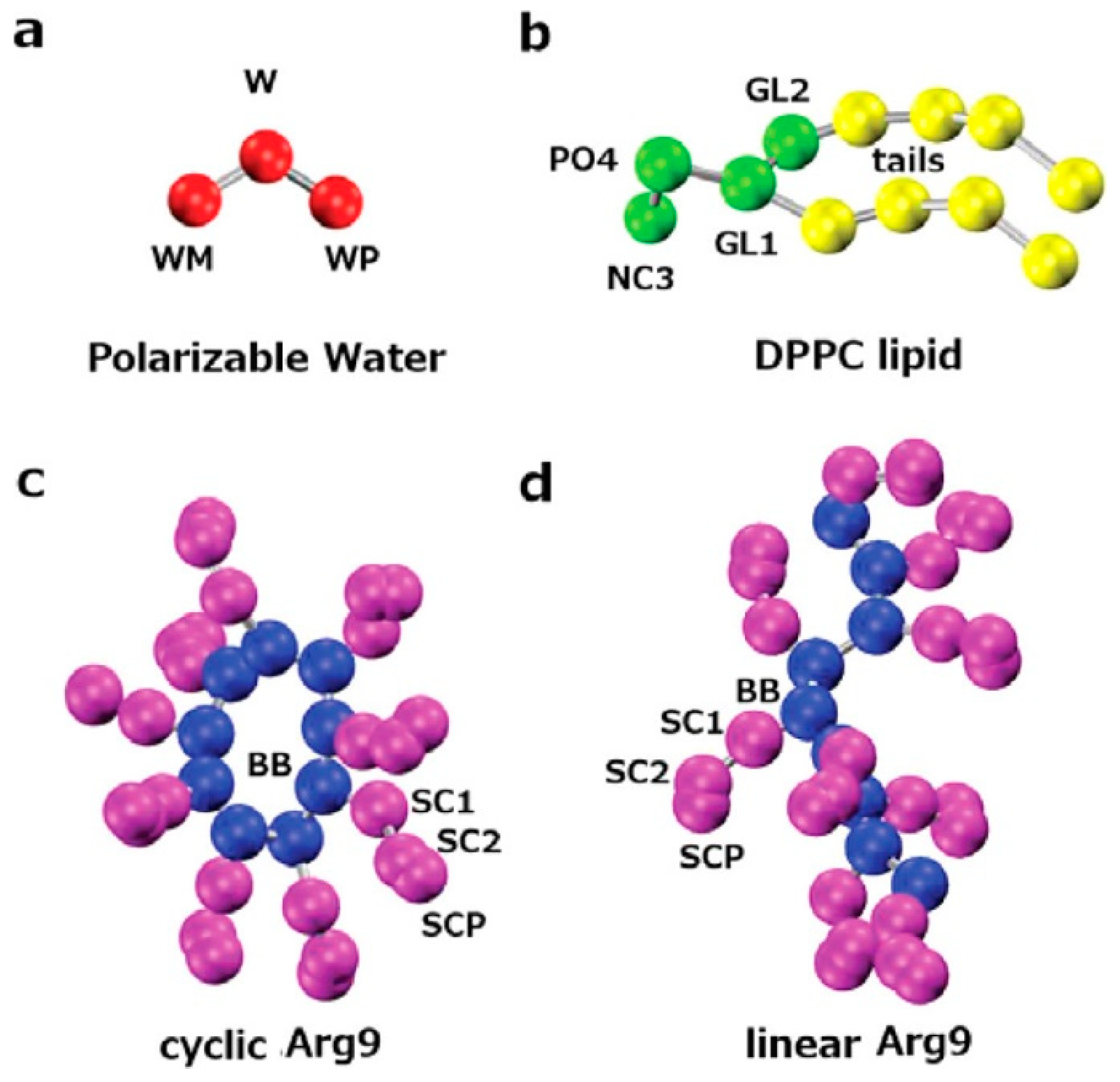
- Hu, Y.; Liu, X.R.; Sinha, S.K.; Patel, S. Trans location Thermodynamics of Linear and Cyclic Nonaarginine into Model DPPC Bilayer via Coarse-Grained Molecular Dynamics Simulation: Implications of Pore Formation and Nonadditivity. J. Phys. Chem. B 2014, 118, 2670–2682. [Google Scholar] [CrossRef]
- Saunders, M.G.; Voth, G.A. Coarse-Graining Methods for Computational Biology. In Annual Review of Biophysics; Dill, K.A., Ed.; Annual Reviews: San Mateo, CA, USA, 2013; Volume 42, pp. 73–93. [Google Scholar]
- Marrink, S.J.; Corradi, V.; Souza, P.C.T.; Ingolfsson, H.I.; Tieleman, D.P.; Sansom, M.S.P. Computational Modeling of Realistic Cell Membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef] [Green Version]
- Lazaridis, T.; Leveritt, J.M.; PeBenito, L. Implicit membrane treatment of buried charged groups: Application to peptide translocation across lipid bilayers. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 2149–2159. [Google Scholar] [CrossRef] [Green Version]
- Nepal, B.; Leveritt, J.; Lazaridis, T. Membrane Curvature Sensing by Amphipathic Helices: Insights from Implicit Membrane Modeling. Biophys. J. 2018, 114, 2128–2141. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Schulten, K. Calculating potentials of mean force from steered molecular dynamics simulations. J. Chem. Phys. 2004, 120, 5946–5961. [Google Scholar] [CrossRef]
- Yesylevskyy, S.; Marrink, S.J.; Mark, A.E. Alternative Mechanisms for the Interaction of the Cell-Penetrating Peptides Penetratin and the TAT Peptide with Lipid Bilayers. Biophys. J. 2009, 97, 40–49. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kstner, J. Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. Wiley Interdiscip. Rev.-Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Reid, L.M.; Verma, C.S.; Essex, J.W. The role of molecular simulations in understanding the mechanisms of cell-penetrating peptides. Drug Discov. Today 2019, 24, 1821–1835. [Google Scholar] [CrossRef]
- Nymeyer, H.; Woolf, T.B.; Garcia, A.E. Folding is not required for bilayer insertion: Replica exchange simulations of an alpha-helical peptide with an explicit lipid bilayer. Proteins-Struct. Funct. Bioinform. 2005, 59, 783–790. [Google Scholar] [CrossRef]
- Alaybeyoglu, B.; Akbulut, B.S.; Ozkirimli, E. pVEC hydrophobic N-terminus is critical for antibacterial activity. J. Pept. Sci. 2018, 24, e3083. [Google Scholar] [CrossRef]
- Babin, V.; Roland, C.; Sagui, C. Adaptively biased molecular dynamics for free energy calculations. J. Chem. Phys. 2008, 128, 134101. [Google Scholar] [CrossRef] [Green Version]
- Gimenez-Dejoz, J.; Numata, K. Molecular dynamics study of the internalization of cell-penetrating peptides containing unnatural amino acids across membranes. Nanoscale Adv. 2022, 4, 397–407. [Google Scholar] [CrossRef]
- Noe, F.; Fischer, S. Transition networks for modeling the kinetics of conformational change in macromolecules. Curr. Opin. Struct. Biol. 2008, 18, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Chodera, J.D.; Swope, W.C.; Pitera, J.W.; Dill, K.A. Long-time protein folding dynamics from short-time molecular dynamics simulations. Multiscale Model. Simul. 2006, 5, 1214–1226. [Google Scholar] [CrossRef] [Green Version]
- Prinz, J.-H.; Wu, H.; Sarich, M.; Keller, B.; Senne, M.; Held, M.; Chodera, J.D.; Schuette, C.; Noe, F. Markov models of molecular kinetics: Generation and validation. J. Chem. Phys. 2011, 134, 174105. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Bowman, G.R.; Huang, X. Dynamics of an Intrinsically Disordered Protein Reveal Metastable Conformations That Potentially Seed Aggregation. J. Am. Chem. Soc. 2013, 135, 16092–16101. [Google Scholar] [CrossRef] [PubMed]
- Noe, F.; Schuette, C.; Vanden-Eijnden, E.; Reich, L.; Weikl, T.R. Constructing the equilibrium ensemble of folding pathways from short off-equilibrium simulations. Proc. Natl. Acad. Sci. USA 2009, 106, 19011–19016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Weng, J.; Wang, W. An unconventional ligand-binding mechanism of substrate-binding proteins: MD simulation and Markov state model analysis of BtuF. J. Comput. Chem. 2019, 40, 1440–1448. [Google Scholar] [CrossRef]
- Sengupta, U.; Carballo-Pacheco, M.; Strodel, B. Automated Markov state models for molecular dynamics simulations of aggregation and self-assembly. J. Chem. Phys. 2019, 150, 115101. [Google Scholar] [CrossRef]
- Weng, J.; Yang, M.; Wang, W.; Xu, X.; Tian, Z. Revealing Thermodynamics and Kinetics of Lipid Self-Assembly by Markov State Model Analysis. J. Am. Chem. Soc. 2020, 142, 21344–21352. [Google Scholar] [CrossRef]
- Huang, K.; Garcia, A.E. Free Energy of Translocating an Arginine-Rich Cell-Penetrating Peptide across a Lipid Bilayer Suggests Pore Formation. Biophys. J. 2013, 104, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Kabelka, I.; Brozek, R.; Vacha, R. Selecting Collective Variables and Free-Energy Methods for Peptide Translocation across Membranes. J. Chem. Inf. Model. 2021, 61, 819–830. [Google Scholar] [CrossRef]
- Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.-J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 2308–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herce, H.D.; Garcia, A.E.; Cardoso, M.C. Fundamental Molecular Mechanism for the Cellular Uptake of Guanidinium-Rich Molecules. J. Am. Chem. Soc. 2014, 136, 17459–17467. [Google Scholar] [CrossRef]
- Mishra, A.; Lai, G.H.; Schmidt, N.W.; Sun, V.Z.; Rodriguez, A.R.; Tong, R.; Tang, L.; Cheng, J.J.; Deming, T.J.; Kamei, D.T.; et al. Translocation of HIV TAT peptide and analogues induced by multiplexed membrane and cytoskeletal interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 16883–16888. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.C.; Sa’adedin, F.; Deme, B.; Rao, P.F.; Bradshaw, J. Insertion of TAT peptide and perturbation of negatively charged model phospholipid bilayer revealed by neutron diffraction. Biochim. Biophys. Acta-Biomembr. 2013, 1828, 1982–1988. [Google Scholar] [CrossRef] [Green Version]
- Pourmousa, M.; Karttunen, M. Early stages of interactions of cell-penetrating peptide penetratin with a DPPC bilayer. Chem. Phys. Lipids 2013, 169, 85–94. [Google Scholar] [CrossRef]
- Yao, C.; Kang, Z.Z.; Yu, B.; Chen, Q.; Liu, Y.C.; Wang, Q. All-Factor Analysis and Correlations on the Transmembrane Process for Arginine-Rich Cell-Penetrating Peptides. Langmuir 2019, 35, 9286–9296. [Google Scholar] [CrossRef]
- Mitchell, D.J.; Kim, D.T.; Steinman, L.; Fathman, C.G.; Rothbard, J.B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef]
- Magzoub, M.; Kilk, K.; Eriksson, L.E.; Langel, U.; Graslund, A. Interaction and structure induction of cell-penetrating peptides in the presence of phospholipid vesicles. Biochim. Biophys. Acta 2001, 1512, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Deshayes, S.; Heitz, A.; Morris, M.C.; Charnet, P.; Divita, G.; Heitz, F. Insight into the mechanism of internalization of the cell-penetrating carrier peptide Pep-1 through conformational analysis. Biochemistry 2004, 43, 1449–1457. [Google Scholar] [CrossRef]
- Dunkin, C.M.; Pokorny, A.; Almeida, P.F.; Lee, H.S. Molecular Dynamics Studies of Transportan 10 (Tp10) Interacting with a POPC Lipid Bilayer. J. Phys. Chem. B 2011, 115, 1188–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.C.; Waring, A.J.; Ruchala, P.; Hong, M. Membrane-Bound Dynamic Structure of an Arginine-Rich Cell-Penetrating Peptide, the Protein Transduction Domain of HIV TAT, from Solid-State NMR. Biochemistry 2010, 49, 6009–6020. [Google Scholar] [CrossRef] [Green Version]
- Song, J.J.; Kai, M.; Zhang, W.; Zhang, J.D.; Liu, L.W.; Zhang, B.Z.; Liu, X.; Wang, R. Cellular uptake of transportan 10 and its analogs in live cells: Selectivity and structure-activity relationship studies. Peptides 2011, 32, 1934–1941. [Google Scholar] [CrossRef]
- Polyansky, A.A.; Volynsky, P.E.; Arseniev, A.S.; Efremov, R.G. Adaptation of a Membrane-active Peptide to Heterogeneous Environment. I. Structural Plasticity of the Peptide. J. Phys. Chem. B 2009, 113, 1107–1119. [Google Scholar] [CrossRef]
- Tsou, L.K.; Tatko, C.D.; Waters, M.L. Simple cation-pi interaction between a phenyl ring and a protonated amine stabilizes an alpha-helix in water. J. Am. Chem. Soc. 2002, 124, 14917–14921. [Google Scholar] [CrossRef]
- Czajlik, A.; Mesko, E.; Penke, B.; Perczel, A. Investigation of penetratin peptides Part 1. The environment dependent conformational properties of penetratin and two of its derivatives. J. Pept. Sci. 2002, 8, 151–171. [Google Scholar] [CrossRef]
- Balayssac, S.; Burlina, F.; Convert, O.; Bolbach, G.; Chassaing, G.; Lequin, O. Comparison of penetratin and other homeodomain-derived cell-penetrating peptides: Interaction in a membrane-mimicking environment and cellular uptake efficiency. Biochemistry 2006, 45, 1408–1420. [Google Scholar] [CrossRef]
- Jobin, M.L.; Vamparys, L.; Deniau, R.; Grelard, A.; Mackereth, C.D.; Fuchs, P.F.J.; Alves, I.D. Biophysical Insight on the Membrane Insertion of an Arginine-Rich Cell-Penetrating Peptide. Int. J. Mol. Sci. 2019, 20, 4441. [Google Scholar] [CrossRef] [Green Version]
- Rothbard, J.B.; Jessop, T.C.; Wender, P.A. Adaptive translocation: The role of hydrogen bonding and membrane potential in the uptake of guanidinium-rich transporters into cells. Adv. Drug Deliv. Rev. 2005, 57, 495–504. [Google Scholar] [CrossRef]
- Tang, M.; Waring, A.J.; Hong, M. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptide from solid-state NMR. J. Am. Chem. Soc. 2007, 129, 11438–11446. [Google Scholar] [CrossRef]
- Lamaziere, A.; Burlina, F.; Wolf, C.; Chassaing, G.; Trugnan, G.; Ayala-Sanmartin, J. Non-Metabolic Membrane Tubulation and Permeability Induced by Bioactive Peptides. PLoS ONE 2007, 2, e201. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Scholtz, J.M. Energetics of polar side-chain interactions in helical peptides: Salt effects on ion pairs and hydrogen bonds. Biochemistry 1998, 37, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Andrew, C.D.; Penel, S.; Jones, G.R.; Doig, A.J. Stabilizing nonpolar/polar side-chain interactions in the alpha-helix. Proteins-Struct. Funct. Genet. 2001, 45, 449–455. [Google Scholar] [CrossRef]
- Walrant, A.; Vogel, A.; Correia, I.; Lequin, O.; Olausson, B.E.S.; Desbat, B.; Sagan, S.; Alves, I.D. Membrane interactions of two arginine-rich peptides with different cell internalization capacities. Biochim. Biophys. Acta-Biomembr. 2012, 1818, 1755–1763. [Google Scholar] [CrossRef]
- He, X.C.; Qu, Z.G.; Xu, F. Simulation study of interaction mechanism between peptide and asymmetric membrane. Mol. Simul. 2017, 43, 34–41. [Google Scholar] [CrossRef]
- Burns, K.E.; Robinson, M.K.; Thevenin, D. Inhibition of Cancer Cell Proliferation and Breast Tumor Targeting of pHLIP-Monomethyl Auristatin E Conjugates. Mol. Pharm. 2015, 12, 1250–1258. [Google Scholar] [CrossRef] [Green Version]
- Viola-Villegas, N.T.; Carlin, S.D.; Ackerstaff, E.; Sevak, K.K.; Divilov, V.; Serganova, I.; Kruchevsky, N.; Anderson, M.; Blasberg, R.G.; Andreev, O.A.; et al. Understanding the pharmacological properties of a metabolic PET tracer in prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 7254–7259. [Google Scholar] [CrossRef] [Green Version]
- Andreev, O.A.; Karabadzhak, A.G.; Weerakkody, D.; Andreev, G.O.; Engelman, D.M.; Reshetnyak, Y.K. pH (low) insertion peptide (pHLIP) inserts across a lipid bilayer as a helix and exits by a different path. Proc. Natl. Acad. Sci. USA 2010, 107, 4081–4086. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.H.; Qian, Z.Y.; Luo, Y.; Zhang, Y.; Mu, Y.G.; Wei, G.H. Membrane Binding and Insertion of a pHLIP Peptide Studied by All-Atom Molecular Dynamics Simulations. Int. J. Mol. Sci. 2013, 14, 14532–14549. [Google Scholar] [CrossRef] [Green Version]
- Ramaker, K.; Henkel, M.; Krause, T.; Roeckendorf, N.; Frey, A. Cell penetrating peptides: A comparative transport analysis for 474 sequence motifs. Drug Deliv. 2018, 25, 928–937. [Google Scholar] [CrossRef]
- Kumara, B.T.; Wijesiri, N.K.; Rathnayake, P.; Ranatunga, R. A Re-evaluation of the Free Energy Profiles for Cell-Penetrating Peptides across DOPC Membranes. Int. J. Pept. Res. Ther. 2021, 27, 2931–2943. [Google Scholar] [CrossRef]
- Ruzza, P.; Biondi, B.; Marchiani, A.; Antolini, N.; Calderan, A. Cell-Penetrating Peptides: A Comparative Study on Lipid Affinity and Cargo Delivery Properties. Pharmaceuticals 2010, 3, 1045–1062. [Google Scholar] [CrossRef] [Green Version]
- Langel, U. Cell-Penetrating Peptides and Transportan. Pharmaceutics 2021, 13, 987. [Google Scholar] [CrossRef]
- Kawamoto, S.; Takasu, M.; Miyakawa, T.; Morikawa, R.; Oda, T.; Futaki, S.; Nagao, H. Inverted micelle formation of cell-penetrating peptide studied by coarse-grained simulation: Importance of attractive force between cell-penetrating peptides and lipid head group. J. Chem. Phys. 2011, 134, 095103. [Google Scholar] [CrossRef]
- Afonin, S.; Frey, A.; Bayerl, S.; Fischer, D.; Wadhwani, P.; Weinkauf, S.; Ulrich, A.S. The cell-penetrating peptide TAT(48–60) induces a non-lamellar phase in DMPC membranes. Chemphyschem 2006, 7, 2134–2142. [Google Scholar] [CrossRef]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, U. Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef]
- Li, L.B.; Vorobyov, I.; Allen, T.W. The Different Interactions of Lysine and Arginine Side Chains with Lipid Membranes. J. Phys. Chem. B 2013, 117, 11906–11920. [Google Scholar] [CrossRef]
- Shimonishi, Y.; Oehlke, J.; Scheller, A.; Janek, K.; Bienert, M. Rapid Translocation of Amphipathic α-Helical and β-Sheet-Forming Peptides through Plasma Membranes of Endothelial Cells; Peptide Science: Present and Future; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Scheller, A.; Oehlke, J.; Wiesner, B.; Dathe, M.; Krause, E.; Beyermann, M.; Melzig, M.; Bienert, M. Structural requirements for cellular uptake of alpha-helical amphipathic peptides. J. Pept. Sci. 1999, 5, 185–194. [Google Scholar] [CrossRef]
- Martin, I.; Teixido, M.; Giralt, E. Design, Synthesis and Characterization of a New Anionic Cell-Penetrating Peptide: SAP(E). Chembiochem 2011, 12, 896–903. [Google Scholar] [CrossRef]
- Khafagy, E.S.; Morishita, M.; Ida, N.; Nishio, R.; Isowa, K.; Takayama, K. Structural requirements of penetratin absorption enhancement efficiency for insulin delivery. J. Control. Release 2010, 143, 302–310. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Langel, U. Recent CPP-based applications in medicine. Expert Opin. Drug Deliv. 2019, 16, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef] [PubMed]
- Farrera-Sinfreu, J.; Giralt, E.; Royo, M.; Albericio, F. Cell-Penetrating Proline-Rich Peptidomimetics. In Peptide Characterization and Application Protocols; Fields, G.B., Ed.; Humana: Atlanta, GA, USA, 2007; Volume 386, pp. 241–267. [Google Scholar]
- Antunes, E.; Azoia, N.G.; Matama, T.; Gomes, A.C.; Cavaco-Paulo, A. The activity of LE10 peptide on biological membranes using molecular dynamics, in vitro and in vivo studies. Colloids Surf. B-Biointerfaces 2013, 106, 240–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.W.; Lin, Z.W.; Chang, W.F.; Chen, Y.F.; Hu, W.W. Development of an indolicidin-derived peptide by reducing membrane perturbation to decrease cytotoxicity and maintain gene delivery ability. Colloids Surf. B-Biointerfaces 2018, 165, 18–27. [Google Scholar] [CrossRef]
- Karle, I.L. Controls exerted by the Aib residue: Helix formation and helix reversal. Biopolymers 2001, 60, 351–365. [Google Scholar] [CrossRef]
- Fujita, S.; Motoda, Y.; Kigawa, T.; Tsuchiya, K.; Numata, K. Peptide-Based Polyion Complex Vesicles That Deliver Enzymes into Intact Plants to Provide Antibiotic Resistance without Genetic Modification. Biomacromolecules 2021, 22, 1080–1090. [Google Scholar] [CrossRef]
- Tuennemann, G.; Ter-Avetisyan, G.; Martin, R.M.; Stoeckl, M.; Herrmann, A.; Cardoso, C. Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J. Pept. Sci. 2008, 14, 469–476. [Google Scholar] [CrossRef]
- Bode, S.A.; Wallbrecher, R.; Brock, R.; van Hest, J.C.M.; Lowik, D.W.P.M. Activation of cell-penetrating peptides by disulfide bridge formation of truncated precursors. Chem. Commun. 2014, 50, 415–417. [Google Scholar] [CrossRef]
- Oskolkov, N.; Arukuusk, P.; Copolovici, D.-M.; Lindberg, S.; Margus, H.; Padari, K.; Pooga, M.; Langel, U. NickFects, Phosphorylated Derivatives of Transportan 10 for Cellular Delivery of Oligonucleotides. Int. J. Pept. Res. Ther. 2011, 17, 147–157. [Google Scholar] [CrossRef]
- Zhang, L.L.; Hoffman, J.A.; Ruoslahti, E. Molecular profiling of heart endothelial cells. Circulation 2005, 112, 1601–1611. [Google Scholar] [CrossRef]
- Stewart, K.M.; Horton, K.L.; Kelley, S.O. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org. Biomol. Chem. 2008, 6, 2242–2255. [Google Scholar] [CrossRef] [PubMed]
- Saini, A.; Jaswal, R.R.; Negi, R.; Nandel, F.S. Insights on the structural characteristics of Vim-TBS (58-81) peptide for future applications as a cell penetrating peptide. Biosci. Trends 2013, 7, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balzeau, J.; Peterson, A.; Eyer, J. The vimentin-tubulin binding site peptide (Vim-TBS.58-81) crosses the plasma membrane and enters the nuclei of human glioma cells. Int. J. Pharm. 2012, 423, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashwanikumar, N.; Plaut, J.S.; Mostofian, B.; Patel, S.; Kwak, P.; Sun, C.; McPhail, K.; Zuckerman, D.M.; Esener, S.C.; Sahay, G. Supramolecular self assembly of nanodrill-like structures for intracellular delivery. J. Control. Release 2018, 282, 76–89. [Google Scholar] [CrossRef]
- Garcia-Sosa, A.T.; Tulp, I.; Langel, K.; Langel, U. Peptide-Ligand Binding Modeling of siRNA with Cell-Penetrating Peptides. Biomed Res. Int. 2014, 2014, 257040. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.; Shirazi, A.N.; Northup, K.; Sullivan, B.; Tiwari, R.K.; Bisoffi, M.; Parang, K. Enhanced Cellular Uptake of Short Polyarginine Peptides through Fatty Acylation and Cyclization. Mol. Pharm. 2014, 11, 2845–2854. [Google Scholar] [CrossRef]
- Hedegaard, S.F.; Bruhn, D.S.; Khandelia, H.; Cardenas, M.; Nielsen, H.M. Shuffled lipidation pattern and degree of lipidation determines the membrane interaction behavior of a linear cationic membrane-active peptide. J. Colloid Interface Sci. 2020, 578, 584–597. [Google Scholar] [CrossRef]
- Akhunzada, M.J.; Chandramouli, B.; Bhattacharjee, N.; Macchi, S.; Cardarelli, F.; Brancato, G. The role of Tat peptide self-aggregation in membrane pore stabilization: Insights from a computational study. Phys. Chem. Chem. Phys. 2017, 19, 27603–27610. [Google Scholar] [CrossRef]
- Hu, J.M.; Lou, Y.M.; Wu, F.M. Improved Intracellular Delivery of Polyarginine Peptides with Cargoes. J. Phys. Chem. B 2019, 123, 2636–2644. [Google Scholar] [CrossRef]
- Grasso, G.; Deriu, M.A.; Prat, M.; Rimondini, L.; Verne, E.; Follenzi, A.; Danani, A. Cell Penetrating Peptide Adsorption on Magnetite and Silica Surfaces: A Computational Investigation. J. Phys. Chem. B 2015, 119, 8239–8246. [Google Scholar] [CrossRef]
- Kubiak-Ossowska, K.; Burley, G.; Patwardhan, S.V.; Mulheran, P.A. Spontaneous Membrane-Translocating Peptide Adsorption at Silica Surfaces: A Molecular Dynamics Study. J. Phys. Chem. B 2013, 117, 14666–14675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, X.B.; Sun, D.L.; Zhou, J. Molecular mechanism of HIV-1 TAT peptide and its conjugated gold nanoparticles translocating across lipid membranes. Phys. Chem. Chem. Phys. 2019, 21, 10300–10310. [Google Scholar] [CrossRef]
- Zhang, S.G.; Holmes, T.C.; Dipersio, C.M.; Hynes, R.O.; Su, X.; Rich, A. Self-complementary oligopeptide matrices support mammalian cell attachment. Biomaterials 1995, 16, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.B.; Wang, W.; Chen, P. Interaction of an ionic complementary peptide with a hydrophobic graphite surface. Protein Sci. 2010, 19, 1639–1648. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.B.; Wang, W.; Chen, P. Adsorption of an Ionic Complementary Peptide on the Hydrophobic Graphite Surface. J. Phys. Chem. C 2010, 114, 454–459. [Google Scholar] [CrossRef]
- Hong, Y.S.; Legge, R.L.; Zhang, S.; Chen, P. Effect of amino acid sequence and pH on nanofiber formation of self-assembling peptides EAK16-II and EAK16-IV. Biomacromolecules 2003, 4, 1433–1442. [Google Scholar] [CrossRef]
- Fung, S.Y.; Yang, H.; Sadatmousavi, P.; Sheng, Y.; Mamo, T.; Nazarian, R.; Chen, P. Amino Acid Pairing for De Novo Design of Self-Assembling Peptides and Their Drug Delivery Potential. Adv. Funct. Mater. 2011, 21, 2456–2464. [Google Scholar] [CrossRef]
- Zhang, L.; Sheng, Y.B.; Yazdi, A.Z.; Sarikhani, K.; Wang, F.; Jiang, Y.S.; Liu, J.W.; Zheng, T.; Wang, W.; Ouyang, P.K.; et al. Surface-assisted assembly of a histidine-rich lipidated peptide for simultaneous exfoliation of graphite and functionalization of graphene nanosheets. Nanoscale 2019, 11, 2999–3012. [Google Scholar] [CrossRef]
- Lu, S.; Cui, W.J.; Li, J.; Sheng, Y.B.; Chen, P. Functional Control of Peptide Amphiphile Assemblies via Modulation of Internal Cohesion and Surface Chemistry Switch. Chem. Eur. J. 2018, 24, 13931–13937. [Google Scholar] [CrossRef]
- Mukherjee, S.; Kar, R.K.; Nanga, R.P.R.; Mroue, K.H.; Ramamoorthy, A.; Bhunia, A. Accelerated molecular dynamics simulation analysis of MSI-594 in a lipid bilayer. Phys. Chem. Chem. Phys. 2017, 19, 19289–19299. [Google Scholar] [CrossRef]
- Hu, Y.; Sinha, S.K.; Patel, S. Investigating Hydrophilic Pores in Model Lipid Bilayers Using Molecular Simulations: Correlating Bilayer Properties with Pore-Formation Thermodynamics. Langmuir 2015, 31, 6615–6631. [Google Scholar] [CrossRef] [Green Version]
- Niemela, P.S.; Ollila, S.; Hyvonen, M.T.; Karttunen, M.; Vattulainen, I. Assessing the nature of lipid raft membranes. PLoS Comput. Biol. 2007, 3, e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharmin, S.; Islam, M.Z.; Karal, M.A.; Shibly, S.U.; Dohra, H.; Yamazaki, M. Effects of Lipid Composition on the Entry of Cell-Penetrating Peptide Oligoarginine into Single Vesicles. Biochemistry 2016, 55, 4154–4165. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Patel, S. Thermodynamics of cell-penetrating HIV1 TAT peptide insertion into PC/PS/CHOL model bilayers through transmembrane pores: The roles of cholesterol and anionic lipids. Soft Matter 2016, 12, 6716–6727. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.W.; Kurum, A.; Kaynak, M.; Bonati, L.; Han, Y.L.; Cencen, V.; Gao, M.; Xie, Y.Q.; Guo, Y.G.; Hannebelle, M.T.M.; et al. Cancer-cell stiffening via cholesterol depletion enhances adoptive T-cell immunotherapy. Nat. Biomed. Eng. 2021, 5, 1411–1425. [Google Scholar] [CrossRef]
- Polyansky, A.A.; Volynsky, P.E.; Arseniev, A.S.; Efremov, R.G. Adaptation of a Membrane-active Peptide to Heterogeneous Environment. II. The Role of Mosaic Nature of the Membrane Surface. J. Phys. Chem. B 2009, 113, 1120–1126. [Google Scholar] [CrossRef]
- Orsi, M.; Essex, J.W. Physical properties of mixed bilayers containing lamellar and nonlamellar lipids: Insights from coarse-grain molecular dynamics simulations. Faraday Discuss. 2013, 161, 249–272. [Google Scholar] [CrossRef]
- Ding, W.; Palaiokostas, M.; Wang, W.; Orsi, M. Effects of Lipid Composition on Bilayer Membranes Quantified by All-Atom Molecular Dynamics. J. Phys. Chem. B 2015, 119, 15263–15274. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.J.; Deserno, M. A Systematically Coarse-Grained Solvent-Free Model for Quantitative Phospholipid Bilayer Simulations. J. Phys. Chem. B 2012, 116, 3907. [Google Scholar] [CrossRef]
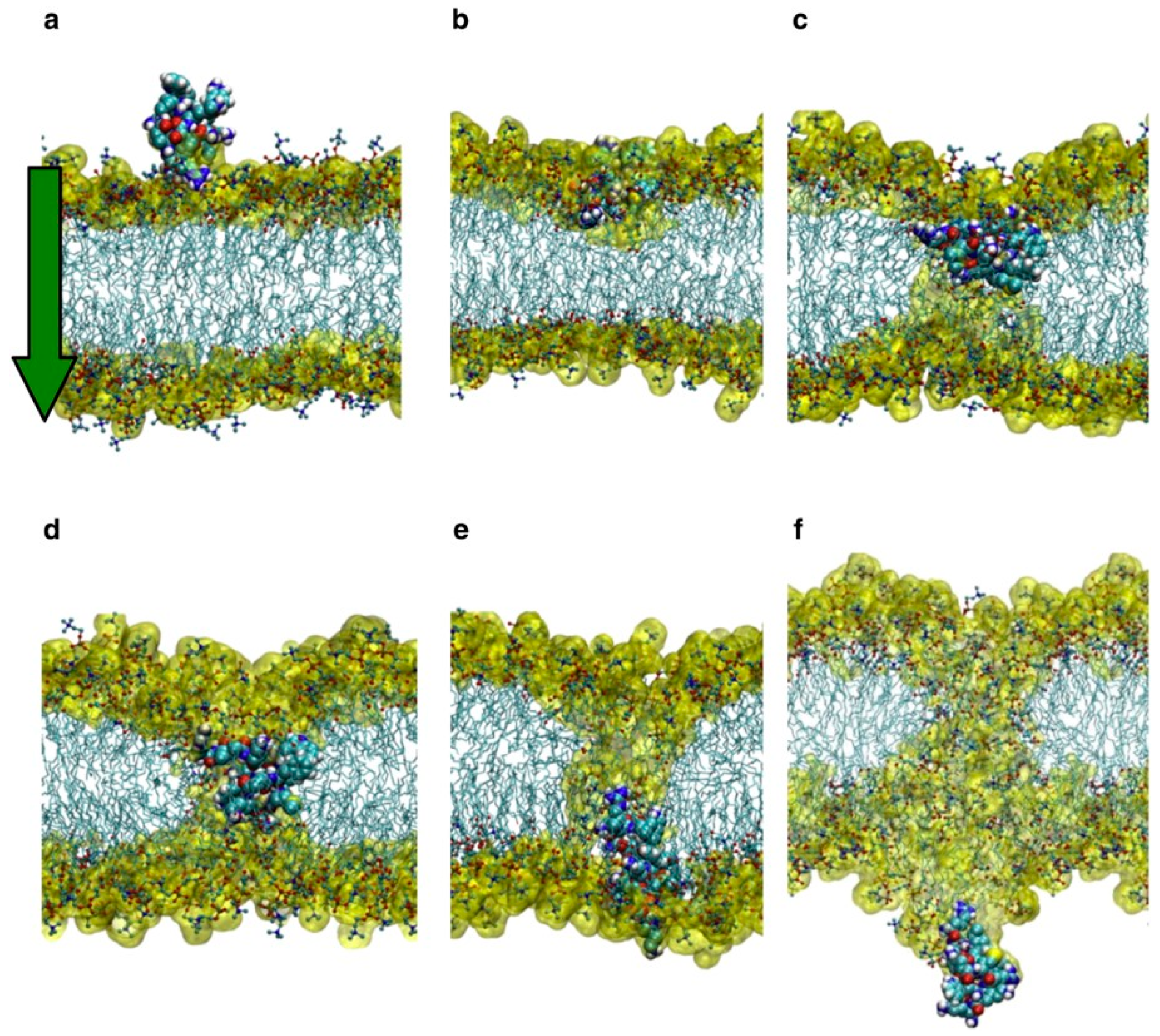
- He, X.C.; Lin, M.; Sha, B.Y.; Feng, S.S.; Shi, X.H.; Qu, Z.G.; Xu, F. Coarse-grained molecular dynamics studies of the translocation mechanism of polyarginines across asymmetric membrane under tension. Sci. Rep. 2015, 5, 12808. [Google Scholar] [CrossRef]
- Gao, X.L.; Hong, S.; Liu, Z.P.; Yue, T.T.; Dobnikar, J.; Zhang, X.R. Membrane potential drives direct translocation of cell-penetrating peptides. Nanoscale 2019, 11, 1949–1958. [Google Scholar] [CrossRef]
- Rao, Y.; Kwok, S.J.J.; Lombardi, J.; Turro, N.J.; Eisenthal, K.B. Label-free probe of HIV-1 TAT peptide binding to mimetic membranes. Proc. Natl. Acad. Sci. USA 2014, 111, 12684–12688. [Google Scholar] [CrossRef] [Green Version]
- Dorairaj, S.; Allen, T.W. On the thermodynamic stability of a charged arginine side chain in a transmembrane helix. Proc. Natl. Acad. Sci. USA 2007, 104, 4943–4948. [Google Scholar] [CrossRef] [Green Version]
- MacCallum, J.L.; Bennett, W.F.D.; Tieleman, D.P. Partitioning of amino acid side chains into lipid bilayers: Results from computer simulations and comparison to experiment. J. Gen. Physiol. 2007, 129, 371–377. [Google Scholar] [CrossRef]
- Grasso, G.; Muscat, S.; Rebella, M.; Morbiducci, U.; Audenino, A.; Danani, A.; Deriu, M.A. Cell penetrating peptide modulation of membrane biomechanics by Molecular dynamics. J. Biomech. 2018, 73, 137–144. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Type | Number of Arginine Residues | Charge | Conformation * | Concentration Dependence ** | Ref. |
|---|---|---|---|---|---|---|---|
| HIV-1 Tat | GRKKRRQRRRPPQ | Cationic | 6 | +8 | random coil | + | [26] |
| Arg9 | RRRRRRRRR | Cationic | 9 | +9 | random coil | + | [40] |
| Penetratin | RQIKIWFQNRRMKWKK | Cationic and Amphipathic | 3 | +7 | α-helix or β-strand | + | [30] |
| TP10 | AGYLLGKINLKALAALAKKIL | Amphipathic | 0 | +4 | α-helix | + | [35] |
| pVEC | LLIILRRRIRKQAHAHSK | Amphipathic | 4 | +8 | β-strand or random coil | + | [41] |
| Pep-1 | KETWWETWWTEWSQPKKKRKV | Amphipathic | 0 | +3 | α-helix | + | [43] |
| CADY | GLWRALWRLLRSLWRLLWRA | Amphipathic | 5 | +5 | α-helix | + | [38] |
| MAP | KLALKLALKALKAALKLA | Amphipathic | 0 | +5 | α-helix | + | [39] |
| SAP(E) | VELPPPVELPPPVELPPP | Amphipathic | 0 | -3 | α-helix | - | [133] |
| K-FGF | AAVLLPVLLAAP | Hydrophobic | 0 | 0 | random coil | + | [44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouyang, J.; Sheng, Y.; Wang, W. Recent Advances of Studies on Cell-Penetrating Peptides Based on Molecular Dynamics Simulations. Cells 2022, 11, 4016. https://doi.org/10.3390/cells11244016
Ouyang J, Sheng Y, Wang W. Recent Advances of Studies on Cell-Penetrating Peptides Based on Molecular Dynamics Simulations. Cells. 2022; 11(24):4016. https://doi.org/10.3390/cells11244016
Chicago/Turabian StyleOuyang, Jun, Yuebiao Sheng, and Wei Wang. 2022. "Recent Advances of Studies on Cell-Penetrating Peptides Based on Molecular Dynamics Simulations" Cells 11, no. 24: 4016. https://doi.org/10.3390/cells11244016
APA StyleOuyang, J., Sheng, Y., & Wang, W. (2022). Recent Advances of Studies on Cell-Penetrating Peptides Based on Molecular Dynamics Simulations. Cells, 11(24), 4016. https://doi.org/10.3390/cells11244016