
Innate Immune Memory in Monocytes and Macrophages: The Potential Therapeutic Strategies for Atherosclerosis

Abstract
:1. Introduction
2. The Mechanisms of Innate Immune Memory Formation
3. Innate Immune Memory Induced Cellular Metabolism Reprogramming Is Involved in Atherosclerosis
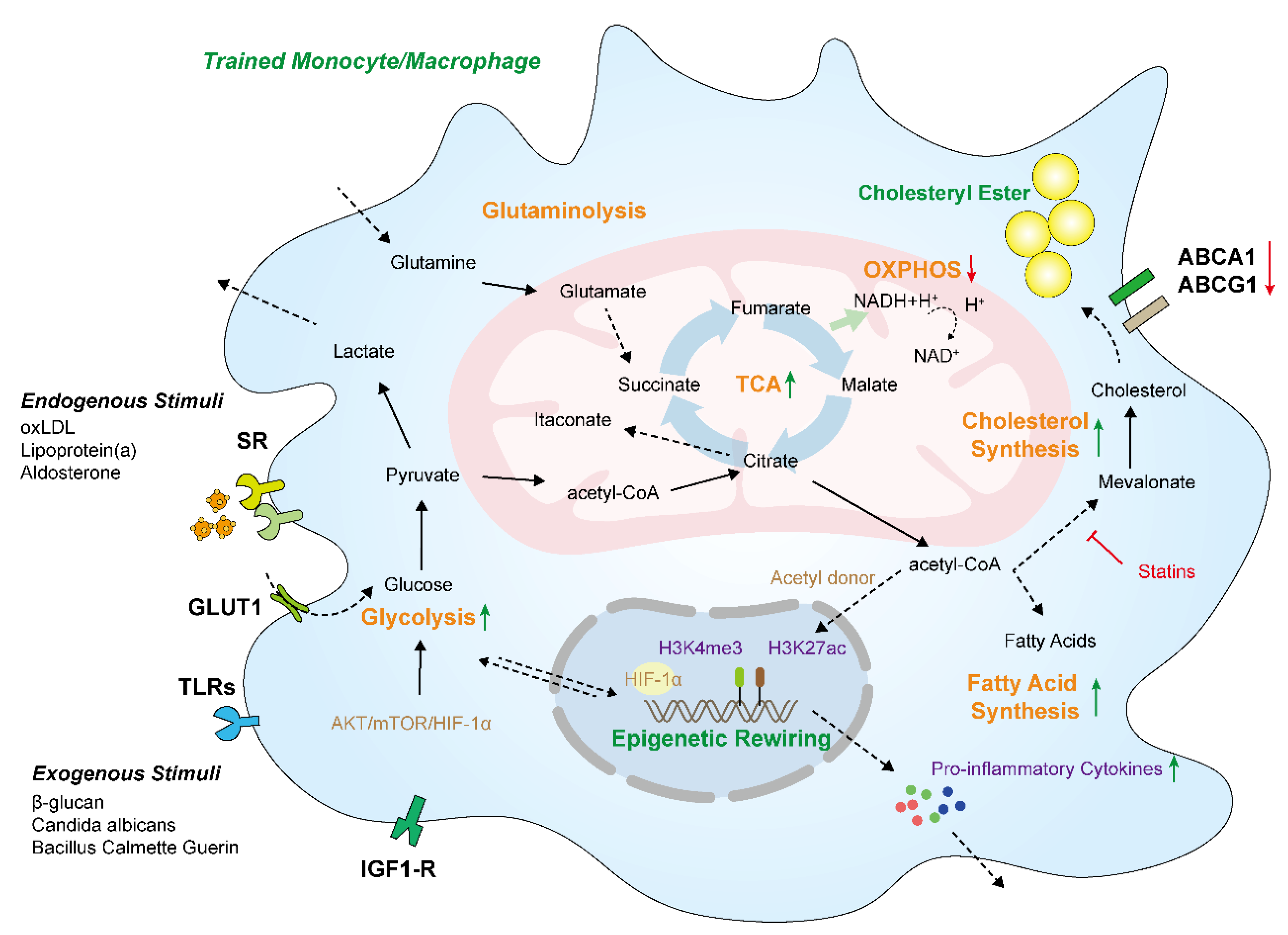
3.1. Glycolysis and Oxidative Phosphorylation
3.2. Tricarboxylic Acid Cycle
3.3. Lipid Metabolism
3.4. Amino Acid Metabolism
4. Innate Immune Memory Mediated Epigenetic Rewiring Is an Important Modulator in Atherosclerosis
4.1. DNA Methylation
4.2. Histone Modifications
4.2.1. Histone Methylation
4.2.2. Histone Acetylation

{kind=link}
{kind=link}
{kind=link}
| Modification | Function in Transcription | Writer | Eraser | Functions in Atherosclerosis |
|---|---|---|---|---|
| H3K4me1 | Positive | KMT2 | KDM1A | Promotes IL-1β expression [91] |
| H3K4me3 | Positive | MLL | KDM5B | Activates proinflammatory genes, matrix metalloproteinase, and ABC transporters [85,86,87,88,89] |
| H3K9me3 | Negative | KMT1 | KDM4A, KDM3 | Enriched at the promoter and suppresses TNFα and IL-6 [12,92] |
| H3K27me3 | Negative | KMT6 | JMJD3, KDM4A | Represses inflammatory genes (IL-6 and TNFα) and suppresses M1 polarization [12,93,94,95] |
| H3K9ac/H3K27ac | Positive | p300, HAT1 | HDAC7, HDAC2 | Enhances Nox5 in macrophages and activates M2 anti-inflammatory genes [109] |
5. Endogenous and Exogenous Atherogenic Stimulations Build Innate Immune Memory in Monocytes and Macrophages
5.1. oxLDL
5.2. Aldosterone
5.3. Cholesterol
5.4. β-Glucan
5.5. Mevalonate
5.6. Lipoprotein(a)
6. Conclusions and Perspective for Therapies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tabas, I.; Lichtman, A.H. Monocyte-macrophages and T cells in atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Wei, Y. A Novel Regulatory Player in the Innate Immune System: Long Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 9535. [Google Scholar] [CrossRef]
- McHeyzer-Williams, L.J.; McHeyzer-Williams, M.G. Antigen-specific memory B cell development. Annu. Rev. Immunol. 2005, 23, 487–513. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Quintin, J.; Van Der Meer, J.W. Trained immunity: A memory for innate host defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, f1098. [Google Scholar] [CrossRef] [Green Version]
- van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C. Oxidized phospholipids on lipoprotein (a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef] [Green Version]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef] [Green Version]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscl. Tthrom. Vas. 2014, 34, 1731–1738. [Google Scholar] [CrossRef]
- van der Heijden, C.D.; Keating, S.T.; Groh, L.; Joosten, L.A.; Netea, M.G.; Riksen, N.P. Aldosterone induces trained immunity: The role of fatty acid synthesis. Cardiovasc. Res. 2020, 116, 317–328. [Google Scholar] [CrossRef]
- Saeed, S.; Quintin, J.; Kerstens, H.H.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.; Ratter, J.; Berentsen, K.; van der Ent, M.A. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekkering, S.; van den Munckhof, I.; Nielen, T.; Lamfers, E.; Dinarello, C.; Rutten, J.; de Graaf, J.; Joosten, L.A.; Netea, M.G.; Gomes, M.E. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis 2016, 254, 228–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, S.T.; Groh, L.; Thiem, K.; Bekkering, S.; Li, Y.; Matzaraki, V.; van der Heijden, C.D.; van Puffelen, J.H.; Lachmandas, E.; Jansen, T. Rewiring of glucose metabolism defines trained immunity induced by oxidized low-density lipoprotein. J. Mol. Med. 2020, 98, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Andrés, J.; Joosten, L.A.; Netea, M.G. Induction of innate immune memory: The role of cellular metabolism. Curr. Opin. Immunol. 2019, 56, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Divangahi, M.; Aaby, P.; Khader, S.A.; Barreiro, L.B.; Bekkering, S.; Chavakis, T.; van Crevel, R.; Curtis, N.; DiNardo, A.R.; Dominguez-Andres, J. Trained immunity, tolerance, priming and differentiation: Distinct immunological processes. Nat. Immunol. 2021, 22, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Jin, F.; Li, J.; Guo, J.; Doeppner, T.R.; Hermann, D.M.; Yao, G.; Dai, Y. Targeting epigenetic modifiers to reprogramme macrophages in non-resolving inflammation-driven atherosclerosis. Eur. Heart J. Open 2021, 1, b22. [Google Scholar] [CrossRef]
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via arginase or nitric oxide synthase: Two competing arginine pathways in macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [Green Version]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [Green Version]
- De Bruin, R.G.; Shiue, L.; Prins, J.; De Boer, H.C.; Singh, A.; Fagg, W.S.; Van Gils, J.M.; Duijs, J.M.; Katzman, S.; Kraaijeveld, A.O. Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat. Commun. 2016, 7, 10846. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Schultze, J.L.; Schmieder, A.; Goerdt, S. Macrophage Activation in Human Diseases; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A. mTOR-and HIF-1α–mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Prados, J.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roosterman, D.; Cottrell, G.S. Rethinking the citric acid cycle: Connecting pyruvate carboxylase and citrate synthase to the flow of energy and material. Int. J. Mol. Sci. 2021, 22, 604. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Yanxiang Guo, J. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Appari, M.; Channon, K.M.; McNeill, E. Metabolic regulation of adipose tissue macrophage function in obesity and diabetes. Antioxid. Redox Sign. 2018, 29, 297–312. [Google Scholar] [CrossRef]
- Tawakol, A.; Singh, P.; Mojena, M.; Pimentel-Santillana, M.; Emami, H.; MacNabb, M.; Rudd, J.H.; Narula, J.; Enriquez, J.A.; Través, P.G. HIF-1α and PFKFB3 mediate a tight relationship between proinflammatory activation and anerobic metabolism in atherosclerotic macrophages. Arterioscl. Tthrom. Vas. 2015, 35, 1463–1471. [Google Scholar] [CrossRef] [Green Version]
- Arts, R.J.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.; Wang, S. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef]
- Arts, R.J.; Blok, B.A.; Aaby, P.; Joosten, L.A.; de Jong, D.; van der Meer, J.W.; Benn, C.S.; van Crevel, R.; Netea, M.G. Long-term in vitro and in vivo effects of γ-irradiated BCG on innate and adaptive immunity. J. Leukocyte Biol. 2015, 98, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, R.J.; Carvalho, A.; La Rocca, C.; Palma, C.; Rodrigues, F.; Silvestre, R.; Kleinnijenhuis, J.; Lachmandas, E.; Gonçalves, L.G.; Belinha, A. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. 2016, 17, 2562–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizawa, T.; Kanter, J.E.; Kramer, F.; Barnhart, S.; Shen, X.; Vivekanandan-Giri, A.; Wall, V.Z.; Kowitz, J.; Devaraj, S.; O Brien, K.D. Testing the role of myeloid cell glucose flux in inflammation and atherosclerosis. Cell Rep. 2014, 7, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palsson McDermott, E.M.; O’Neill, L.A. The Warburg effect then and now: From cancer to inflammatory diseases. Bioessays 2013, 35, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.V.; Domiguéz-Andrés, J.; Netea, M.G. The role of cell metabolism in innate immune memory. J. Innate Immun. 2022, 14, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, Y.; Lagache, S.M.; Schnack, L.; Godfrey, R.; Kahles, F.; Bruemmer, D.; Waltenberger, J.; Findeisen, H.M. mTOR-dependent oxidative stress regulates oxLDL-induced trained innate immunity in human monocytes. Front. Immunol. 2019, 9, 3155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groh, L.A.; Ferreira, A.V.; Helder, L.; van der Heijden, C.D.; Novakovic, B.; van de Westerlo, E.; Matzaraki, V.; Moorlag, S.J.; de Bree, L.C.; Koeken, V.A. oxLDL-induced trained immunity is dependent on mitochondrial metabolic reprogramming. Immunometabolism 2021, 3, e210025. [Google Scholar] [CrossRef]
- Fensterheim, B.A.; Young, J.D.; Luan, L.; Kleinbard, R.R.; Stothers, C.L.; Patil, N.K.; McAtee-Pereira, A.G.; Guo, Y.; Trenary, I.; Hernandez, A. The TLR4 agonist monophosphoryl lipid A drives broad resistance to infection via dynamic reprogramming of macrophage metabolism. J. Immunol. 2018, 200, 3777–3789. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; O’Neill, L.A. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol. 2016, 46, 13–21. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.; Kolbe, C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J. Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via ATP-citrate lyase. Immunity 2019, 51, 997–1011. [Google Scholar] [CrossRef]
- Sugimoto, M.; Sakagami, H.; Yokote, Y.; Onuma, H.; Kaneko, M.; Mori, M.; Sakaguchi, Y.; Soga, T.; Tomita, M. Non-targeted metabolite profiling in activated macrophage secretion. Metabolomics 2012, 8, 624–633. [Google Scholar] [CrossRef]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Wang, X.; Xie, Y.; Cai, X.; Yu, N.; Hu, Y.; Zheng, Z. 4-Octyl itaconate activates Nrf2 signaling to inhibit pro-inflammatory cytokine production in peripheral blood mononuclear cells of systemic lupus erythematosus patients. Cell. Physiol. Biochem. 2018, 51, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Andrés, J.; Novakovic, B.; Li, Y.; Scicluna, B.P.; Gresnigt, M.S.; Arts, R.J.; Oosting, M.; Moorlag, S.J.; Groh, L.A.; Zwaag, J. The itaconate pathway is a central regulatory node linking innate immune tolerance and trained immunity. Cell Metab. 2019, 29, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Ye, F.; Xue, W.; Zhou, Z.; Kang, Y.J. Copper regulation of hypoxia-inducible factor-1 activity. Mol. Pharmacol. 2009, 75, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Legrand-Poels, S.; Esser, N.; L Homme, L.; Scheen, A.; Paquot, N.; Piette, J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem. Pharmacol. 2014, 92, 131–141. [Google Scholar] [CrossRef]
- Freigang, S.; Ampenberger, F.; Weiss, A.; Kanneganti, T.; Iwakura, Y.; Hersberger, M.; Kopf, M. Fatty acid–induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat. Immunol. 2013, 14, 1045–1053. [Google Scholar] [CrossRef]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Bloc’h, J.L.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef]
- Lachmandas, E.; Boutens, L.; Ratter, J.M.; Hijmans, A.; Hooiveld, G.J.; Joosten, L.A.; Rodenburg, R.J.; Fransen, J.A.; Houtkooper, R.H.; Van Crevel, R. Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat. Microbiol. 2016, 2, 16246. [Google Scholar] [CrossRef]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 2018, 172, 147–161. [Google Scholar] [CrossRef]
- Riksen, N.P.; Netea, M.G. Immunometabolic control of trained immunity. Mol. Asp. Med. 2021, 77, 100897. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Arts, R.J.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.D.; Li, Y.; Popa, C.D.; Ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T. Metabolic induction of trained immunity through the mevalonate pathway. Cell 2018, 172, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohrabi, Y.; Sonntag, G.V.; Braun, L.C.; Lagache, S.M.; Liebmann, M.; Klotz, L.; Godfrey, R.; Kahles, F.; Waltenberger, J.; Findeisen, H.M. LXR activation induces a proinflammatory trained innate immunity-phenotype in human monocytes. Front. Immunol. 2020, 11, 353. [Google Scholar] [CrossRef]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Rom, O.; Grajeda-Iglesias, C.; Najjar, M.; Abu-Saleh, N.; Volkova, N.; Dar, D.E.; Hayek, T.; Aviram, M. Atherogenicity of amino acids in the lipid-laden macrophage model system in vitro and in atherosclerotic mice: A key role for triglyceride metabolism. J. Nutr. Biochem. 2017, 45, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Cao, X. Epigenetic remodeling in innate immunity and inflammation. Annu. Rev. Immunol. 2021, 39, 279–311. [Google Scholar] [CrossRef]
- Greenberg, M.V.; Bourc His, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Iguchi-Ariga, S.M.; Schaffner, W. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Gene. Dev. 1989, 3, 612–619. [Google Scholar] [CrossRef] [Green Version]
- Comb, M.; Goodman, H.M. CpG methylation inhibits proenkephalin gene expression and binding of the transcription factor AP-2. Nucleic Acids Res. 1990, 18, 3975–3982. [Google Scholar] [CrossRef]
- Falzon, M.; Kuff, E.L. Binding of the transcription factor EBP-80 mediates the methylation response of an intracisternal A-particle long terminal repeat promoter. Mol. Cell. Biol. 1991, 11, 117–125. [Google Scholar] [PubMed] [Green Version]
- Cobo, I.; Tanaka, T.N.; Mangalhara, K.C.; Lana, A.; Yeang, C.; Han, C.; Schlachetzki, J.; Challcombe, J.; Fixsen, B.R.; Sakai, M. DNA methyltransferase 3 alpha and TET methylcytosine dioxygenase 2 restrain mitochondrial DNA-mediated interferon signaling in macrophages. Immunity 2022, 55, 1386–1401. [Google Scholar] [CrossRef] [PubMed]
- Chandramouly, G. Gadd45 in DNA Demethylation and DNA Repair. In Gadd45 Stress Sensor Genes; Springer: Cham, Switzerland, 2022; pp. 55–67. [Google Scholar]
- Wu, S.C.; Zhang, Y. Active DNA demethylation: Many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2010, 11, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needham, B.L.; Smith, J.A.; Zhao, W.; Wang, X.; Mukherjee, B.; Kardia, S.L.; Shively, C.A.; Seeman, T.E.; Liu, Y.; Diez Roux, A.V. Life course socioeconomic status and DNA methylation in genes related to stress reactivity and inflammation: The multi-ethnic study of atherosclerosis. Epigenetics 2015, 10, 958–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballestar, E.; Sawalha, A.H.; Lu, Q. Clinical value of DNA methylation markers in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2020, 16, 514–524. [Google Scholar] [CrossRef]
- Papanicolau-Sengos, A.; Aldape, K. DNA methylation profiling: An emerging paradigm for cancer diagnosis. Annu. Rev. Pathol-Mech. 2022, 17, 295–321. [Google Scholar] [CrossRef]
- Chi, G.C.; Liu, Y.; MacDonald, J.W.; Reynolds, L.M.; Enquobahrie, D.A.; Fitzpatrick, A.L.; Kerr, K.F.; J. Budoff, M.; Lee, S.; Siscovick, D. Epigenome-wide analysis of long-term air pollution exposure and DNA methylation in monocytes: Results from the Multi-Ethnic Study of Atherosclerosis. Epigenetics 2022, 17, 297–313. [Google Scholar] [CrossRef]
- Jiang, D.; Sun, M.; You, L.; Lu, K.; Gao, L.; Hu, C.; Wu, S.; Chang, G.; Tao, H.; Zhang, D. DNA methylation and hydroxymethylation are associated with the degree of coronary atherosclerosis in elderly patients with coronary heart disease. Life Sci. 2019, 224, 241–248. [Google Scholar] [CrossRef]
- Chawla, A. Control of macrophage activation and function by PPARs. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Yu, J.; Qiu, Y.; Yang, J.; Bian, S.; Chen, G.; Deng, M.; Kang, H.; Huang, L. DNMT1-PPARγ pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice. Sci. Rep. 2016, 6, 30053. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; Rossi, A.E.; Alvarez, M.G.; Swanson, K.E.; Deveaux, H.K.; Earley, J.U.; Hadhazy, M.; Vohra, R.; Walter, G.A.; Pytel, P. Dysferlin and myoferlin regulate transverse tubule formation and glycerol sensitivity. Am. J. Pathol. 2014, 184, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Morreé, A.; Flix, B.; Bagaric, I.; Wang, J.; Van Den Boogaard, M.; Moursel, L.G.; Frants, R.R.; Illa, I.; Gallardo, E.; Toes, R. Dysferlin regulates cell adhesion in human monocytes. J. Biol. Chem. 2013, 288, 14147–14157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; He, D.; Xiang, Y.; Wang, C.; Liang, B.; Li, B.; Qi, D.; Deng, Q.; Yu, H.; Lu, Z. DYSF promotes monocyte activation in atherosclerotic cardiovascular disease as a DNA methylation-driven gene. Transl. Res. 2022, 247, 19–38. [Google Scholar] [CrossRef]
- Stillman, B. Histone modifications: Insights into their influence on gene expression. Cell 2018, 175, 6–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Hughes, A.L.; Kelley, J.R.; Klose, R.J. Understanding the interplay between CpG island-associated gene promoters and H3K4 methylation. BBA-Gene Regul. Mech. 2020, 1863, 194567. [Google Scholar] [CrossRef]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.; Peng, W.; Zhang, M.Q. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef]
- Guo, C.; Dong, G.; Liang, X.; Dong, Z. Epigenetic regulation in AKI and kidney repair: Mechanisms and therapeutic implications. Nat. Rev. Nephrol. 2019, 15, 220–239. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, K.; Li, T.; Run, C.; Chen, Y. The engagement of histone lysine methyltransferases with nucleosomes: Structural basis, regulatory mechanisms, and therapeutic implications. In Protein Cell; Oxford University Press: New York, NY, USA, 2022. [Google Scholar]
- Kittan, N.A.; Allen, R.M.; Dhaliwal, A.; Cavassani, K.A.; Schaller, M.; Gallagher, K.A.; Carson IV, W.F.; Mukherjee, S.; Grembecka, J.; Cierpicki, T. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS ONE 2013, 8, e78045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Fang, F.; Dai, X.; Xu, H.; Qi, X.; Fang, M.; Xu, Y. MKL1 defines the H3K4Me3 landscape for NF-κB dependent inflammatory response. Sci. Rep. 2017, 7, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leopold Wager, C.M.; Hole, C.R.; Campuzano, A.; Castro-Lopez, N.; Cai, H.; Caballero Van Dyke, M.C.; Wozniak, K.L.; Wang, Y.; Wormley Jr, F.L. IFN-γ immune priming of macrophages in vivo induces prolonged STAT1 binding and protection against Cryptococcus neoformans. PLoS Pathog. 2018, 14, e1007358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanucchi, S.; Domínguez-Andrés, J.; Joosten, L.A.; Netea, M.G.; Mhlanga, M.M. The intersection of epigenetics and metabolism in trained immunity. Immunity 2021, 54, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Neele, A.E.; Hoeksema, M.A.; De Heij, F.; Boshuizen, M.C.; Van Der Velden, S.; De Boer, V.C.; Reedquist, K.A.; De Winther, M.P. Inhibiting epigenetic enzymes to improve atherogenic macrophage functions. Biochem. Biophys. Res. Commun. 2014, 455, 396–402. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, B.; Liang, P.; Tong, Z.; Liu, M.; Lv, Q.; Liu, Y.; Liu, X.; Tang, Y.; Xiao, X. Nucleolin protects macrophages from oxLDL-induced foam cell formation through up-regulating ABCA1 expression. Biochem. Biophys. Res. Commun. 2017, 486, 364–371. [Google Scholar] [CrossRef]
- Xie, L.; Ding, N.; Zhang, H.; Liu, K.; Xiong, J.; Ma, S.; Yang, A.; Zhang, H.; Jiang, Y. SNF5 promotes IL-1β expression via H3K4me1 in atherosclerosis induced by homocysteine. Int. J. Biochem. Cell Biol. 2021, 135, 105974. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Stiekema, L.C.; Moens, S.B.; Verweij, S.L.; Novakovic, B.; Prange, K.; Versloot, M.; van Lennep, J.E.R.; Stunnenberg, H.; de Winther, M. Treatment with statins does not revert trained immunity in patients with familial hypercholesterolemia. Cell Metab. 2019, 30, 1–2. [Google Scholar] [CrossRef]
- Davis, F.M.; Tsoi, L.C.; Melvin, W.J.; DenDekker, A.; Wasikowski, R.; Joshi, A.D.; Wolf, S.; Obi, A.T.; Billi, A.C.; Xing, X. Inhibition of macrophage histone demethylase JMJD3 protects against abdominal aortic aneurysms. J. Exp. Med. 2021, 218, e20201839. [Google Scholar] [CrossRef]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, F.M.; Tsoi, L.; Joshi, A.D.A.; Audu, C.; Melvin, W.J.; Kunkel, S.; Eliason, J.; Gudojonsson, J.; Gallagher, K. JMJD3 Influences Macrophage-Mediated Inflammation in Abdominal Aortic Aneurysms. J. Vasc. Surg. 2020, 72, e296. [Google Scholar] [CrossRef]
- Yan, Q.; Sun, L.; Zhu, Z.; Wang, L.; Li, S.; Richard, D.Y. Jmjd3-mediated epigenetic regulation of inflammatory cytokine gene expression in serum amyloid A-stimulated macrophages. Cell. Signal. 2014, 26, 1783–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; Takeuchi, O.; Vandenbon, A.; Yasuda, K.; Tanaka, Y.; Kumagai, Y.; Miyake, T.; Matsushita, K.; Okazaki, T.; Saitoh, T. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010, 11, 936–944. [Google Scholar] [CrossRef]
- Agger, K.; Nishimura, K.; Miyagi, S.; Messling, J.; Rasmussen, K.D.; Helin, K. The KDM4/JMJD2 histone demethylases are required for hematopoietic stem cell maintenance. Blood 2019, 134, 1154–1158. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, S.; Yao, G.; Yu, D.; Chen, K.; Tong, Q.; Ye, L.; Wu, C.; Sun, Y.; Li, H. Identification of the histone lysine demethylase KDM4A/JMJD2A as a novel epigenetic target in M1 macrophage polarization induced by oxidized LDL. Oncotarget 2017, 8, 114442. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Oh, S.; Yoo, H.; Kwon, Y. The key role of DNA methylation and histone acetylation in epigenetics of atherosclerosis. J. Lipid Atheroscler. 2020, 9, 419. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, J.; Liu, Z.; Jiang, A.; Li, S.; Wu, D.; Zhang, Y.; Zhu, X.; Zhou, E.; Wei, Z. Sodium butyrate alleviates lipopolysaccharide-induced inflammatory responses by down-regulation of NF-κB, NLRP3 signaling pathway, and activating histone acetylation in bovine macrophages. Front. Vet. Sci. 2020, 7, 579674. [Google Scholar] [CrossRef]
- Hu, L.; Yu, Y.; Huang, H.; Fan, H.; Hu, L.; Yin, C.; Li, K.; Fulton, D.J.; Chen, F. Epigenetic regulation of interleukin 6 by histone acetylation in macrophages and its role in paraquat-induced pulmonary fibrosis. Front. Immunol. 2017, 7, 696. [Google Scholar] [CrossRef] [Green Version]
- Igolkina, A.A.; Zinkevich, A.; Karandasheva, K.O.; Popov, A.A.; Selifanova, M.V.; Nikolaeva, D.; Tkachev, V.; Penzar, D.; Nikitin, D.M.; Buzdin, A. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 histone tags suggest distinct regulatory evolution of open and condensed chromatin landmarks. Cells 2019, 8, 1034. [Google Scholar] [CrossRef]
- Kim, M.; Yun, J. Molecular mechanism of the protective effect of zerumbone on lipopolysaccharide-induced inflammation of THP-1 cell-derived macrophages. J. Med. Food 2019, 22, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Vlad, M.; Manea, S.; Lazar, A.; Raicu, M.; Muresian, H.; Simionescu, M.; Manea, A. Histone acetyltransferase-dependent pathways mediate upregulation of NADPH oxidase 5 in human macrophages under inflammatory conditions: A potential mechanism of reactive oxygen species overproduction in atherosclerosis. Oxid. Med. Cell. Longev. 2019, 2019, 3201062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, K.D.; Shakespear, M.R.; Curson, J.E.; Murthy, A.M.; Iyer, A.; Hodson, M.P.; Ramnath, D.; Tillu, V.A.; von Pein, J.B.; Reid, R.C. Class IIa histone deacetylases drive toll-like receptor-inducible glycolysis and macrophage inflammatory responses via pyruvate kinase M2. Cell Rep. 2020, 30, 2712–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Yu, X.; Chen, J.; Hu, M.; Zhang, Y.; Lin, H.; Tang, W.; He, P.; Ouyang, X. Histone Deacetylase 3: A Potential Therapeutic Target for Atherosclerosis. Aging Dis. 2022, 13, 773–786. [Google Scholar] [CrossRef]
- Bai, L.; Li, Z.; Zhao, S.; Zhang, J.; Fan, J.; Shyy, J.Y.; Liu, E. Mediator 1 Deficiency in Macrophage Increases Atherosclerosis via Modulation of Macrophage Polarization. Circulation 2017, 136, A18022. [Google Scholar]
- Groh, L.A.; Verel, D.E.; van der Heijden, C.D.; Matzaraki, V.; Moorlag, S.J.; de Bree, L.C.; Koeken, V.A.; Mourits, V.P.; Keating, S.T.; van Puffelen, J.H. Immune modulatory effects of progesterone on oxLDL-induced trained immunity in monocytes. J. Leukocyte Biol. 2022, 112, 279–288. [Google Scholar] [CrossRef]
- Christ, A.; Günther, P.; Lauterbach, M.A.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 2018, 172, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Quintin, J.; Saeed, S.; Martens, J.H.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.; Wijmenga, C. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Thanassoulis, G. Lipoprotein (a) in calcific aortic valve disease: From genomics to novel drug target for aortic stenosis. J. Lipid Res. 2016, 57, 917–924. [Google Scholar] [CrossRef] [Green Version]
- Stiekema, L.C.; Prange, K.H.; Hoogeveen, R.M.; Verweij, S.L.; Kroon, J.; Schnitzler, J.G.; Dzobo, K.E.; Cupido, A.J.; Tsimikas, S.; Stroes, E.S. Potent lipoprotein (a) lowering following apolipoprotein (a) antisense treatment reduces the pro-inflammatory activation of circulating monocytes in patients with elevated lipoprotein (a). Eur. Heart J. 2020, 41, 2262–2271. [Google Scholar] [CrossRef] [PubMed]
- McGraw, A.P.; Bagley, J.; Chen, W.S.; Galayda, C.; Nickerson, H.; Armani, A.; Caprio, M.; Carmeliet, P.; Jaffe, I.Z. Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism. J. Am. Heart Assoc. 2013, 2, e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Heijden, C.D.; Deinum, J.; Joosten, L.A.; Netea, M.G.; Riksen, N.P. The mineralocorticoid receptor as a modulator of innate immunity and atherosclerosis. Cardiovasc. Res. 2018, 114, 944–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.; Sano, S.; Muralidharan, S.; Rius, C. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Dricu, A.; Wang, M.; Hjertman, M.; Malec, M.; Blegen, H.; Wejde, J.; Carlberg, M.; Larsson, O. Mevalonate-regulated mechanisms in cell growth control: Role of dolichyl phosphate in expression of the insulin-like growth factor-1 receptor (IGF-1R) in comparison to Ras prenylation and expression of c-myc. Glycobiology 1997, 7, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Kronenberg, F.; Utermann, G. Lipoprotein (a): Resurrected by genetics. J. Intern. Med. 2013, 273, 6–30. [Google Scholar] [CrossRef]
- Kumari, A.; Kristensen, K.K.; Ploug, M.; Winther, A.L. The importance of lipoprotein lipase regulation in atherosclerosis. Biomedicines 2021, 9, 782. [Google Scholar] [CrossRef]
- Zubair, K.; You, C.; Kwon, G.; Kang, K. Two faces of macrophages: Training and tolerance. Biomedicines 2021, 9, 1596. [Google Scholar] [CrossRef]
- Novakovic, B.; Habibi, E.; Wang, S.; Arts, R.J.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J. β-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell 2016, 167, 1354–1368. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Weis, S.; Netea, M.G.; Wetzker, R. Remembering pathogen dose: Long-term adaptation in innate immunity. Trends Immunol. 2018, 39, 438–445. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Gao, Y.; Shou, S.; Chai, Y. The roles of macrophage polarization in the host immune response to sepsis. Int. Immunopharmacol. 2021, 96, 107791. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.; Najafi, M. Modulation of the tumor microenvironment (TME) by melatonin. Eur. J. Pharmacol. 2021, 907, 174365. [Google Scholar] [CrossRef] [PubMed]


| Atherosclerosis-Associated Stimuli | Metabolic Reprograming | Epigenetic Reprograming | Signaling Pathways |
|---|---|---|---|
| oxLDL [9,13,110] | Glycolysis, OXPHOS, TCA, cholesterol metabolism, | H3K4me3 | TLRs-mediated inflammatory response, mTOR/HIF-1α axis |
| Aldosterone [10] | Fatty acid biosynthesis | H3K4me3 | TLR2-mediated IL-6 and TNFα pathway, ROS production, fatty acid synthesis pathway |
| Cholesterol [92,111] | OXPHOS, glycolysis, amino acid synthesis | H3K4me3, H3K9me3 | IFN signaling pathway, IL-1 and NLRP3 pathway |
| β-glucan [31,112] | Cholesterol synthesis, glycolysis, glutaminolysis | H3K4me3, H3K27me3, H3K27Ac | Dectin-1/Raf-1 pathway, AKT/mTOR/HIF-1α pathway, TLR2-mediated IL-6 and TNFα pathway |
| Mevalonate [53] | Cholesterol synthesis, glycolysis, TCA | H3K4me3, H3K27Ac | IGF1-R-mTOR signaling pathway |
| Lipoprotein(a) [25,113,114] | - | - | TLR-mediated inflammatory response |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Z.; Wang, L.; Liu, H.; Xie, Y. Innate Immune Memory in Monocytes and Macrophages: The Potential Therapeutic Strategies for Atherosclerosis. Cells 2022, 11, 4072. https://doi.org/10.3390/cells11244072
Guo Z, Wang L, Liu H, Xie Y. Innate Immune Memory in Monocytes and Macrophages: The Potential Therapeutic Strategies for Atherosclerosis. Cells. 2022; 11(24):4072. https://doi.org/10.3390/cells11244072
Chicago/Turabian StyleGuo, Zhigang, Lixue Wang, Hongjian Liu, and Yuhuai Xie. 2022. "Innate Immune Memory in Monocytes and Macrophages: The Potential Therapeutic Strategies for Atherosclerosis" Cells 11, no. 24: 4072. https://doi.org/10.3390/cells11244072