

Pharmacological Targeting of the RAGE-NFκB Signalling Axis Impedes Monocyte Activation under Diabetic Conditions through the Repression of SHP-2 Tyrosine Phosphatase Function
 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Reagents
2.1.2. Clinical Cohorts and Ethical Approval
2.2. Methods
2.2.1. Isolation of Primary Human and Murine Monocytes
2.2.2. Animals
2.2.3. Cell Culture and Treatment
2.2.4. Monocyte Chemokinesis Analysis
2.2.5. Immunoprecipitation and Phosphatase Activity Measurement
2.2.6. Western Blotting
2.2.7. ELISA-based Cytokine Measurement
2.2.8. FACS Staining
2.2.9. Electrophoretic Mobility Shift Assays (EMSA)
2.2.10. Gene Silencing by siRNA
2.2.11. RNA Isolation, qPCR and Gene Expression Analysis
2.2.12. RT-PCR Analysis of RAGE mRNA Splice Variants
2.2.13. Study Design, Blinding and Statistical Analysis
2.2.14. Data Availability
3. Results
3.1. T2DM Milieu and AGE Precursor, Methylglyoxal, Induces Upregulation of SHP-2 Expression and Promotes Random Migration of Monocytes
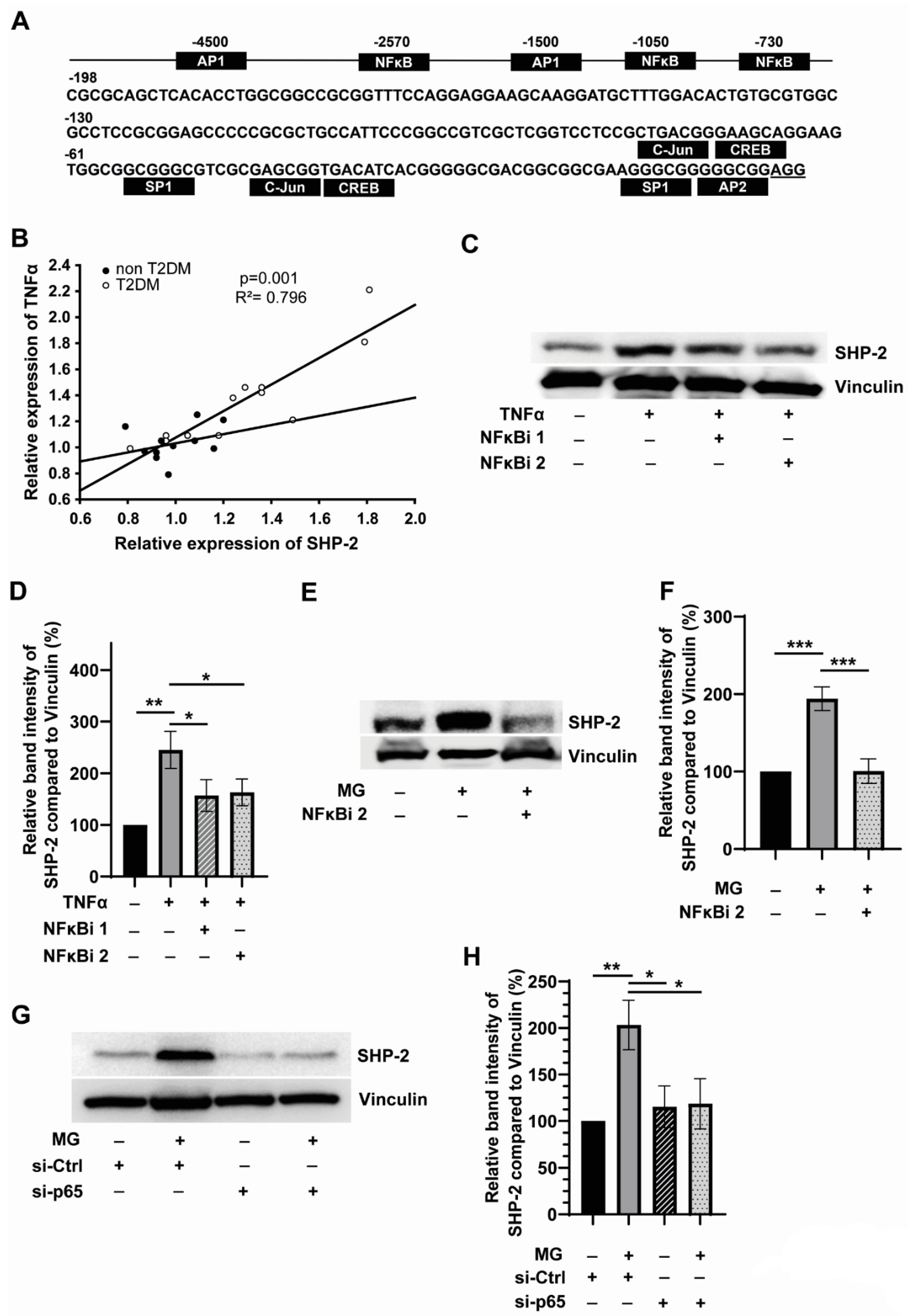
3.2. SHP-2 Expression Is Linked to Inflammation and Is Regulated by NFκB in Monocytes
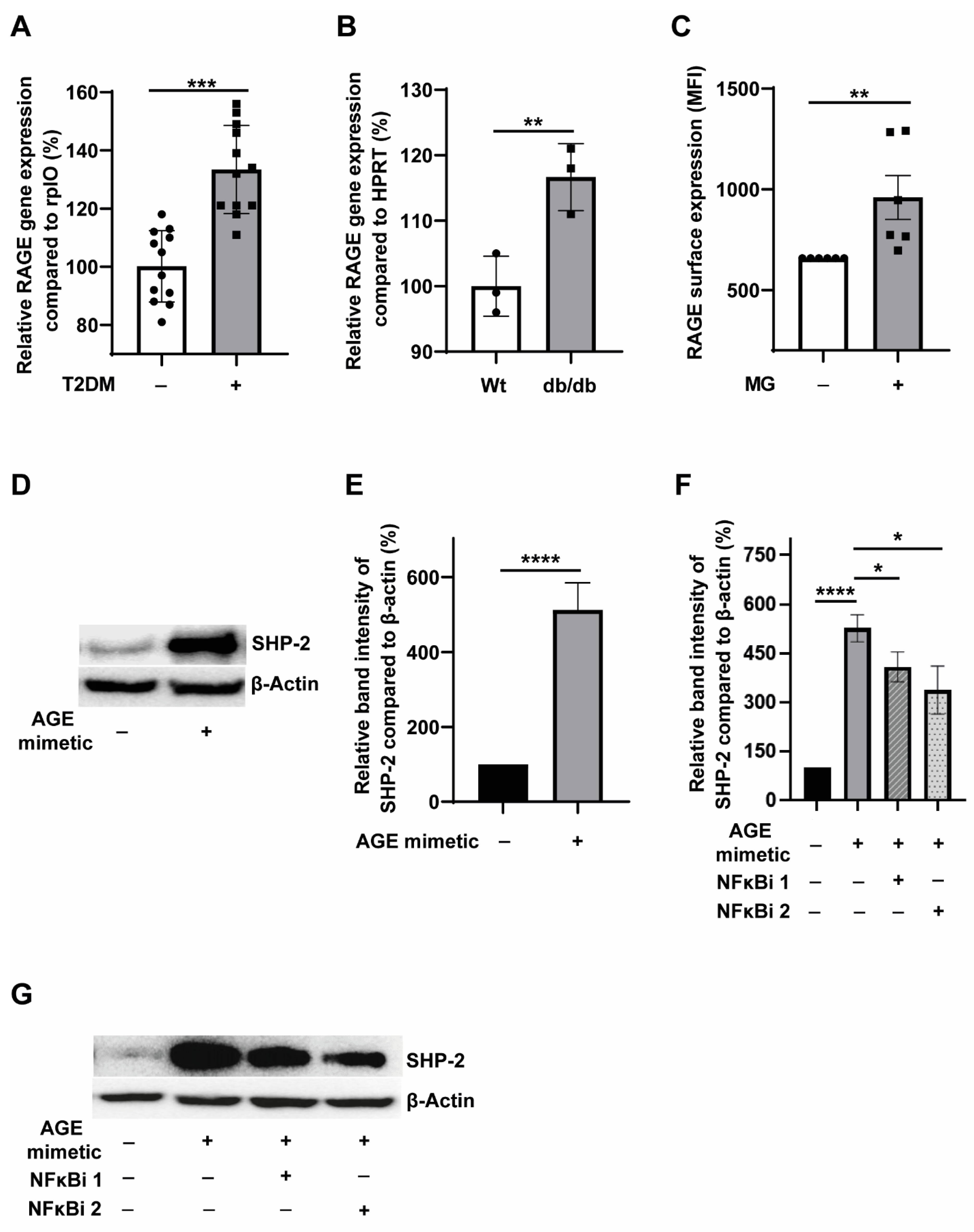
3.3. T2DM Milieu Induces RAGE Expression in Monocytes, and AGE-RAGE Signalling Potentiates SHP-2 Expression through the NFκB Pathway
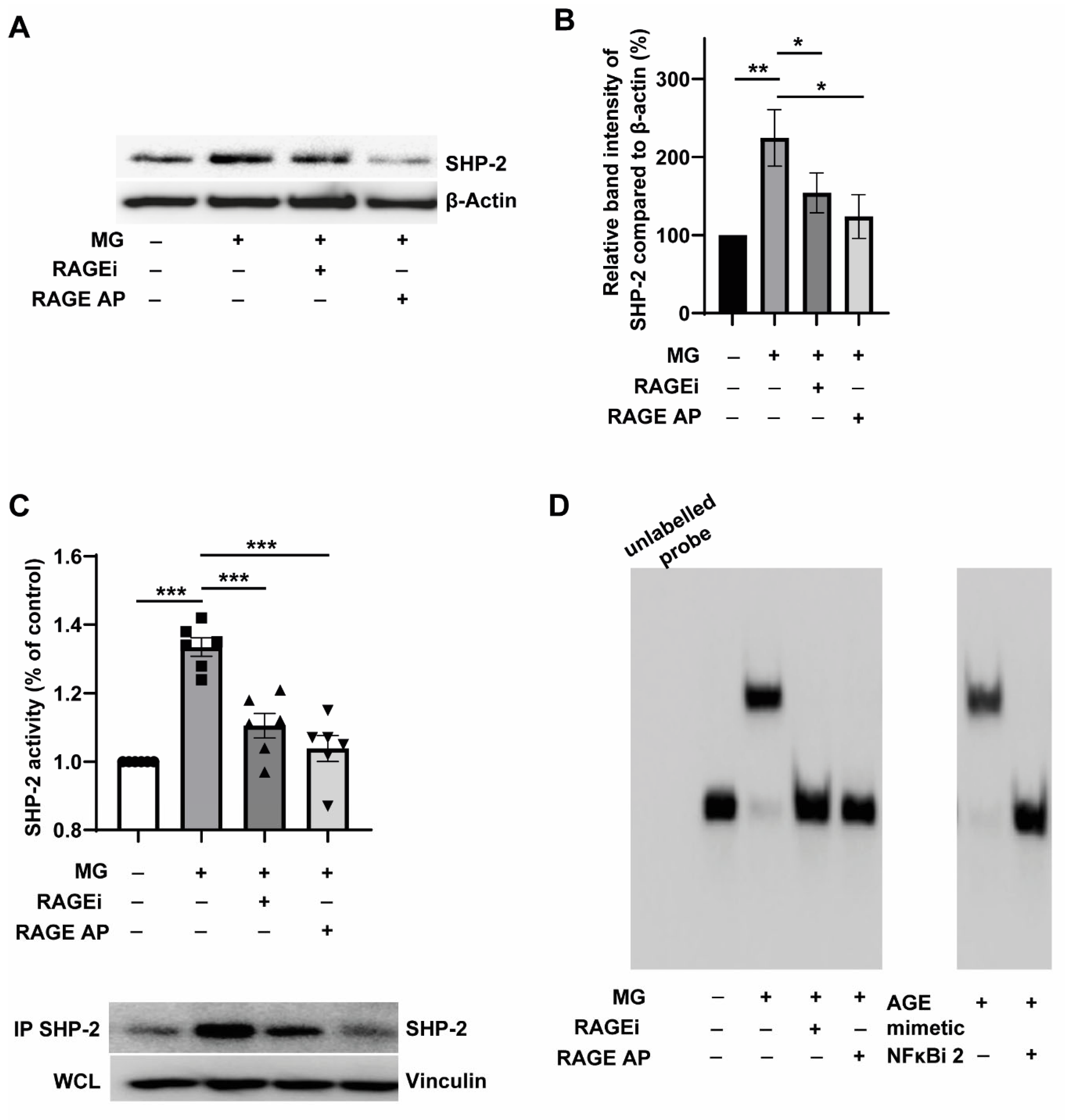
3.4. Pharmacological Targeting of RAGE Reverses MG-Induced SHP-2 Expression and Activity through the Inhibition of NFκB-p65 Binding to the SHP-2 Promoter
3.5. RAGE Ligation Promotes, and RAGE Inhibition Attenuates the Random Motility of Monocytes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flynn, M.C.; Pernes, G.; Lee, M.K.S.; Nagareddy, P.R.; Murphy, A.J. Monocytes, Macrophages, and Metabolic Disease in Atherosclerosis. Front. Pharmacol. 2019, 10, 666. [Google Scholar] [CrossRef] [PubMed]
- Flint, A.J.; Rexrode, K.M.; Hu, F.B.; Glynn, R.J.; Caspard, H.; Manson, J.E.; Willett, W.C.; Rimm, E.B. Body mass index, waist circumference, and risk of coronary heart disease: A prospective study among men and women. Obes. Res. Clin. Pract. 2010, 4, e171–e181. [Google Scholar] [CrossRef] [PubMed]
- Zalai, C.V.; Kolodziejczyk, M.D.; Pilarski, L.; Christov, A.; Nation, P.N.; Lundstrom-Hobman, M.; Tymchak, W.; Dzavik, V.; Humen, D.P.; Kostuk, W.J.; et al. Increased circulating monocyte activation in patients with unstable coronary syndromes. J. Am. Coll. Cardiol. 2001, 38, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Pande, R.L.; Brown, J.; Buck, S.; Redline, W.; Doyle, J.; Plutzky, J.; Creager, M.A. Association of monocyte tumor necrosis factor alpha expression and serum inflammatory biomarkers with walking impairment in peripheral artery disease. J. Vasc. Surg. 2015, 61, 155–161. [Google Scholar] [CrossRef]
- Carr, S.C.; Farb, A.; Pearce, W.H.; Virmani, R.; Yao, J.S. Activated inflammatory cells are associated with plaque rupture in carotid artery stenosis. Surgery 1997, 122, 757–763. [Google Scholar] [CrossRef]
- Jin, S.Y.; Kim, E.K.; Ha, J.M.; Lee, D.H.; Kim, J.S.; Kim, I.Y.; Song, S.H.; Shin, H.K.; Kim, C.; Bae, S.S. Insulin regulates monocyte trans-endothelial migration through surface expression of macrophage-1 antigen. Biochim. Biophys. Acta 2014, 1842, 1539–1548. [Google Scholar] [CrossRef]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Tjaden, K.; Adam, C.; Godfrey, R.; Hanley, P.J.; Pardali, E.; Waltenberger, J. Low density lipoprotein interferes with intracellular signaling of monocytes resulting in impaired chemotaxis and enhanced chemokinesis. Int. J. Cardiol. 2018, 255, 160–165. [Google Scholar] [CrossRef]
- Dorenkamp, M.; Muller, J.P.; Shanmuganathan, K.S.; Schulten, H.; Muller, N.; Loffler, I.; Müller, U.A.; Wolf, G.; Böhmer, F.-D.; Godfrey, R.; et al. Hyperglycaemia-induced methylglyoxal accumulation potentiates VEGF resistance of diabetic monocytes through the aberrant activation of tyrosine phosphatase SHP-2/SRC kinase signalling axis. Sci. Rep. 2018, 8, 14684. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef] [Green Version]
- Berlanga, J.; Cibrian, D.; Guillen, I.; Freyre, F.; Alba, J.S.; Lopez-Saura, P.; Merino, N.; Aldama, A.; Quintela, A.M.; Triana, M.E.; et al. Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of cutaneous wounds. Clin. Sci. 2005, 109, 83–95. [Google Scholar] [CrossRef]
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; Van Obberghen, E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 2006, 55, 1289–1299. [Google Scholar] [CrossRef]
- Moraru, A.; Wiederstein, J.; Pfaff, D.; Fleming, T.; Miller, A.K.; Nawroth, P.; Teleman, A.A. Elevated Levels of the Reactive Metabolite Methylglyoxal Recapitulate Progression of Type 2 Diabetes. Cell Metab. 2018, 27, 926–934.e8. [Google Scholar] [CrossRef]
- Hemling, P.; Zibrova, D.; Strutz, J.; Sohrabi, Y.; Desoye, G.; Schulten, H.; Findeisen, H.; Heller, R.; Godfrey, R.; Waltenberger, J. Hyperglycemia-induced endothelial dysfunction is alleviated by thioredoxin mimetic peptides through the restoration of VEGFR-2-induced responses and improved cell survival. Int. J. Cardiol. 2020, 308, 73–81. [Google Scholar] [CrossRef]
- Wu, L.; Juurlink, B.H. Increased methylglyoxal and oxidative stress in hypertensive rat vascular smooth muscle cells. Hypertension 2002, 39, 809–814. [Google Scholar] [CrossRef]
- Bhattacharyya, N.; Pal, A.; Patra, S.; Haldar, A.K.; Roy, S.; Ray, M. Activation of macrophages and lymphocytes by methylglyoxal against tumor cells in the host. Int. Immunopharmacol. 2008, 8, 1503–1512. [Google Scholar] [CrossRef]
- Qu, C.K. The SHP-2 tyrosine phosphatase: Signaling mechanisms and biological functions. Cell Res. 2000, 10, 279–288. [Google Scholar] [CrossRef]
- Tajan, M.; de Rocca Serra, A.; Valet, P.; Edouard, T.; Yart, A. SHP2 sails from physiology to pathology. Eur. J. Med. Genet. 2015, 58, 509–525. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Niu, R. Functions of Shp2 in cancer. J. Cell. Mol. Med. 2015, 19, 2075–2083. [Google Scholar] [CrossRef]
- Chen, J.; Cao, Z.; Guan, J. SHP2 inhibitor PHPS1 protects against atherosclerosis by inhibiting smooth muscle cell proliferation. BMC Cardiovasc. Disord. 2018, 18, 72. [Google Scholar] [CrossRef]
- Paccoud, R.; Saint-Laurent, C.; Piccolo, E.; Tajan, M.; Dortignac, A.; Pereira, O.; Le Gonidec, S.; Baba, I.; Gélineau, A.; Askia, H.; et al. SHP2 drives inflammation-triggered insulin resistance by reshaping tissue macrophage populations. Sci. Transl. Med. 2021, 13, 591. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, X.; Wang, Y.; Yang, Y.; Sun, D.; Li, H.; Chen, L. Targeting SHP2 as a therapeutic strategy for inflammatory diseases. Eur. J. Med. Chem. 2021, 214, 113264. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, R.; Arora, D.; Bauer, R.; Stopp, S.; Muller, J.P.; Heinrich, T.; Böhmer, S.-A.; Dagnell, M.; Schnetzke, U.; Scholl, S.; et al. Cell transformation by FLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumor suppressor protein-tyrosine phosphatase DEP-1/PTPRJ. Blood 2012, 119, 4499–4511. [Google Scholar] [CrossRef]
- Lieuw-a-Fa, M.L.; Schalkwijk, C.G.; Engelse, M.; van Hinsbergh, V.W. Interaction of Nepsilon(carboxymethyl)lysine- and methylglyoxal-modified albumin with endothelial cells and macrophages. Splice variants of RAGE may limit the responsiveness of human endothelial cells to AGEs. Thromb. Haemost. 2006, 95, 320–328. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.; Cuny, J.; Nawroth, G.; Djuric, Z.; Humpert, P.M.; Zeier, M.; Bierhaus, A.; Nawroth, P.P. Is diabetes an acquired disorder of reactive glucose metabolites and their intermediates? Diabetologia 2012, 55, 1151–1155. [Google Scholar] [CrossRef]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- Chen, L.; Sung, S.S.; Yip, M.L.; Lawrence, H.R.; Ren, Y.; Guida, W.C.; Sebti, S.M.; Lawrence, N.J.; Wu, J. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol. Pharmacol. 2006, 70, 562–570. [Google Scholar] [CrossRef]
- Paneni, F.; Mocharla, P.; Akhmedov, A.; Costantino, S.; Osto, E.; Volpe, M.; Lüscher, T.F.; Cosentino, F. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ. Res. 2012, 111, 278–289. [Google Scholar] [CrossRef]
- Moller, D.E. Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol. Metab. 2000, 11, 212–217. [Google Scholar] [CrossRef]
- Ramasamy, R.; Vannucci, S.J.; Yan, S.S.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef]
- Soro-Paavonen, A.; Watson, A.M.; Li, J.; Paavonen, K.; Koitka, A.; Calkin, A.C.; Barit, D.; Coughlan, M.T.; Drew, B.G.; Lancaster, G.I.; et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 2008, 57, 2461–2469. [Google Scholar] [CrossRef]
- Burke, A.P.; Kolodgie, F.D.; Zieske, A.; Fowler, D.R.; Weber, D.K.; Varghese, P.J.; Farb, A.; Virmani, R. Morphologic findings of coronary atherosclerotic plaques in diabetics: A postmortem study. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1266–1271. [Google Scholar] [CrossRef]
- Ostendorp, T.; Leclerc, E.; Galichet, A.; Koch, M.; Demling, N.; Weigle, B.; Heizmann, C.W.; Kroneck, P.M.H.; Fritz, G. Structural and functional insights into RAGE activation by multimeric S100B. EMBO J. 2007, 26, 3868–3878. [Google Scholar] [CrossRef]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef]
- Okamoto, T.; Yamagishi, S.; Inagaki, Y.; Amano, S.; Koga, K.; Abe, R.; Takeuchi, M.; Ohno, S.; Yoshimura, A.; Makita, Z. Angiogenesis induced by advanced glycation end products and its prevention by cerivastatin. FASEB J. 2002, 16, 1928–1930. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.; et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Gomez, S.B.; Schmidt, A.M.; Logsdon, C.D. S100P-derived RAGE antagonistic peptide reduces tumor growth and metastasis. Clin. Cancer Res. 2012, 18, 4356–4364. [Google Scholar] [CrossRef]
- Williams, J.W.; Zaitsev, K.; Kim, K.W.; Ivanov, S.; Saunders, B.T.; Schrank, P.R.; Kim, K.; Elvington, A.; Kim, S.H.; Tucker, C.G. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 2020, 21, 1194–1204. [Google Scholar] [CrossRef]
- Gu, L.; Okada, Y.; Clinton, S.K.; Gerard, C.; Sukhova, G.K.; Libby, P.; Rollins, B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell. 1998, 2, 275–281. [Google Scholar] [CrossRef]
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Veillard, N.R.; Kwak, B.; Pelli, G.; Mulhaupt, F.; James, R.W.; Proudfoot, A.E.; Mach, F. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ. Res. 2004, 94, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.H.; Sukhova, G.K.; Yang, J.T.; Wang, B.; Xie, T.; Fu, H.; Zhang, Y.; Satoskar, A.R.; David, J.R.; Metz, C.N.; et al. Macrophage migration inhibitory factor deficiency impairs atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2004, 109, 3149–3153. [Google Scholar] [CrossRef] [PubMed]
- Kislinger, T.; Tanji, N.; Wendt, T.; Qu, W.; Lu, Y.; Ferran, L.J., Jr.; Taguchi, A.; Olson, K.; Bucciarelli, L.; Goova, M.; et al. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Raman, K.G.; Lee, K.J.; Lu, Y.; Ferran, L.J., Jr.; Chow, W.S.; Stern, D.; Schmidt, A.M. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998, 4, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Bucciarelli, L.G.; Wendt, T.; Qu, W.; Lu, Y.; Lalla, E.; Rong, L.L.; Goova, M.T.; Moser, B.; Kislinger, T.; Lee, D.C.; et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 2002, 106, 2827–2835. [Google Scholar] [CrossRef]
- Xiao, P.; Zhang, H.; Zhang, Y.; Zheng, M.; Liu, R.; Zhao, Y.; Zhang, X.; Cheng, H.; Cao, Q.; Ke, Y. Phosphatase Shp2 exacerbates intestinal inflammation by disrupting macrophage responsiveness to interleukin-10. J. Exp. Med. 2019, 216, 337–349. [Google Scholar] [CrossRef]
- Giri, H.; Muthuramu, I.; Dhar, M.; Rathnakumar, K.; Ram, U.; Dixit, M. Protein tyrosine phosphatase SHP2 mediates chronic insulin-induced endothelial inflammation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1943–1950. [Google Scholar] [CrossRef]
- McNair, E.; Qureshi, M.; Prasad, K.; Pearce, C. Atherosclerosis and the Hypercholesterolemic AGE-RAGE Axis. Int. J. Angiol. 2016, 25, 110–116. [Google Scholar]
- Chung, S.S.; Ho, E.C.; Lam, K.S.; Chung, S.K. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. 2003, 14, S233–S236. [Google Scholar] [CrossRef]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Polyol pathway and RAGE: A central metabolic and signaling axis in diabetic complications. Expert Rev. Endocrinol. Metab. 2010, 5, 65–75. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorenkamp, M.; Nasiry, M.; Semo, D.; Koch, S.; Löffler, I.; Wolf, G.; Reinecke, H.; Godfrey, R. Pharmacological Targeting of the RAGE-NFκB Signalling Axis Impedes Monocyte Activation under Diabetic Conditions through the Repression of SHP-2 Tyrosine Phosphatase Function. Cells 2023, 12, 513. https://doi.org/10.3390/cells12030513
Dorenkamp M, Nasiry M, Semo D, Koch S, Löffler I, Wolf G, Reinecke H, Godfrey R. Pharmacological Targeting of the RAGE-NFκB Signalling Axis Impedes Monocyte Activation under Diabetic Conditions through the Repression of SHP-2 Tyrosine Phosphatase Function. Cells. 2023; 12(3):513. https://doi.org/10.3390/cells12030513
Chicago/Turabian StyleDorenkamp, Marc, Madina Nasiry, Dilvin Semo, Sybille Koch, Ivonne Löffler, Gunter Wolf, Holger Reinecke, and Rinesh Godfrey. 2023. "Pharmacological Targeting of the RAGE-NFκB Signalling Axis Impedes Monocyte Activation under Diabetic Conditions through the Repression of SHP-2 Tyrosine Phosphatase Function" Cells 12, no. 3: 513. https://doi.org/10.3390/cells12030513
APA StyleDorenkamp, M., Nasiry, M., Semo, D., Koch, S., Löffler, I., Wolf, G., Reinecke, H., & Godfrey, R. (2023). Pharmacological Targeting of the RAGE-NFκB Signalling Axis Impedes Monocyte Activation under Diabetic Conditions through the Repression of SHP-2 Tyrosine Phosphatase Function. Cells, 12(3), 513. https://doi.org/10.3390/cells12030513