
Mitochondrial Respiratory Complexes as Targets of Drugs: The PPAR Agonist Example

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Drug Treatment
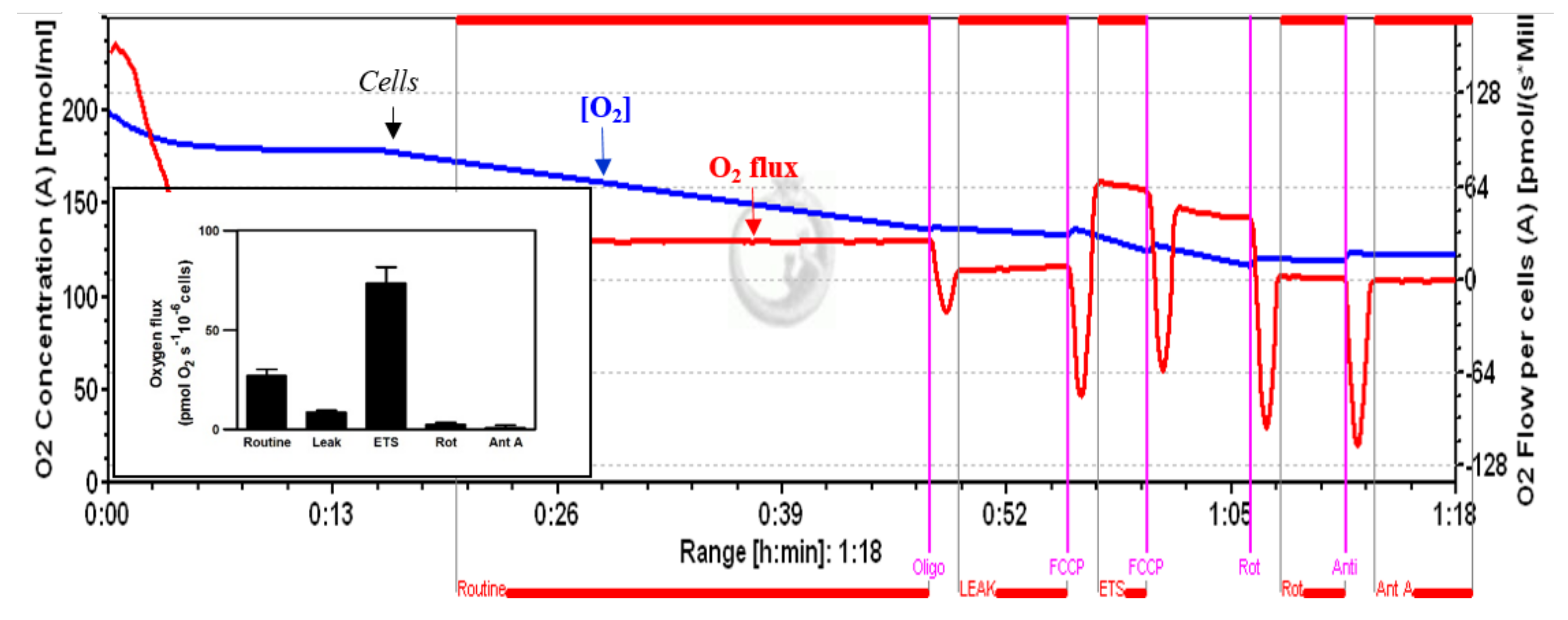
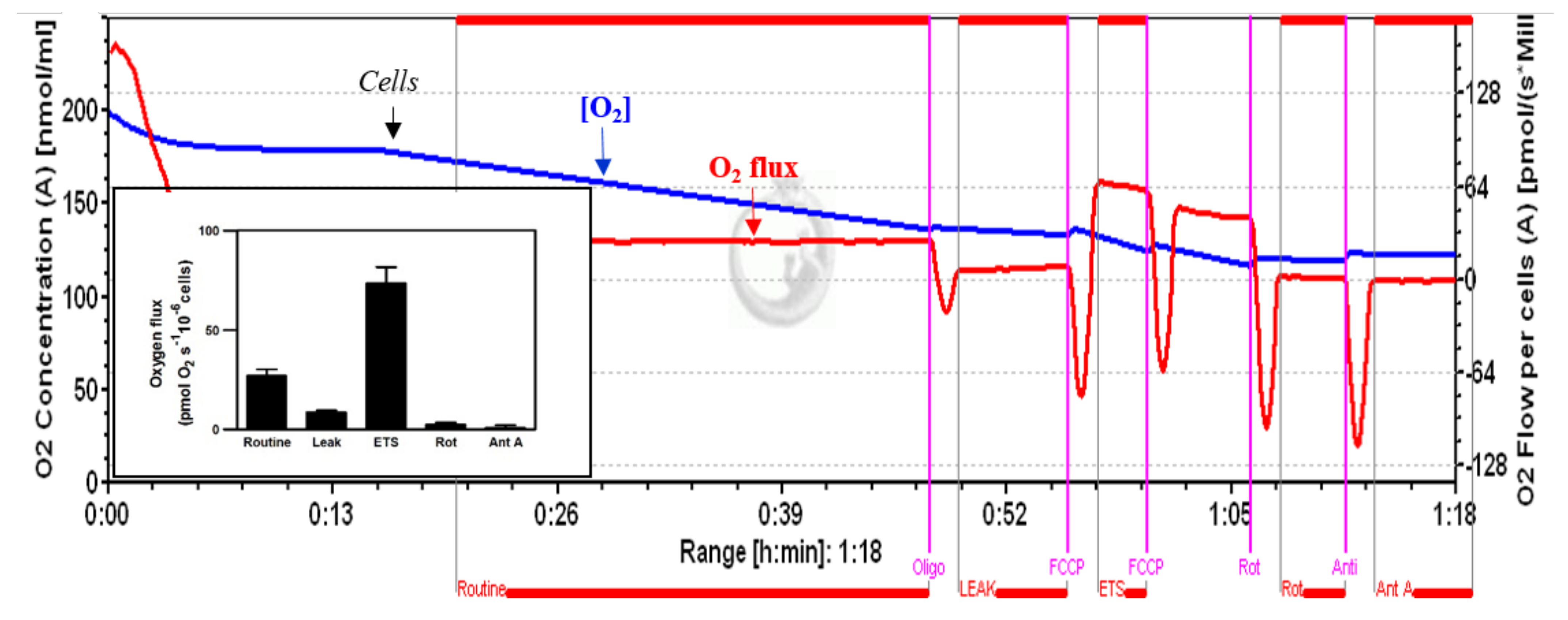
2.4. High-Resolution Respirometry
2.4.1. Permeabilized Cells
2.4.2. Intact Cells
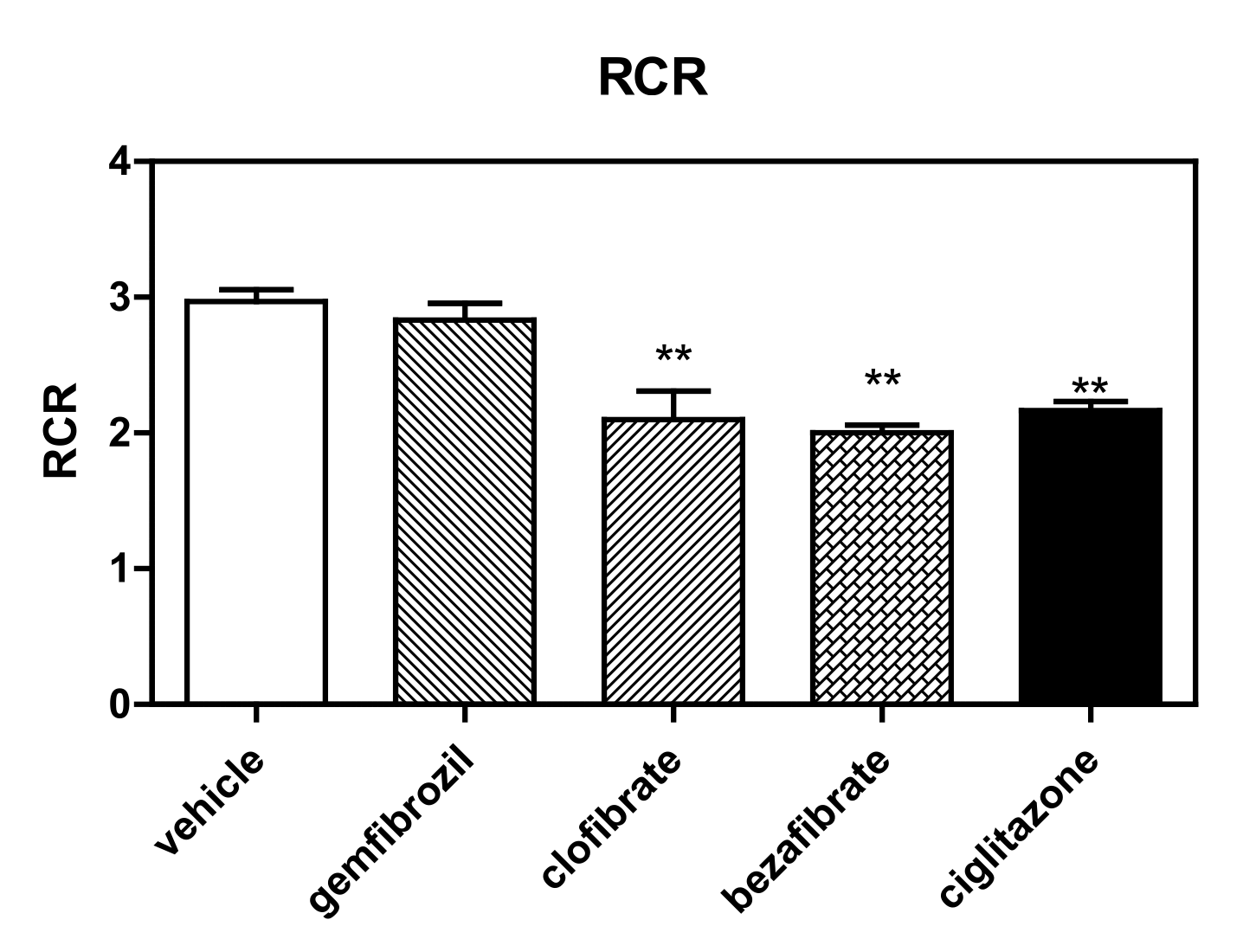
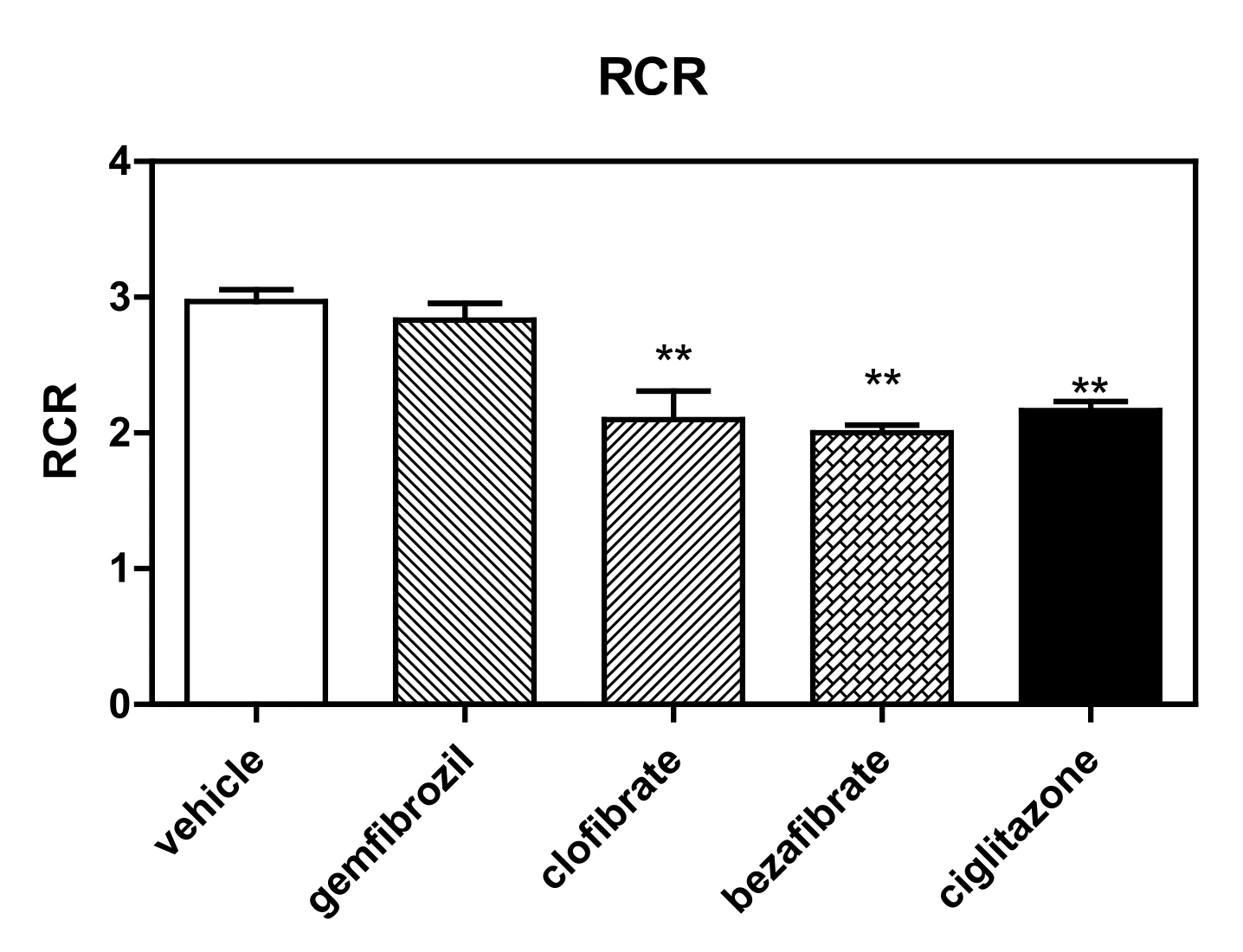
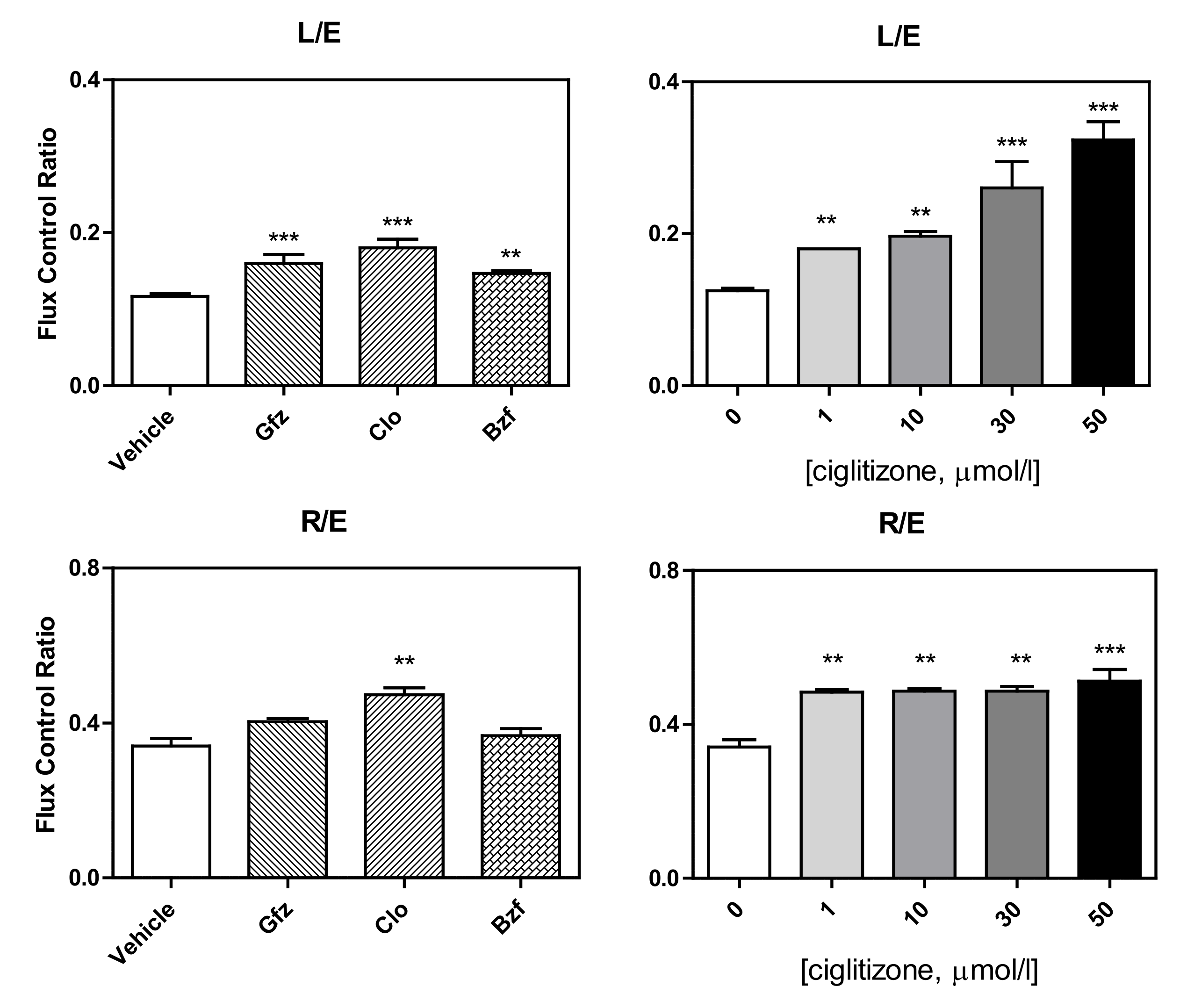
2.4.3. Respiratory Control Ratios
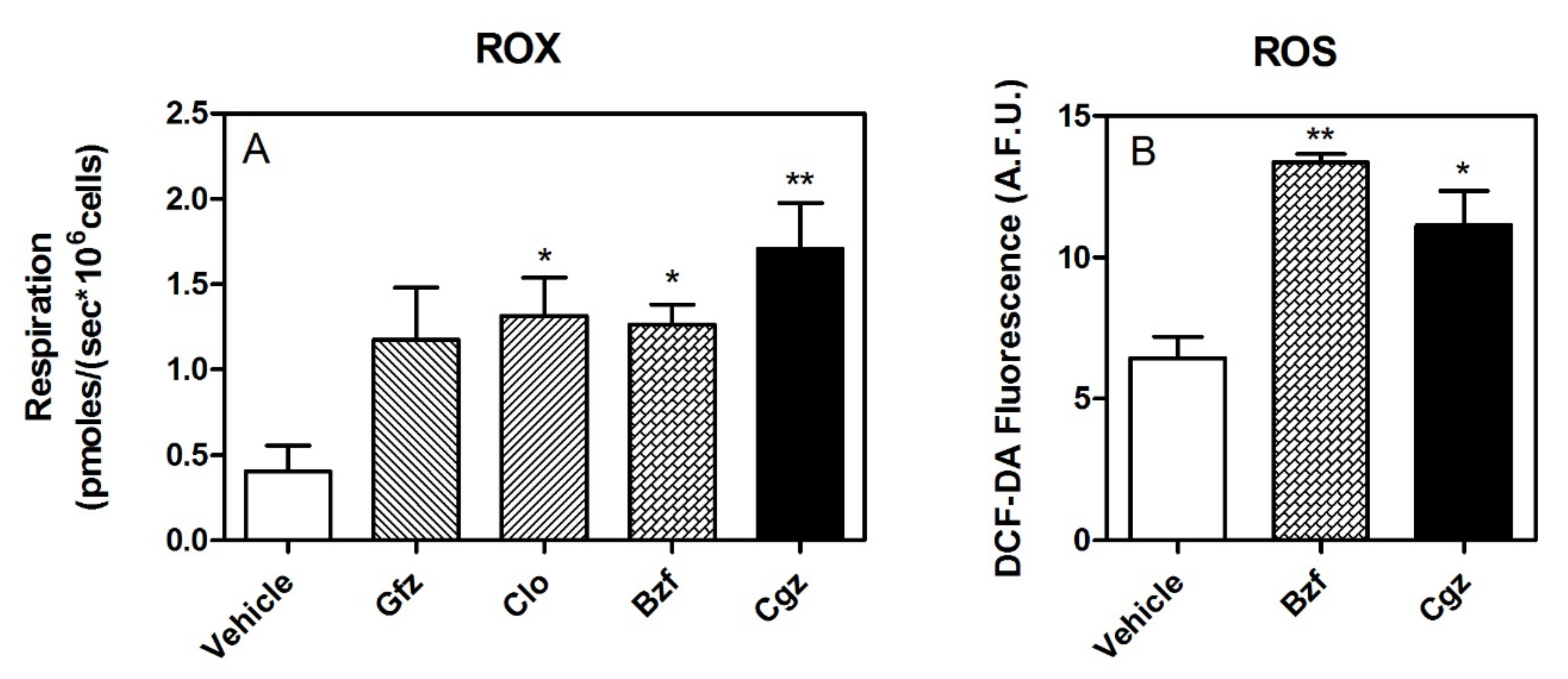
2.5. Assay for ROS
2.6. Proteomic Analysis
2.6.1. Protein Purification and Label-Free Differential Proteomic Shotgun Analysis
2.6.2. Bioinformatics Analysis
2.7. Statistical Analysis
3. Results
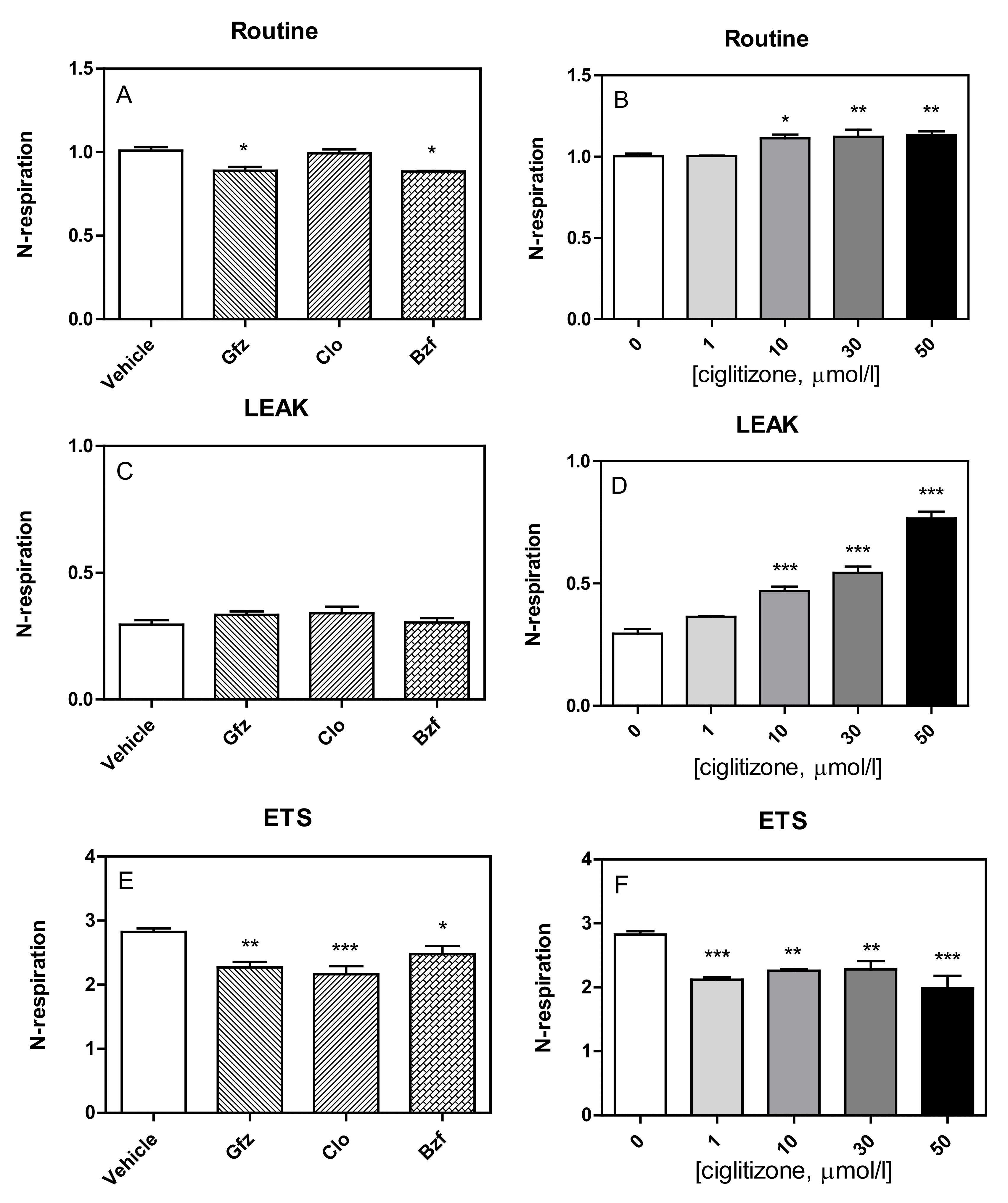
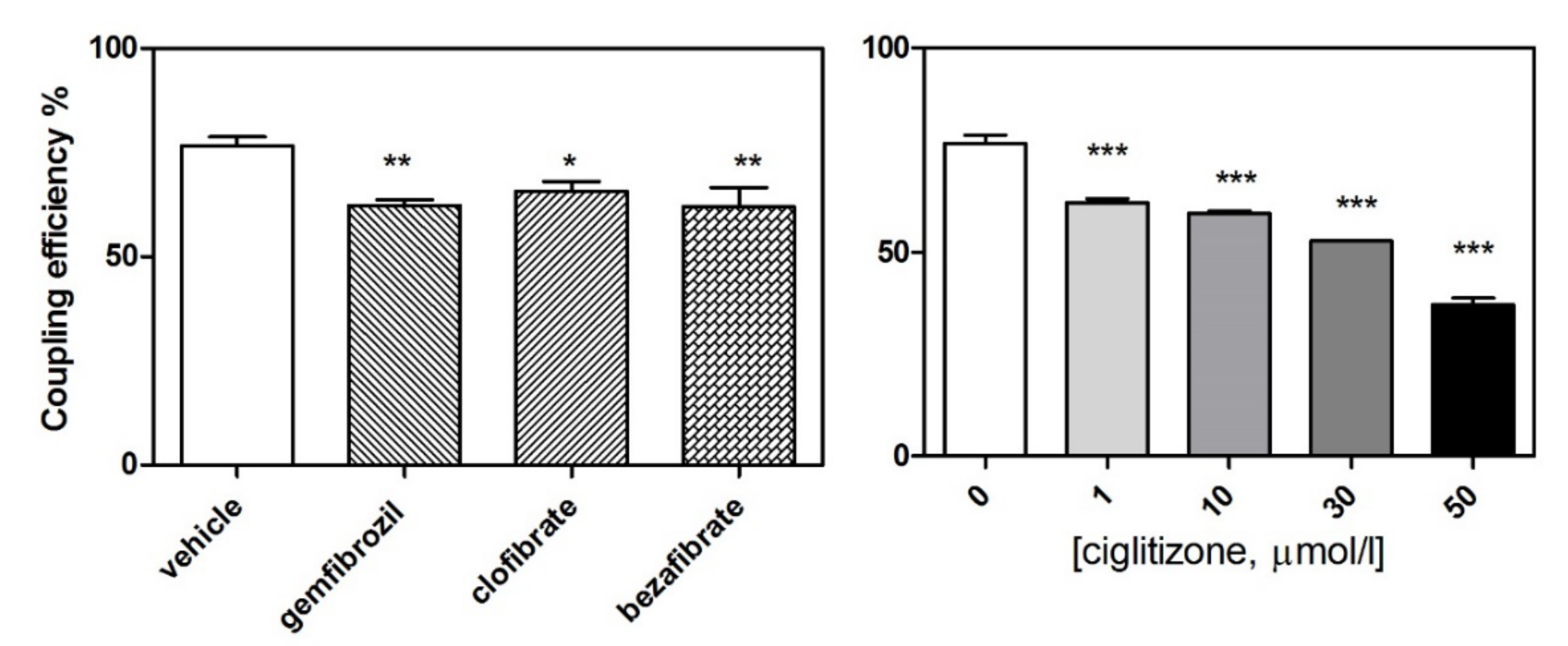
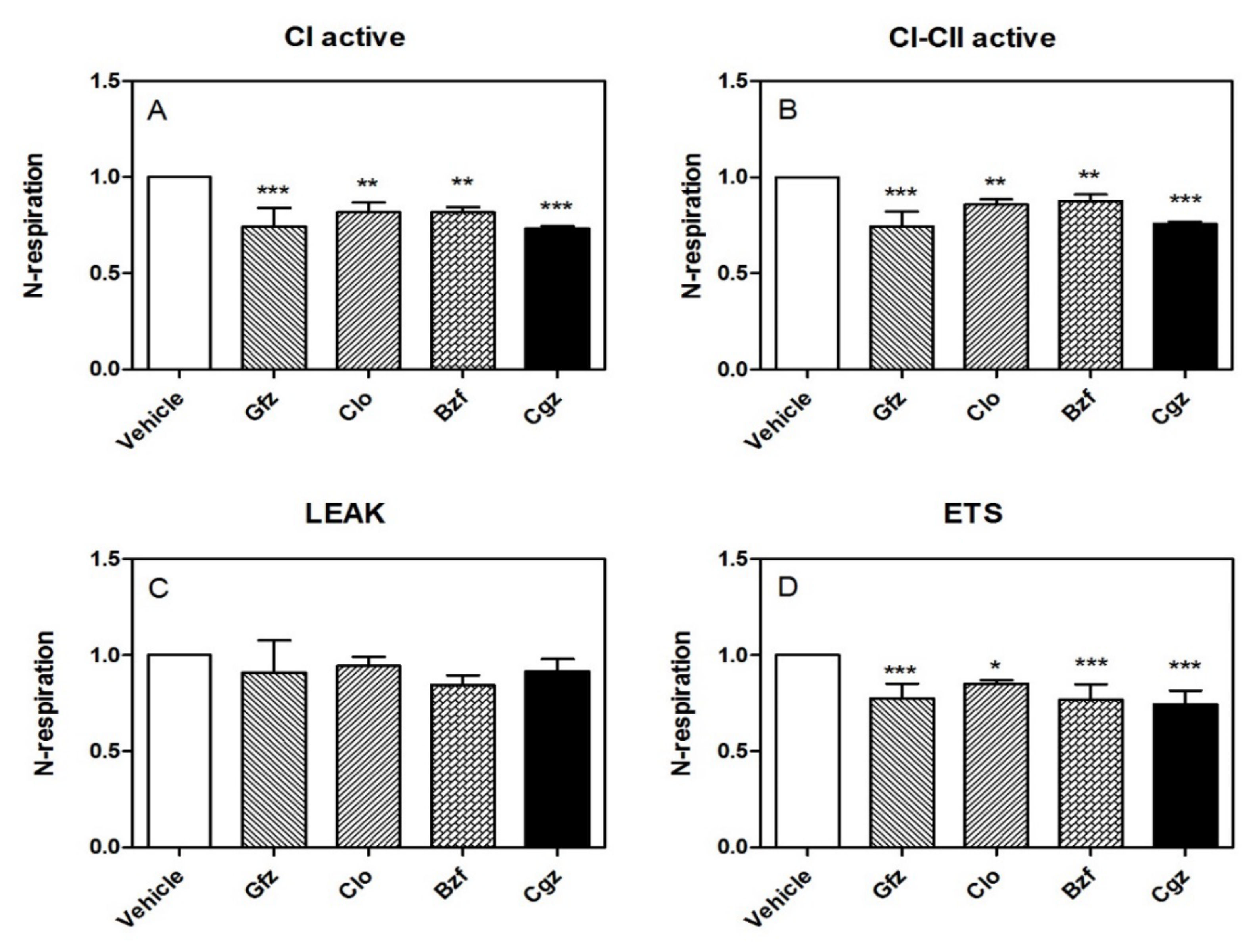
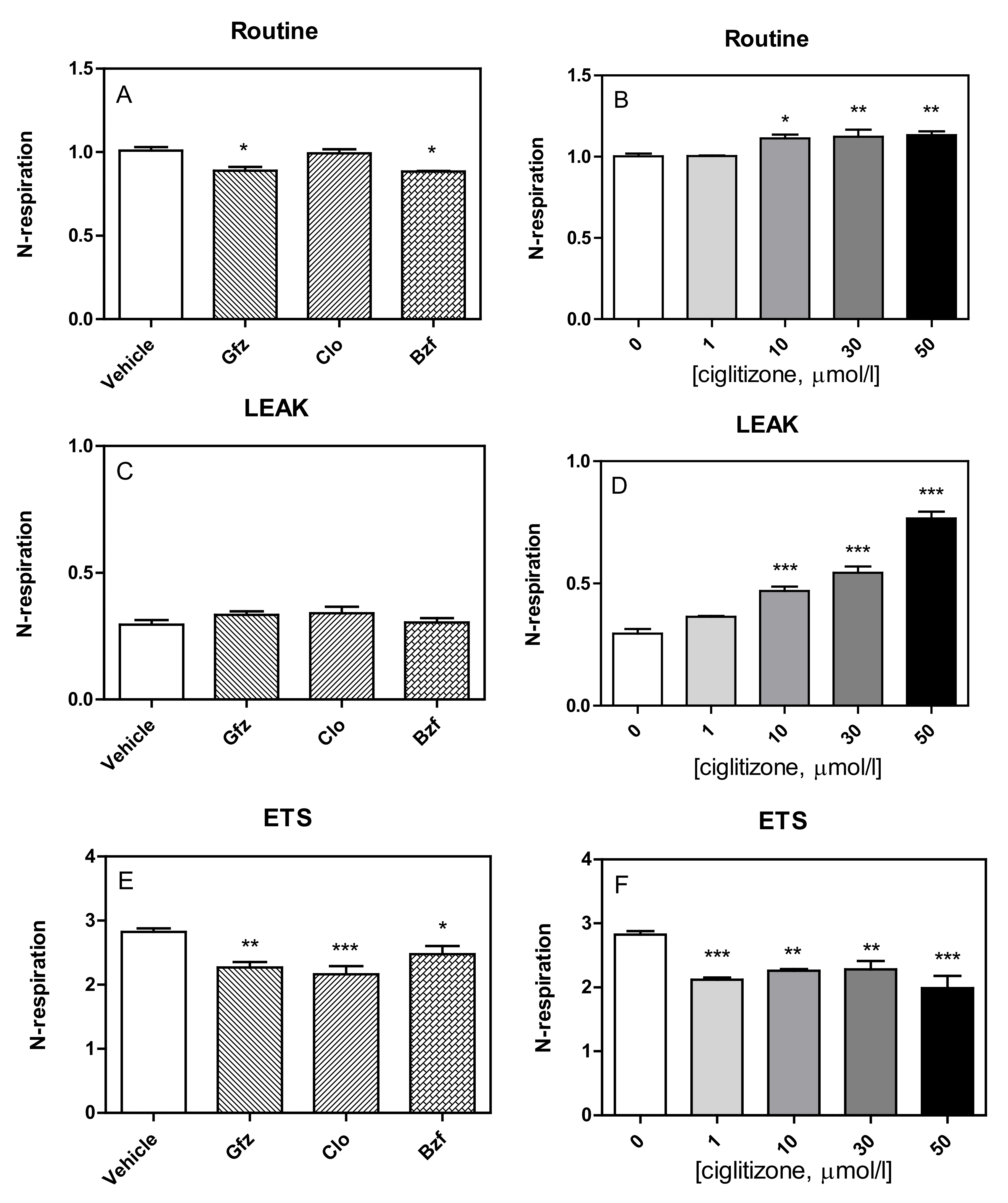
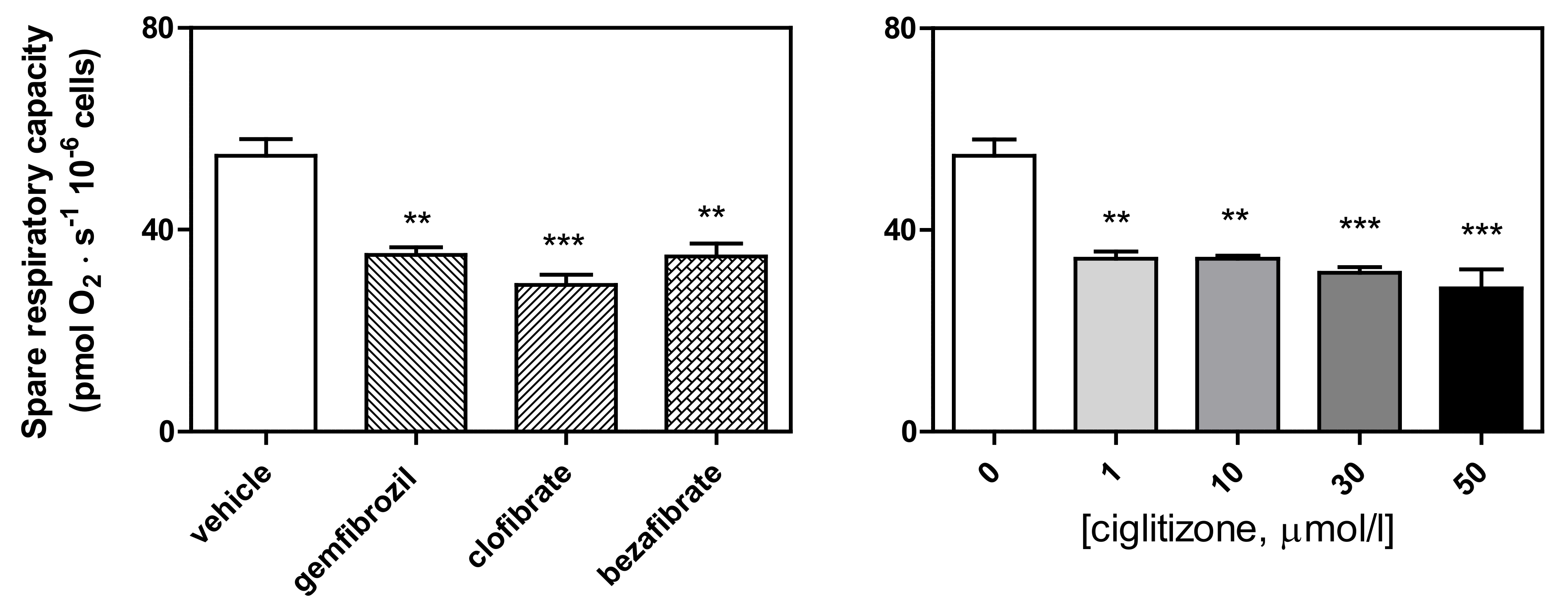
3.1. High-Resolution Respirometry (HRR) of Permeabilized Cells
3.2. HRR of Intact Cells
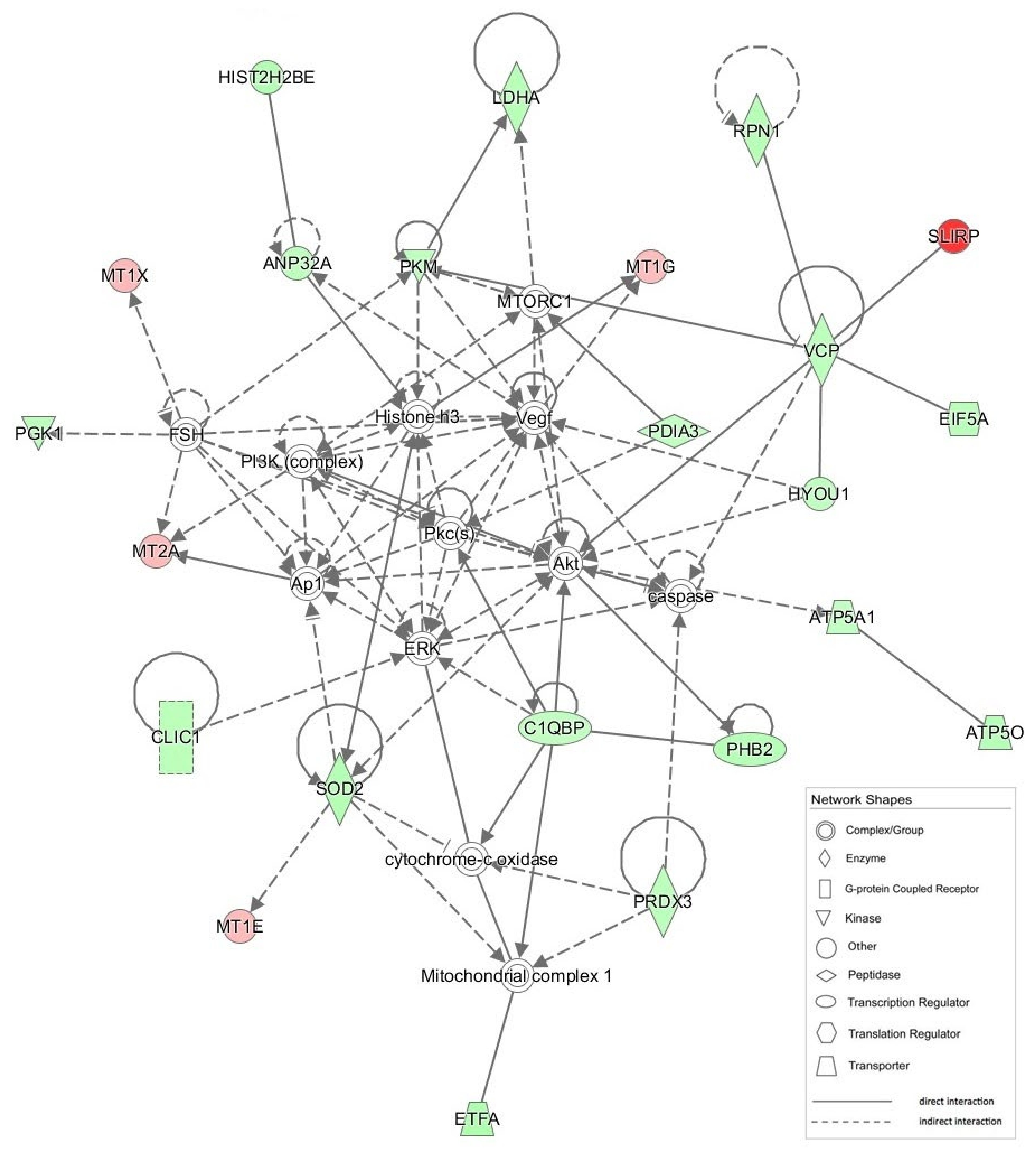
3.3. Shotgun Proteomic Profiling of Differentiated HepG2 Cells by Label-Free nUPLC-MSE-Bioinformatics Analyses of Differentially Expressed Proteins
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Asp. Med. 2004, 25, 365–451. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Botta, G.; Martorana, G.E.; Giardina, B. The role of mitochondria in pharmacotoxicology: A reevaluation of an old, newly emerging topic. Am. J. Physiol. Cell Physiol. 2007, 293, C12–C21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; Vacca, R.A.; de Bari, L. 3-Bromopyruvate induces rapid human prostate cancer cell death by affecting cell energy metabolism, GSH pool and the glyoxalase system. J. Bioenerg. Biomembr. 2015, 47, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Kunjithapatham, R.; Geschwind, J.F. Anticancer efficacy of the metabolic blocker 3-bromopyruvate: Specific molecular targeting. Anticancer Res. 2013, 33, 13–20. [Google Scholar] [PubMed]
- Qin, J.Z.; Xin, H.; Nickoloff, B.J. 3-bromopyruvate induces necrotic cell death in sensitive melanoma cell lines. Biochem. Biophys. Res. Commun. 2010, 396, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Verhoeven, H.A.; Lee, M.J.; Corbin, D.J.; Vogl, T.J.; Pedersen, P.L. A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: From bench side to bedside. J. Bioenerg. Biomembr. 2012, 44, 163–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira da Silva, A.P.; El-Bacha, T.; Kyaw, N.; dos Santos, R.S.; da-Silva, W.S.; Almeida, F.C.; Da Poian, A.T.; Galina, A. Inhibition of energy-producing pathways of HepG2 cells by 3-bromopyruvate. Biochem. J. 2009, 417, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, N.P.; Banerjee, S. Mitochondrial dependency in progression of acute myeloid leukemia. Mitochondrion 2015, 21, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S. Mitochondria as pharmacological targets. Br. J. Pharmacol. 2010, 160, 217–219. [Google Scholar] [CrossRef]
- Szewczyk, A.; Wojtczak, L. Mitochondria as a pharmacological target. Pharmacol. Rev. 2002, 54, 101–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scatena, R. Mitochondria and drugs. Adv. Exp. Med. Biol. 2012, 942, 329–346. [Google Scholar] [CrossRef]
- Mani, S.; Swargiary, G.; Singh, K.K. Natural Agents Targeting Mitochondria in Cancer. Int. J. Mol. Sci. 2020, 21, 6992. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-Targeted Drugs. Curr. Mol. Pharmacol. 2019, 12, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddrey, W.C. Drug-induced hepatotoxicity. J. Clin. Gastroenterol. 2005, 39, S83–S89. [Google Scholar] [CrossRef] [PubMed]
- Meadows, M. Serious liver injury. Leading reason for drug removals, restrictions. FDA Consum. 2001, 35, 8–9. [Google Scholar] [PubMed]
- Nesto, R.W.; Bell, D.; Bonow, R.O.; Fonseca, V.; Grundy, S.M.; Horton, E.S.; Le Winter, M.; Porte, D.; Semenkovich, C.F.; Smith, S.; et al. Thiazolidinedione use, fluid retention, and congestive heart failure: A consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004, 27, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Gale, E.A. The second time as farce: Rosiglitazone and the regulators. Nat. Rev. Endocrinol. 2011, 7, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.M. Drug-induced hepatotoxicity. N. Engl. J. Med. 2003, 349, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Crunkhorn, S. Diabetes: Bypassing the side effects of thiazolidinediones? Nat. Rev. Drug Discov. 2007, 6, 517. [Google Scholar] [CrossRef]
- Rizos, C.V.; Elisaf, M.S.; Mikhailidis, D.P.; Liberopoulos, E.N. How safe is the use of thiazolidinediones in clinical practice? Expert Opin. Drug Saf. 2009, 8, 15–32. [Google Scholar] [CrossRef]
- Scatena, R.; Bottoni, P.; Vincenzoni, F.; Messana, I.; Martorana, G.E.; Nocca, G.; De Sole, P.; Maggiano, N.; Castagnola, M.; Giardina, B. Bezafibrate induces a mitochondrial derangement in human cell lines: A PPAR-independent mechanism for a peroxisome proliferator. Chem. Res. Toxicol. 2003, 16, 1440–1447. [Google Scholar] [CrossRef]
- Brunmair, B.; Staniek, K.; Gras, F.; Scharf, N.; Althaym, A.; Clara, R.; Roden, M.; Gnaiger, E.; Nohl, H.; Waldhäusl, W.; et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: A common mechanism contributing to their antidiabetic actions? Diabetes 2004, 53, 1052–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scatena, R.; Bottoni, P.; Martorana, G.E.; Ferrari, F.; De Sole, P.; Rossi, C.; Giardina, B. Mitochondrial respiratory chain dysfunction, a non-receptor-mediated effect of synthetic PPAR-ligands: Biochemical and pharmacological implications. Biochem. Biophys. Res. Commun. 2004, 319, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Martorana, G.E.; Vincenzoni, F.; Botta, G.; Pastore, P.; Giardina, B. Mitochondria, ciglitazone and liver: A neglected interaction in biochemical pharmacology. Eur. J. Pharmacol. 2007, 567, 50–58. [Google Scholar] [CrossRef]
- Ramirez, T.; Strigun, A.; Verlohner, A.; Huener, H.A.; Peter, E.; Herold, M.; Bordag, N.; Mellert, W.; Walk, T.; Spitzer, M.; et al. Prediction of liver toxicity and mode of action using metabolomics in vitro in HepG2 cells. Arch. Toxicol. 2018, 92, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E. Capacity of oxidative phosphorylation in human skeletal muscle: New perspectives of mitochondrial physiology. Int. J. Biochem. Cell Biol. 2009, 41, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Pieroni, L.; Finamore, F.; Ronci, M.; Mattoscio, D.; Marzano, V.; Mortera, S.L.; Quattrucci, S.; Federici, G.; Romano, M.; Urbani, A. Proteomics investigation of human platelets in healthy donors and cystic fibrosis patients by shotgun nUPLC-MSE and 2DE: A comparative study. Mol. Biosyst. 2011, 7, 630–639. [Google Scholar] [CrossRef]
- Pieroni, L.; Levi Mortera, S.; Greco, V.; Sirolli, V.; Ronci, M.; Felaco, P.; Fucci, G.; De Fulviis, S.; Massoud, R.; Condò, S.; et al. Biocompatibility assessment of haemodialysis membrane materials by proteomic investigations. Mol. Biosyst. 2015, 11, 1633–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control. An Introduction to OXPHOS Analysis. Mitochondr Physiol Network 19.12, 4th ed.; Oroboros MiPNet Publications: Innsbruck, Austria, 2014; p. 80. [Google Scholar]
- Wallace, K.B. Mitochondrial off targets of drug therapy. Trends Pharmacol. Sci. 2008, 29, 361–366. [Google Scholar] [CrossRef]
- Brunmair, B.; Lest, A.; Staniek, K.; Gras, F.; Scharf, N.; Roden, M.; Nohl, H.; Waldhäusl, W.; Fürnsinn, C. Fenofibrate impairs rat mitochondrial function by inhibition of respiratory complex I. J. Pharmacol. Exp. Ther. 2004, 311, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.Y.; Xiao, C.X.; Zhao, W.X.; Xiao, L.; Chen, X.; Li, P.; Wang, X.M. Enoyl-coenzyme A hydratase short chain 1 silencing attenuates the proliferation of hepatocellular carcinoma by inhibiting epidermal growth factor signaling in vitro and in vivo. Mol. Med. Rep. 2015, 12, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Soltys, B.J.; Kang, D.; Gupta, R.S. Localization of P32 protein (gC1q-R) in mitochondria and at specific extramitochondrial locations in normal tissues. Histochem. Cell Biol. 2000, 114, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Muta, T.; Kang, D.; Kitajima, S.; Fujiwara, T.; Hamasaki, N. p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J. Biol. Chem. 1997, 272, 24363–24370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogal, V.; Richardson, A.D.; Karmali, P.P.; Scheffler, I.E.; Smith, J.W.; Ruoslaht, E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell. Biol. 2010, 30, 1303–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGee, A.M.; Baines, C.P. Complement 1q-binding protein inhibits the mitochondrial permeability transition pore and protects against oxidative stress-induced death. Biochem. J. 2011, 433, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, K.; Morisaki, T.; Setoyama, D.; Sasaki, K.; Yagi, M.; Igami, K.; Mizuguchi, S.; Uchiumi, T.; Fukui, Y.; Kang, D. Mitochondrial p32/C1qbp Is a Critical Regulator of Dendritic Cell Metabolism and Maturation. Cell Rep. 2018, 25, 1800–1815.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenenbaum, A.; Fisman, E.Z.; Motro, M. Rhabdomyolysis and lipid-lowering drugs. JAMA 2005, 293, 1448. [Google Scholar] [CrossRef] [PubMed]
- Floyd, J.S.; Barbehenn, E.; Lurie, P.; Wolfe, S.M. Case series of liver failure associated with rosiglitazone and pioglitazone. Pharmacoepidemiol. Drug Saf. 2009, 18, 1238–1243. [Google Scholar] [CrossRef]
- Tamraz, B.; Fukushima, H.; Wolfe, A.R.; Kaspera, R.; Totah, R.A.; Floyd, J.S.; Ma, B.; Chu, C.; Marciante, K.D.; Heckbert, S.R.; et al. OATP1B1-related drug–drug and drug–gene interactions as potential risk factors for cerivastatin-induced rhabdomyolysis. Pharm. Genom. 2013, 23, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- dos Santos, A.G.; Guardia, A.C.; Pereira, T.S.; Ataíde, E.C.; Mei, M.; Udo, M.E.; Boin, I.F.; Stucchi, R.S. Rhabdomyolysis as a clinical manifestation of association with ciprofibrate, sirolimus, cyclosporine, and pegylated interferon-α in liver-transplanted patients: A case report and literature review. Transpl. Proc. 2014, 46, 1887–1888. [Google Scholar] [CrossRef]
- Hedrington, M.S.; Davis, S.N. Peroxisome proliferator-activated receptor alpha-mediated drug toxicity in the liver. Expert Opin. Drug Metab. Toxicol. 2018, 14, 671–677. [Google Scholar] [CrossRef]
- Gale, E.A. Lessons from the glitazones: A story of drug development. Lancet 2001, 357, 1870–1875. [Google Scholar] [CrossRef]
- Youssef, J.; Badr, M. Extraperoxisomal targets of peroxisome proliferators: Mitochondrial, microsomal, and cytosolic effects. Implications for health and disease. Crit. Rev. Toxicol. 1998, 28, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Julie, N.L.; Julie, I.M.; Kende, A.I.; Wilson, G.L. Mitochondrial dysfunction and delayed hepatotoxicity: Another lesson from troglitazone. Diabetologia 2008, 51, 2108–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divakaruni, A.S.; Wiley, S.E.; Rogers, G.W.; Andreyev, A.Y.; Petrosyan, S.; Loviscach, M.; Wall, E.A.; Yadava, N.; Heuck, A.P.; Ferrick, D.A.; et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc. Natl. Acad. Sci. USA 2013, 110, 5422–5427. [Google Scholar] [CrossRef] [Green Version]
- Dzhekova-Stojkova, S.; Bogdanska, J.; Stojkova, Z. Peroxisome proliferators: Their biological and toxicological effects. Clin. Chem. Lab. Med. 2001, 39, 468–474. [Google Scholar] [CrossRef]
- Kostapanos, M.S.; Elisaf, M.S.; Mikhailidis, D.P. Pioglitazone and cancer: Angel or demon? Curr. Pharm. Des. 2013, 19, 4913–4929. [Google Scholar] [CrossRef] [PubMed]
- Amend, K.L.; Landon, J.; Thyagarajan, V.; Niemcryk, S.; McAfee, A. Incidence of hospitalized rhabdomyolysis with statin and fibrate use in an insured US population. Ann. Pharmacother. 2011, 45, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Herdeiro, M.T.; Soares, S.; Silva, T.; Roque, F.; Figueiras, A. Impact of rosiglitazone safety alerts on oral antidiabetic sales trends: A countrywide study in Portugal. Fundam. Clin. Pharmacol. 2016, 30, 440–449. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
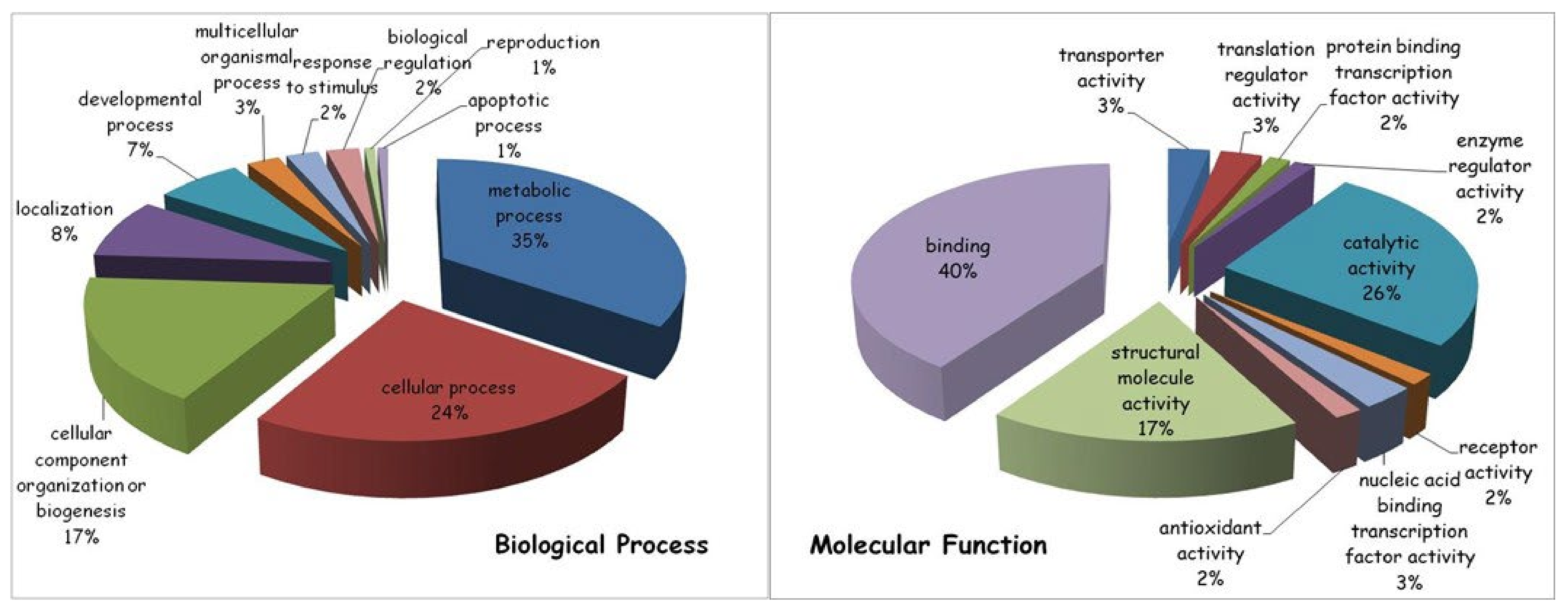{kind=link}
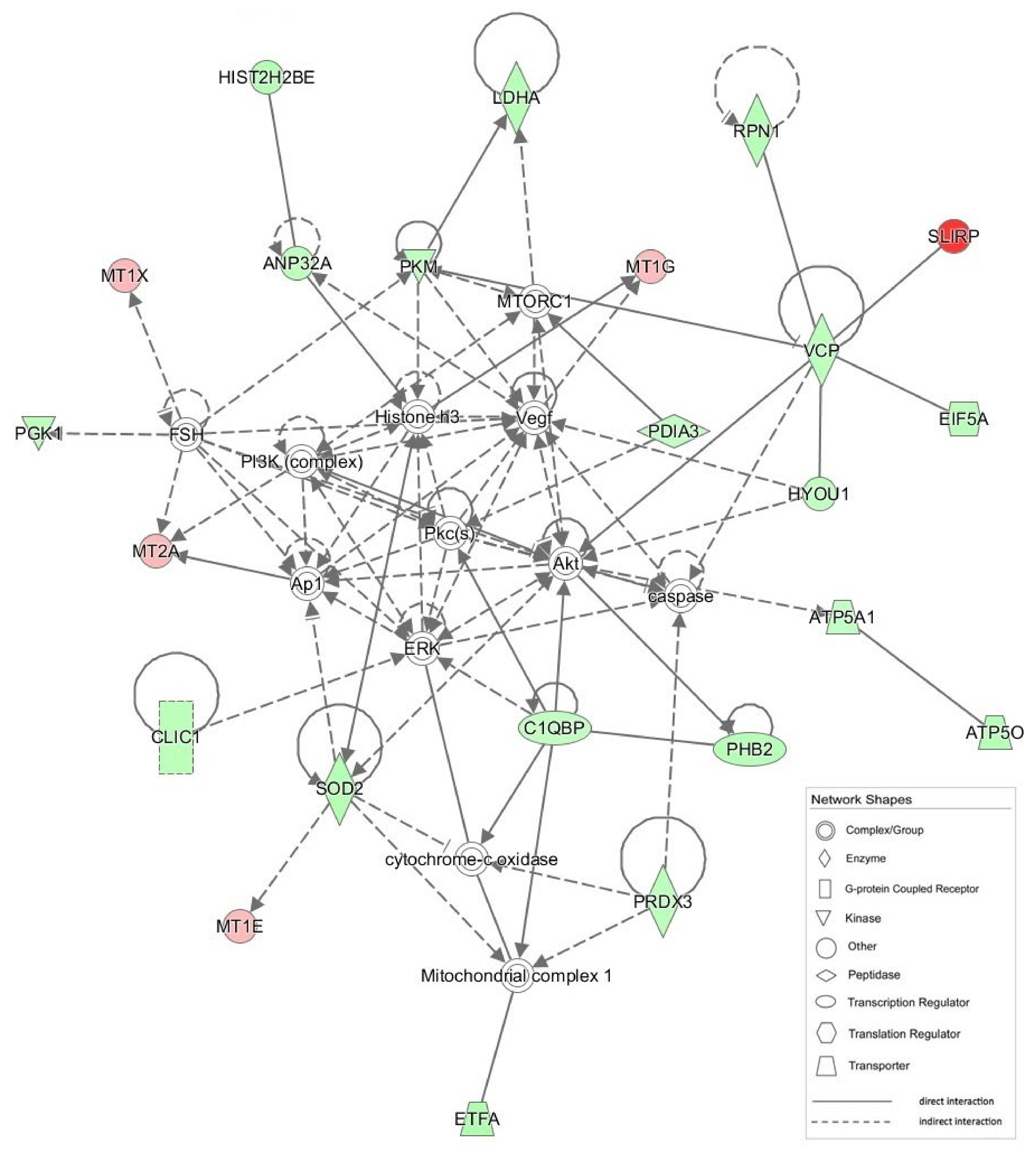{kind=link}
| Tox List | Proteins | p Value |
|---|---|---|
| Mitochondrial dysfunction | ATP5A1, ATP50, PRDX3, SOD2, VDAC2 | 2.74 × 104 |
| Cell death | ANXA5, HYOU1, LDHA, PPIA, PRDX3, SOD2, VCP | 1.51 × 103 |
| Fatty acid metabolism | ACAT2, DHRS2, ECHS1 | 4.62 × 103 |
| Alteration transmembrane potential of mitochondria and mitochondrial membrane | PRDX3, SOD2 | 1.27 × 102 |
| Oxidative stress | PRDX3, SOD2 | 1.58 × 102 |
| ID | Molecules in Network | Score | Focus Molecules | Top Diseases and Functions |
|---|---|---|---|---|
| 1 | Akt, ANP32A, Ap1, ATP5A1, ATP5O, C1QBP, caspase, CLIC1, cytocrome-c oxidase, EIF5A, ERK, ETFA, FSH, HIST2H2BE, Histone H3, HYOU1, LDHA, Mitochondrial complex 1, MT1E, MT1G, MT1X, MT2A, MTORC1, PDIA3, PGK1, PHB2, PI3K (complex), Pkc(s), PKM, PRDX3, RPN1, SLIRP, SOD2, VCP, Vegf | 51 | 23 | Neurological Disease, Skeletal and Muscular Disorders, Hereditary Disorder |
| 2 | ACAT2, Actin, ADRB, ANXA5, CD3, CFL1, DHRS2, DHX9, ERK1/2, F Actin, HNRNPK, Hsp90, KHDRBS1, KRT8, MAP2K1/2, MATR3, NACA, NCL, PDGF BB, PFN1, PPIA, Rnr, RPS12, RPS3A, RPSA, Rsk, TCR, TIMM13, TUBB, TUBB6, TUBB2A, TUBB2B, tubulin complex, tubulin (family), VDAC2 | 48 | 21 | Infectious Diseases, Developmental Disorder, Neurological Disease |
| 3 | AKT1, ALDOC, APBB3, APP, ATPAF2, ECE1, ECHS1, EED, EMG1, ERP29, FERMT2, FN1, GNRH2, HIRIP3, HIST1H2BB, HIST1H2BD, HIS1H2BK, HIST1H2BM, HIST2H2BN, HIST2H2BF, IGF2BP2, ITGA4, MAPK1, MBNL1, OSTF1, Ptk, RPLP2, RPS12, RPS16, RPS4Y1, SHOC2, TOMM22, VARS, YWHAZ, ZMAT3 | 20 | 11 | Cell Morphology, Reproductive System Development and Function, Lipid Metabolism |
| 4 | 26s Proteasome, Alpha actin, APLF, ARRB2, ASF1B, CDC34, CDKN2A, CUL1, CYP2EI, DHX9, GTF2E2, HIST1H2AG, HIST1H2BA, HIST1H2BH, HIST1H2BJ, HIST1H2BL, HIST1H2BO, HIST3H2BB, Histone h4, JMJD6, Jnk, MAP3K13, NFkB (complex), P38 MAPK, PAG1, PARP10, PTGES, Ras, RBMXL2, RNA polymerase II, RPLP2, RPS12, SIGIRR, TBP, ZMAT3 | 15 | 9 | Cancer, Organismal Injury and Abnormalities, Reproductive System Disease |
| 5 | H2BFS, MT1M, PAN2 | 5 | 2 | Psychological Disorders, Antimicrobial Response, Inflammatory Response |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bottoni, P.; Pontoglio, A.; Scarà, S.; Pieroni, L.; Urbani, A.; Scatena, R. Mitochondrial Respiratory Complexes as Targets of Drugs: The PPAR Agonist Example. Cells 2022, 11, 1169. https://doi.org/10.3390/cells11071169
Bottoni P, Pontoglio A, Scarà S, Pieroni L, Urbani A, Scatena R. Mitochondrial Respiratory Complexes as Targets of Drugs: The PPAR Agonist Example. Cells. 2022; 11(7):1169. https://doi.org/10.3390/cells11071169
Chicago/Turabian StyleBottoni, Patrizia, Alessandro Pontoglio, Salvatore Scarà, Luisa Pieroni, Andrea Urbani, and Roberto Scatena. 2022. "Mitochondrial Respiratory Complexes as Targets of Drugs: The PPAR Agonist Example" Cells 11, no. 7: 1169. https://doi.org/10.3390/cells11071169
APA StyleBottoni, P., Pontoglio, A., Scarà, S., Pieroni, L., Urbani, A., & Scatena, R. (2022). Mitochondrial Respiratory Complexes as Targets of Drugs: The PPAR Agonist Example. Cells, 11(7), 1169. https://doi.org/10.3390/cells11071169