
Alteration of Mitochondrial Integrity as Upstream Event in the Pathophysiology of SOD1-ALS

 , ,
, ,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Characteristics
2.2. Karyotyping
2.3. Genotyping
2.4. Mycoplasma Testing
2.5. Generation, Gene Editing and Differentiation of Human iPSC Cell Lines to MNs in Microfluidic Chambers (MFCs)
2.6. Live Imaging in MFCs
2.7. Organelle Tracking and Shape Analysis
2.8. Stress Granule Induction and Protein Aggregation
2.9. DNA Damage
2.10. Immunofluorescence Stainings
2.11. Image Quantification
2.12. Measurements of ATP in MNs
2.13. Cell Extracts SOD1 Analysis
2.14. Quantification of Disordered and Total SOD1 by ELISA
2.15. Quantification of SOD1 in Detergent-Insoluble Aggregates
2.16. Statistical Analyses of ELISA and Western Blotting
3. Results
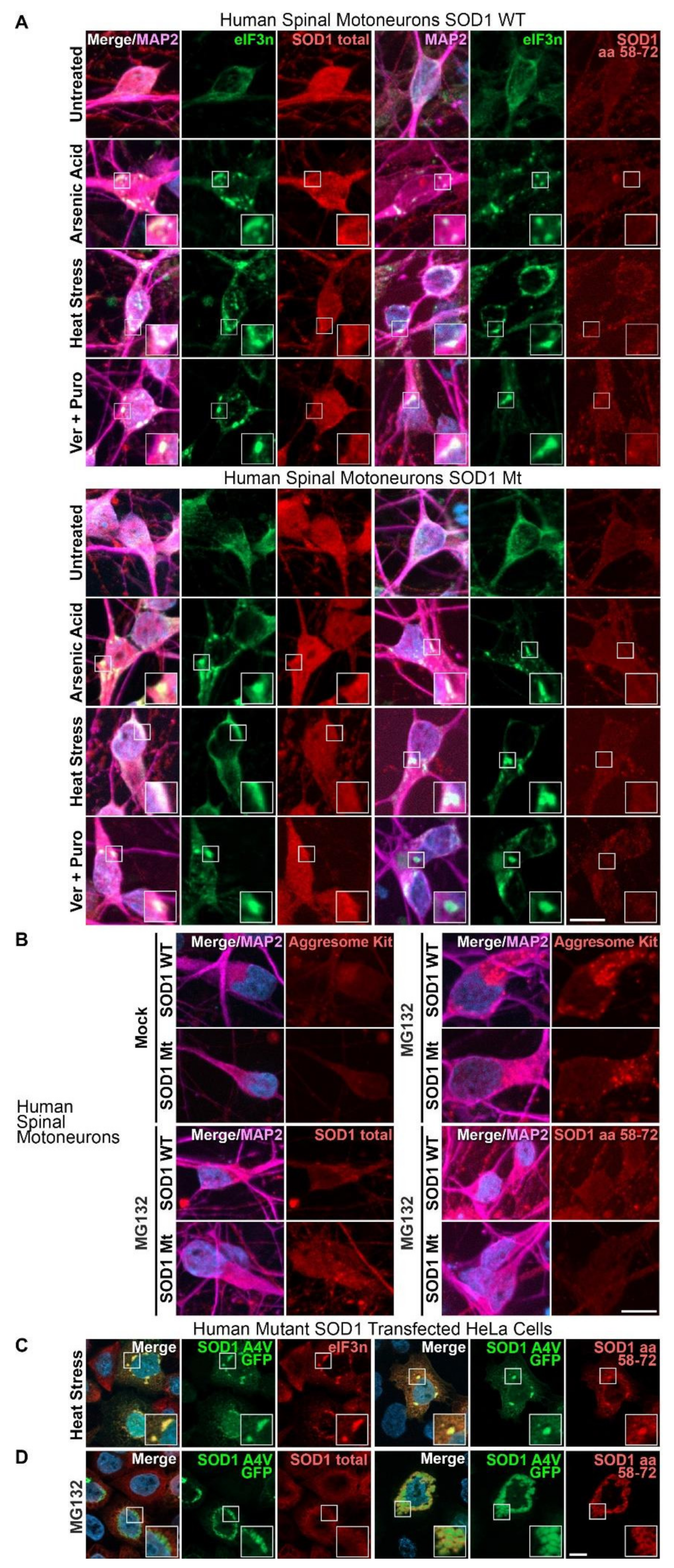
3.1. Aggregates of SOD1 Are Not Detected in iPSC-Derived MNs from SOD1-ALS Patients
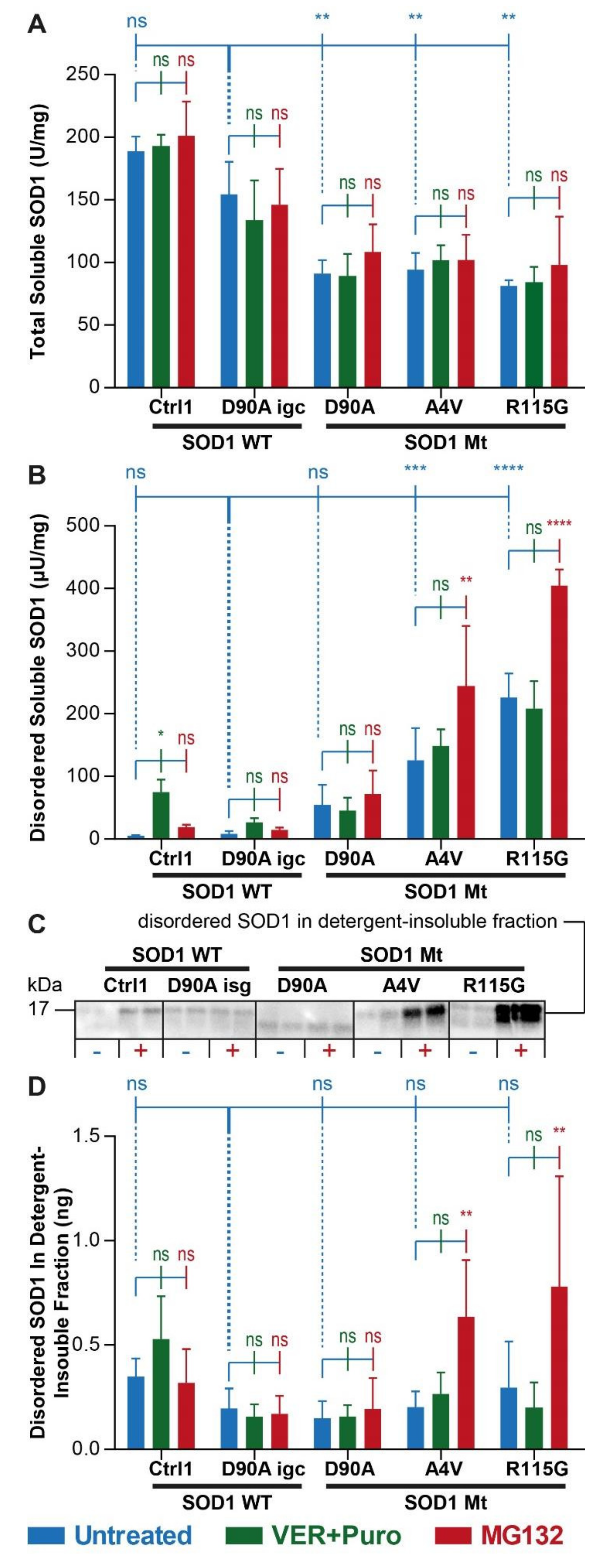
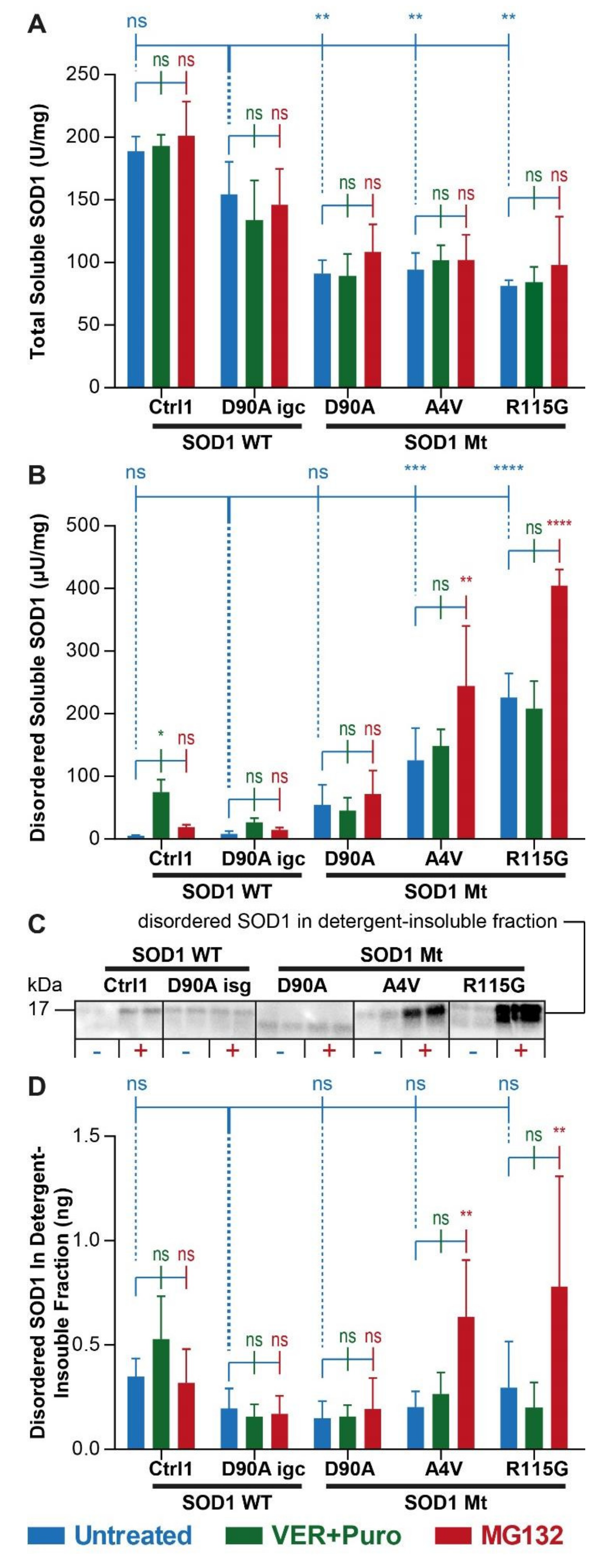
3.2. Augmented Levels of Disordered SOD1 upon Stress in SOD1 Mutant MNs
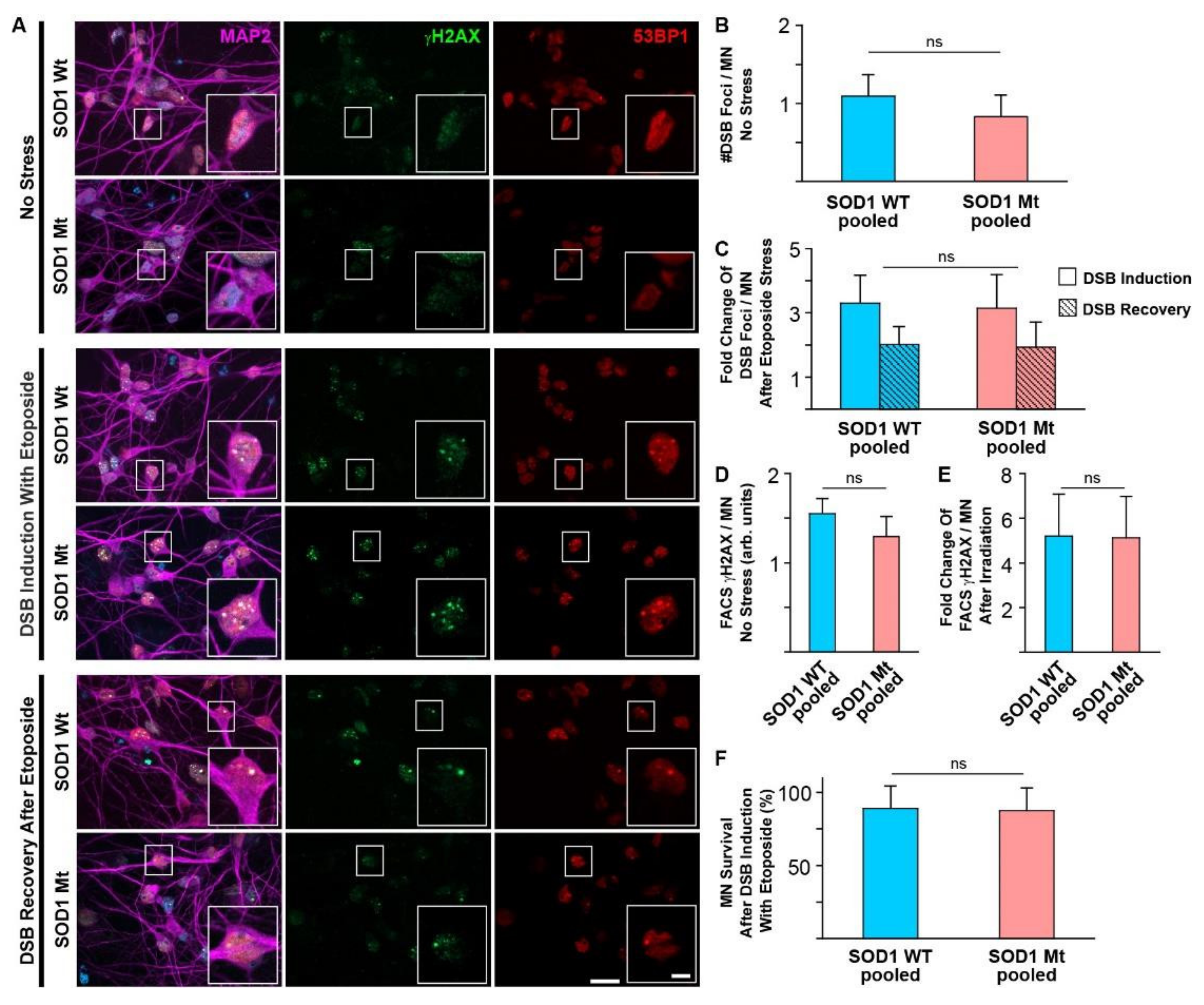
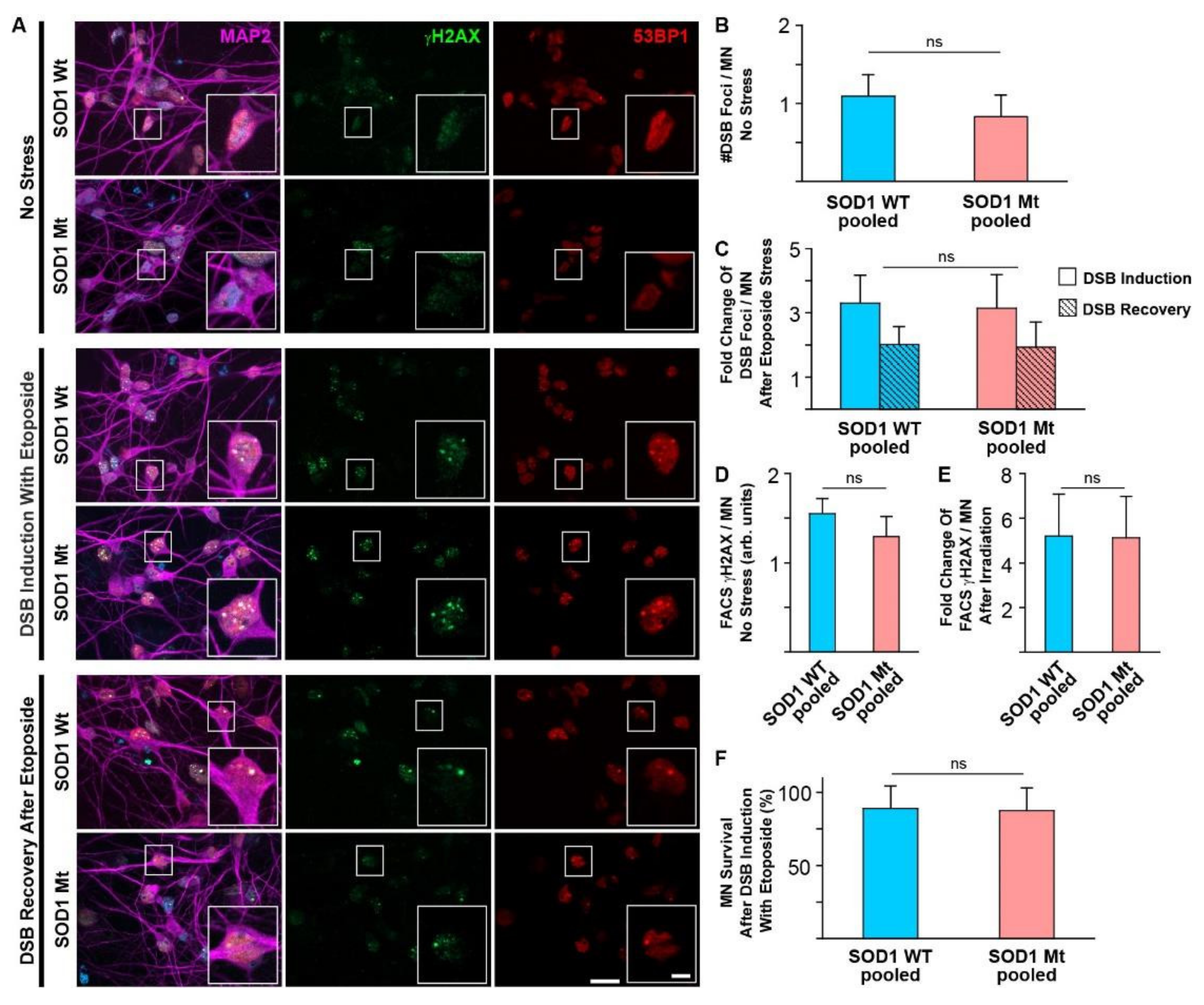
3.3. No Signs of Elevated DNA Damage or Impaired DNA Repair in SOD1 Mutant MNs
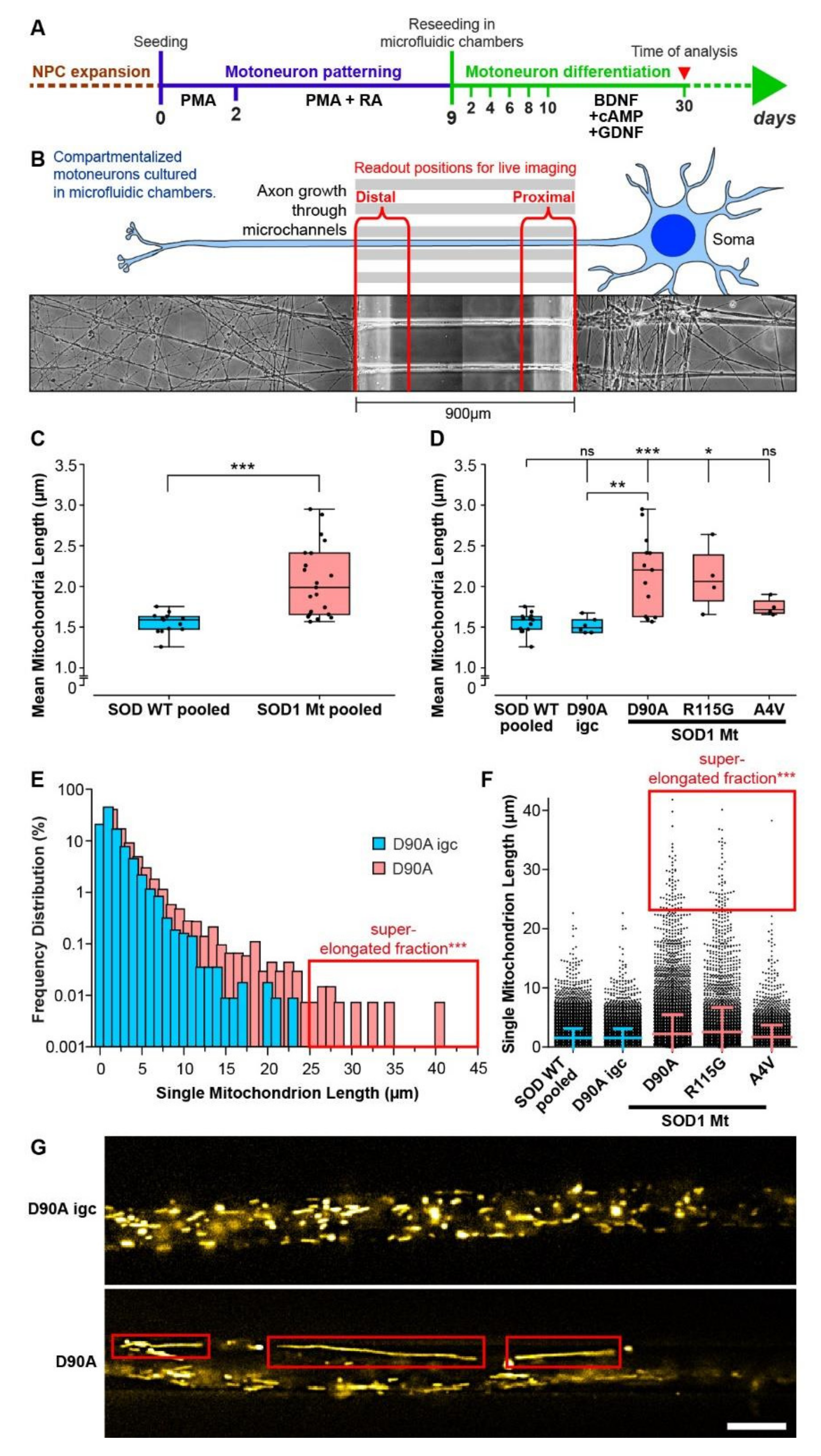
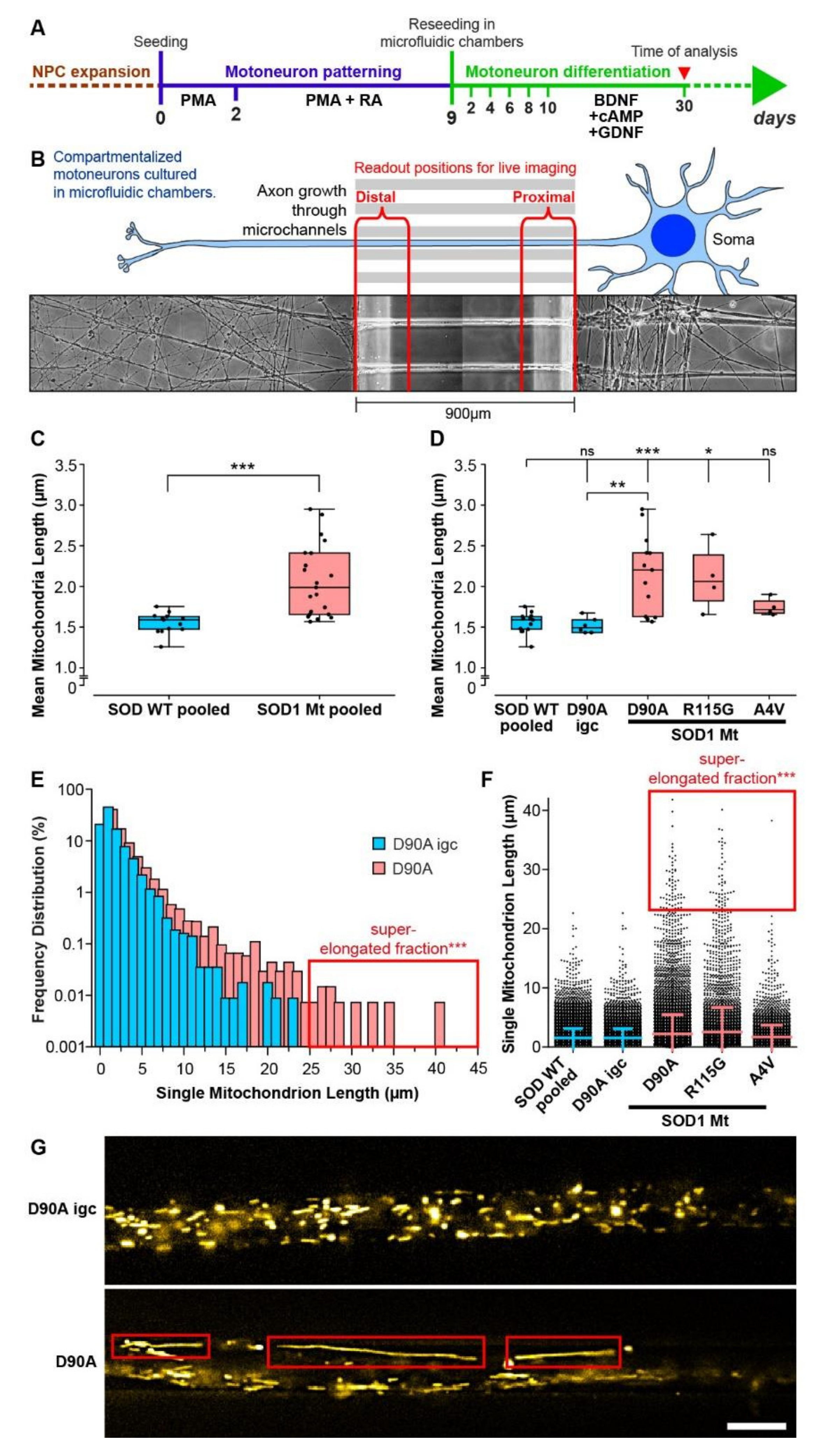
3.4. Elongated Mitochondrial Morphology in SOD1 Mutant MNs
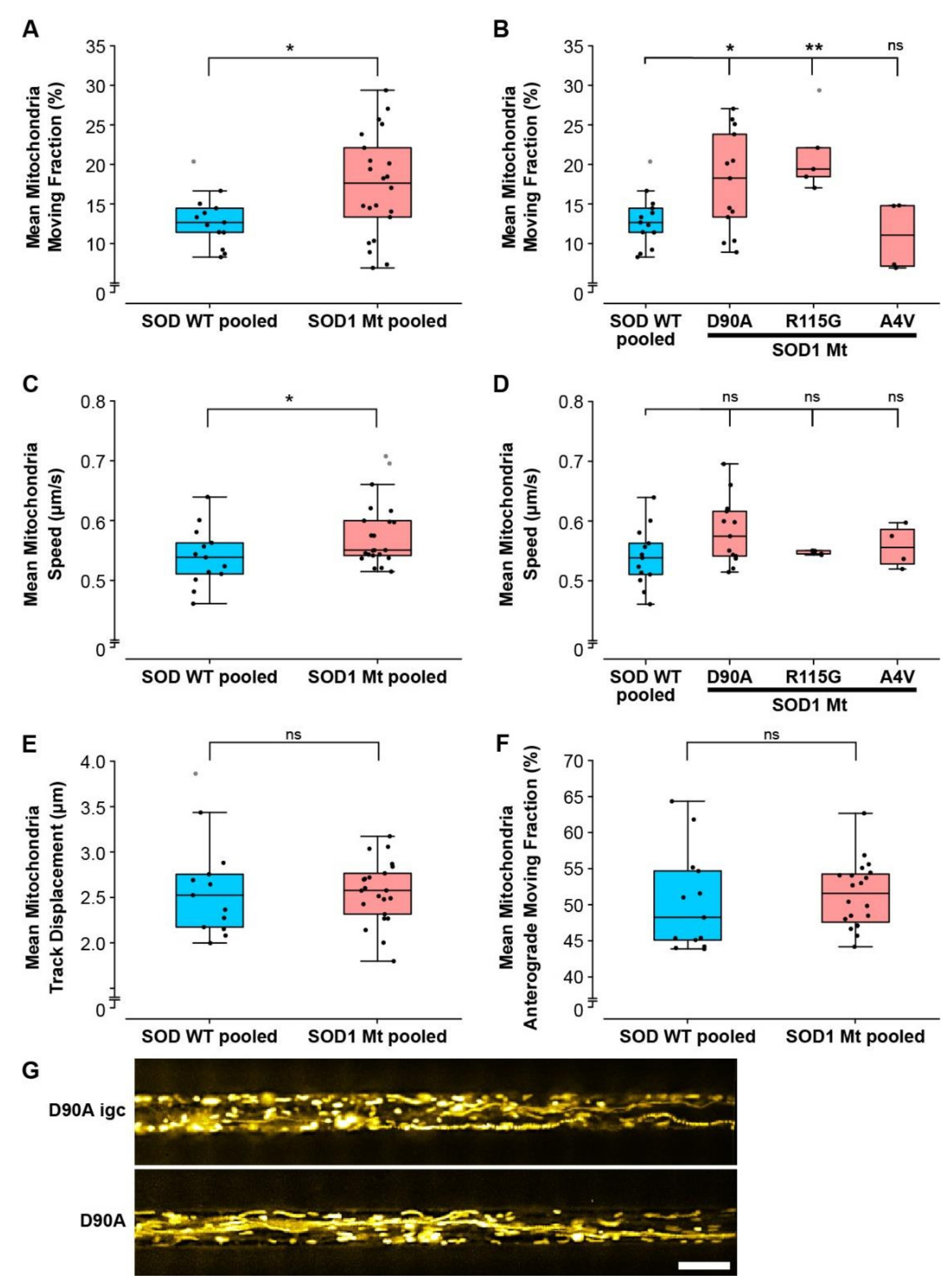
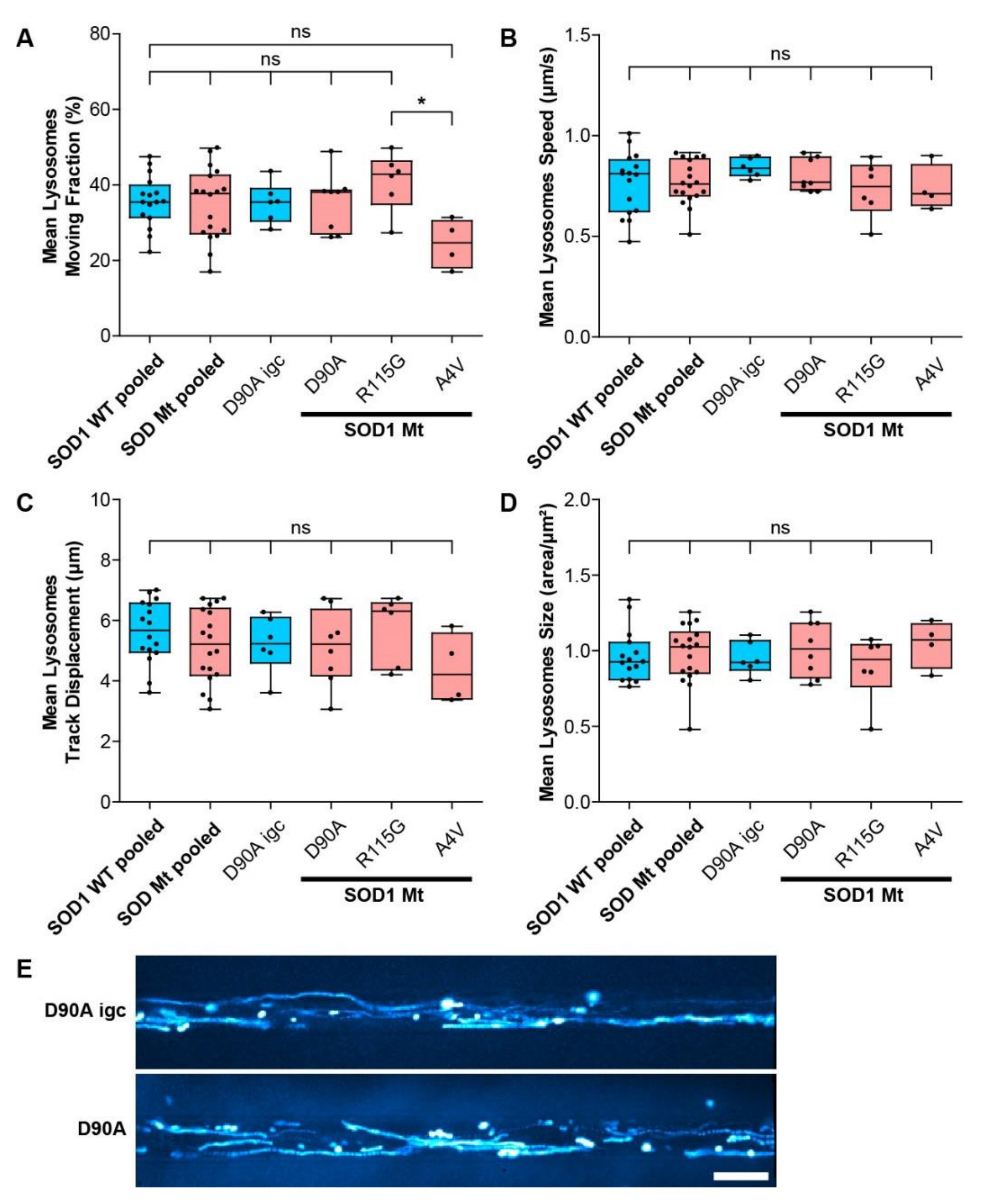
3.5. Mitochondrial but Not Lysosomal Trafficking Phenotypes in SOD1 MNs
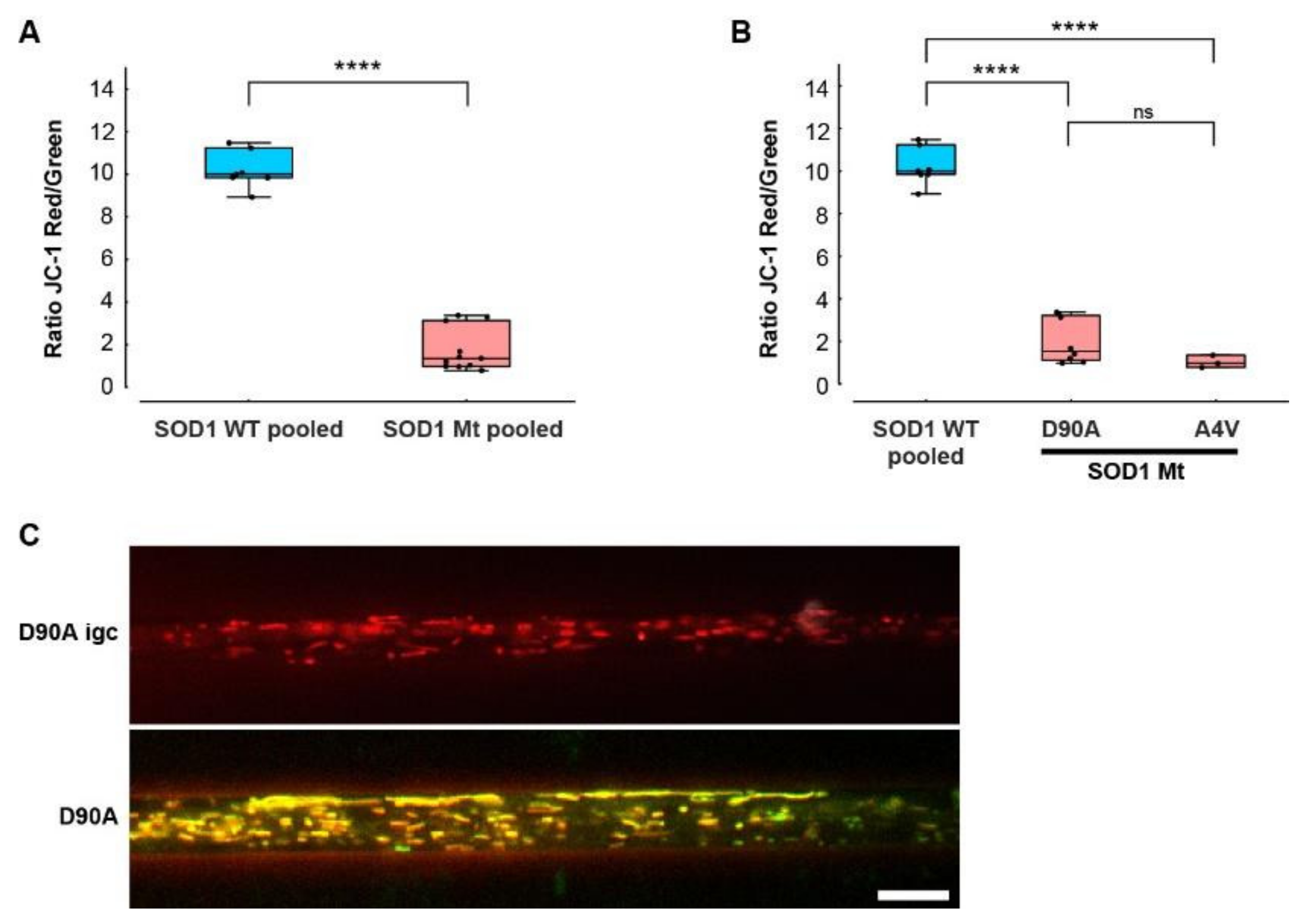
3.6. Hypopolarization of Mitochondria in SOD1 Mt MNs
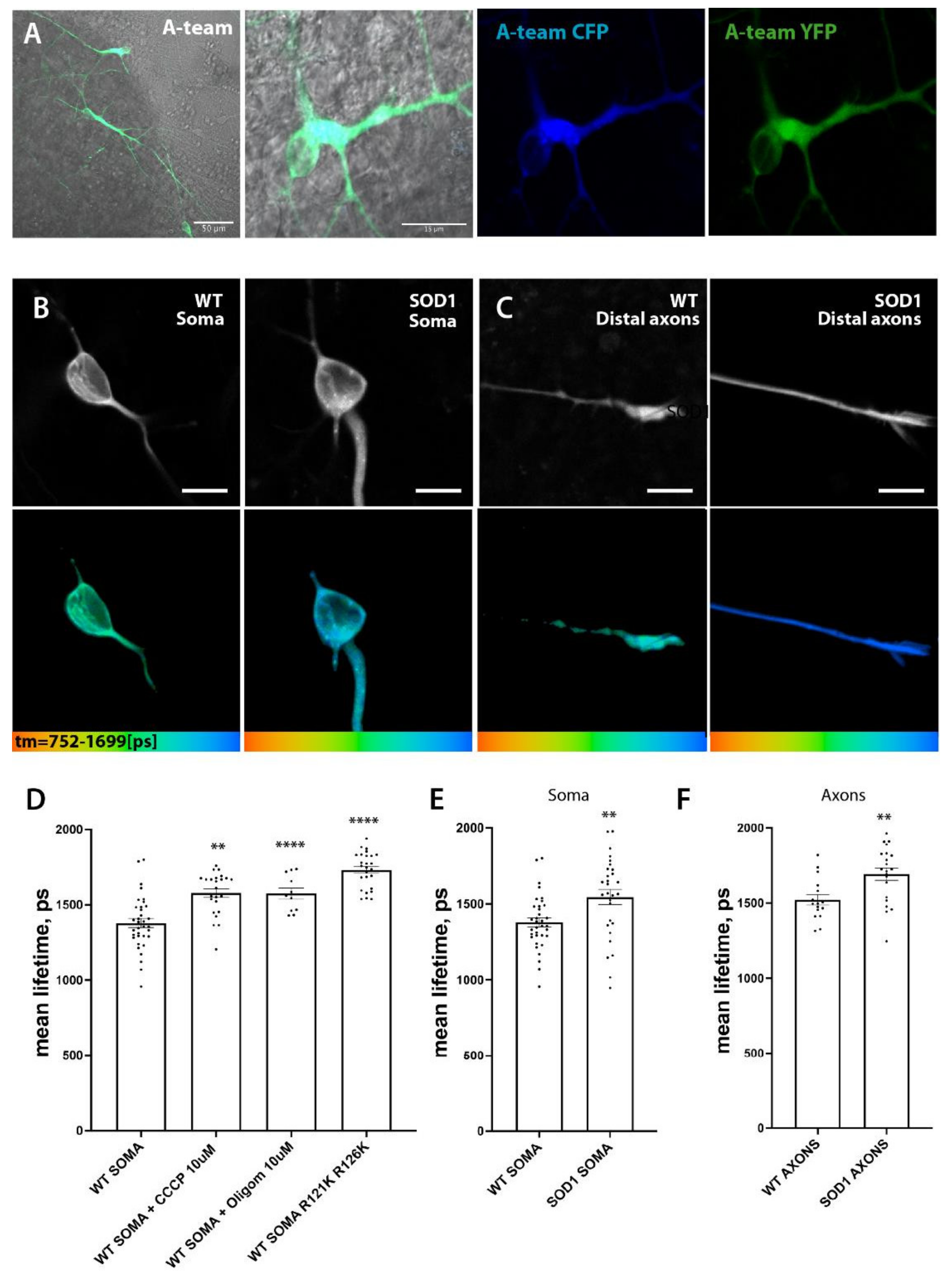
3.7. SOD1 Mt MN Have Reduced Levels of ATP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Nilsson, P.; Keranen, M.L.; Forsgren, L.; Hagglund, J.; Karlsborg, M.; Ronnevi, L.O.; Gredal, O.; Marklund, S.L. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia. Brain J. Neurol. 1997, 120 Pt 10, 1723–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, P.M.; Forsgren, L.; Binzer, M.; Nilsson, P.; Ala-Hurula, V.; Keranen, M.L.; Bergmark, L.; Saarinen, A.; Haltia, T.; Tarvainen, I.; et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain J. Neurol. 1996, 119 Pt 4, 1153–1172. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.M.; Nilsson, P.; Ala-Hurula, V.; Keranen, M.L.; Tarvainen, I.; Haltia, T.; Nilsson, L.; Binzer, M.; Forsgren, L.; Marklund, S.L. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat. Genet. 1995, 10, 61–66. [Google Scholar] [CrossRef]
- Bali, T.; Self, W.; Liu, J.; Siddique, T.; Wang, L.H.; Bird, T.D.; Ratti, E.; Atassi, N.; Boylan, K.B.; Glass, J.D.; et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J. Neurol. Neurosurg. Psychiatry 2017, 88, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Patrikios, J.S.; Marie, P. Contribution à l’étude des Formes Cliniques et de l’anatomie Pathologique de la Sclérose Latérale Amyotrophique: étude Clinique de 21 cas de Sclérose Latérale Amyotrophique dont 5 avec Autopsie. Diploma Thesis, Université de Paris, Paris, France, 1918. [Google Scholar]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2017, 710, 132933. [Google Scholar] [CrossRef]
- Indo, H.P.; Yen, H.C.; Nakanishi, I.; Matsumoto, K.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Said Ahmed, M.; Hung, W.Y.; Zu, J.S.; Hockberger, P.; Siddique, T. Increased reactive oxygen species in familial amyotrophic lateral sclerosis with mutations in SOD1. J. Neurol. Sci. 2000, 176, 88–94. [Google Scholar] [CrossRef]
- Rizzardini, M.; Mangolini, A.; Lupi, M.; Ubezio, P.; Bendotti, C.; Cantoni, L. Low levels of ALS-linked Cu/Zn superoxide dismutase increase the production of reactive oxygen species and cause mitochondrial damage and death in motor neuron-like cells. J. Neurol. Sci. 2005, 232, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D. Edaravone: A new drug approved for ALS. Cell 2017, 171, 725. [Google Scholar] [CrossRef] [PubMed]
- Keskin, I.; Birve, A.; Berdynski, M.; Hjertkvist, K.; Rofougaran, R.; Nilsson, T.K.; Glass, J.D.; Marklund, S.L.; Andersen, P.M. Comprehensive analysis to explain reduced or increased SOD1 enzymatic activity in ALS patients and their relatives. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Elpers, C.; Reunert, J.; McCormick, M.L.; Mohr, J.; Biskup, S.; Schwartz, O.; Rust, S.; Gruneberg, M.; Seelhofer, A.; et al. SOD1 deficiency: A novel syndrome distinct from amyotrophic lateral sclerosis. Brain J. Neurol. 2019, 142, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Nordstrom, U.; Tsiakas, K.; Johannsen, J.; Volk, A.E.; Bierhals, T.; Zetterstrom, P.; Marklund, S.L.; Hempel, M.; Santer, R. Phenotype in an Infant with SOD1 Homozygous Truncating Mutation. N. Engl. J. Med. 2019, 381, 486–488. [Google Scholar] [CrossRef]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef]
- Vande Velde, C.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Bel Hadj, S.; Zandona, A.; Julien, J.P.; Shah, S.B.; Cleveland, D.W. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS ONE 2011, 6, e22031. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Jeong, S.Y.; Lim, W.C.; Kim, S.; Park, Y.Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 2007, 282, 22977–22983. [Google Scholar] [CrossRef] [Green Version]
- Kaasik, A.; Safiulina, D.; Choubey, V.; Kuum, M.; Zharkovsky, A.; Veksler, V. Mitochondrial swelling impairs the transport of organelles in cerebellar granule neurons. J. Biol. Chem. 2007, 282, 32821–32826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siklos, L.; Engelhardt, J.; Harati, Y.; Smith, R.G.; Joo, F.; Appel, S.H. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotropic lateral sclerosis. Ann. Neurol. 1996, 39, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Gunther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Forman, M.S.; Trojanowski, J.Q.; Lee, V.M. Neurodegenerative diseases: A decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 2004, 10, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Takikawa, M.; Nakashima, K.; Hirano, A.; Cleveland, D.W.; Kusaka, H.; Shibata, N.; Kato, M.; Nakano, I.; Ohama, E. New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: Inclusions containing SOD1 in neurons and astrocytes. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 163–184. [Google Scholar] [CrossRef]
- Kato, S. Amyotrophic lateral sclerosis models and human neuropathology: Similarities and differences. Acta Neuropathol. 2008, 115, 97–114. [Google Scholar] [CrossRef]
- Ekhtiari Bidhendi, E.; Bergh, J.; Zetterstrom, P.; Forsberg, K.; Pakkenberg, B.; Andersen, P.M.; Marklund, S.L.; Brannstrom, T. Mutant superoxide dismutase aggregates from human spinal cord transmit amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 136, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Ayers, J.I.; Fromholt, S.E.; O’Neal, V.M.; Diamond, J.H.; Borchelt, D.R. Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol. 2016, 131, 103–114. [Google Scholar] [CrossRef]
- Tandan, R.; Robison, S.H.; Munzer, J.S.; Bradley, W.G. Deficient DNA repair in amyotrophic lateral sclerosis cells. J. Neurol. Sci. 1987, 79, 189–203. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naujock, M.; Stanslowsky, N.; Bufler, S.; Naumann, M.; Reinhardt, P.; Sterneckert, J.; Kefalakes, E.; Kassebaum, C.; Bursch, F.; Lojewski, X.; et al. 4-Aminopyridine Induced Activity Rescues Hypoexcitable Motor Neurons from Amyotrophic Lateral Sclerosis Patient-Derived Induced Pluripotent Stem Cells. Stem Cells 2016, 34, 1563–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, A.; Glaß, H.; Naumann, M.; Kreiter, N.; Japtok, J.; Sczech, R.; Hermann, A. High content organelle trafficking enables disease state profiling as powerful tool for disease modelling. Sci. Data 2018, 5, 180241. [Google Scholar] [CrossRef] [Green Version]
- Bursch, F.; Kalmbach, N.; Naujock, M.; Staege, S.; Eggenschwiler, R.; Abo-Rady, M.; Japtok, J.; Guo, W.; Hensel, N.; Reinhardt, P.; et al. Altered calcium dynamics and glutamate receptor properties in iPSC-derived motor neurons from ALS patients with C9orf72, FUS, SOD1 or TDP43 mutations. Hum. Mol. Genet. 2019, 28, 2835–2850. [Google Scholar] [CrossRef]
- Kreiter, N.; Pal, A.; Lojewski, X.; Corcia, P.; Naujock, M.; Reinhardt, P.; Sterneckert, J.; Petri, S.; Wegner, F.; Storch, A.; et al. Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol. Dis. 2018, 115, 167–181. [Google Scholar] [CrossRef]
- Japtok, J.; Lojewski, X.; Naumann, M.; Klingenstein, M.; Reinhardt, P.; Sterneckert, J.; Putz, S.; Demestre, M.; Boeckers, T.M.; Ludolph, A.C.; et al. Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol. Dis. 2015, 82, 420–429. [Google Scholar] [CrossRef]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schondorf, D.C.; Wagner, L.; Glatza, M.; Hoing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Glaß, H.; Pal, A.; Reinhardt, P.; Sterneckert, J.; Wegner, F.; Storch, A.; Hermann, A. Defective mitochondrial and lysosomal trafficking in chorea-acanthocytosis is independent of Src-kinase signaling. Mol. Cell. Neurosci. 2018, 92, 137–148. [Google Scholar] [CrossRef]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Hoing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef]
- Glaß, H.; Neumann, P.; Pal, A.; Reinhardt, P.; Storch, A.; Sterneckert, J.; Hermann, A. Combined Dendritic and Axonal Deterioration Are Responsible for Motoneuronopathy in Patient-Derived Neuronal Cell Models of Chorea-Acanthocytosis. Int. J. Mol. Sci. 2020, 21, 1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bounedjah, O.; Desforges, B.; Wu, T.D.; Pioche-Durieu, C.; Marco, S.; Hamon, L.; Curmi, P.A.; Guerquin-Kern, J.L.; Pietrement, O.; Pastre, D. Free mRNA in excess upon polysome dissociation is a scaffold for protein multimerization to form stress granules. Nucleic Acids Res. 2014, 42, 8678–8691. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef]
- Wallrabe, H.; Svindrych, Z.; Alam, S.R.; Siller, K.H.; Wang, T.; Kashatus, D.; Hu, S.; Periasamy, A. Segmented cell analyses to measure redox states of autofluorescent NAD(P)H, FAD & Trp in cancer cells by FLIM. Sci. Rep. 2018, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Keskin, I.; Forsgren, E.; Lehmann, M.; Andersen, P.M.; Brännström, T.; Lange, D.J.; Synofzik, M.; Nordström, U.; Zetterström, P.; Marklund, S.L.; et al. The molecular pathogenesis of superoxide dismutase 1-linked ALS is promoted by low oxygen tension. Acta Neuropathol. 2019, 138, 85–101. [Google Scholar] [CrossRef] [Green Version]
- Zetterström, P.; Andersen, P.M.; Brännström, T.; Marklund, S.L. Misfolded superoxide dismutase-1 in CSF from amyotrophic lateral sclerosis patients. J. Neurochem. 2011, 117, 91–99. [Google Scholar] [CrossRef]
- Marklund, S.L.; Andersen, P.M.; Forsgren, L.; Nilsson, P.; Ohlsson, P.I.; Wikander, G.; Oberg, A. Normal binding and reactivity of copper in mutant superoxide dismutase isolated from amyotrophic lateral sclerosis patients. J. Neurochem. 1997, 69, 675–681. [Google Scholar] [CrossRef] [Green Version]
- Bentmann, E.; Haass, C.; Dormann, D. Stress granules in neurodegeneration--lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J. 2013, 280, 4348–4370. [Google Scholar] [CrossRef]
- Lu, L.; Wang, S.; Zheng, L.; Li, X.; Suswam, E.A.; Zhang, X.; Wheeler, C.G.; Nabors, L.B.; Filippova, N.; King, P.H. Amyotrophic lateral sclerosis-linked mutant SOD1 sequesters Hu antigen R (HuR) and TIA-1-related protein (TIAR): Implications for impaired post-transcriptional regulation of vascular endothelial growth factor. J. Biol. Chem. 2009, 284, 33989–33998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal, J.; Kuang, L.; Barnett, K.R.; Zhu, B.Z.; Shissler, S.C.; Korotkov, K.V.; Hayward, L.J.; Kasarskis, E.J.; Zhu, H. ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 2016, 132, 563–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergh, J.; Zetterstrom, P.; Andersen, P.M.; Brannstrom, T.; Graffmo, K.S.; Jonsson, P.A.; Lang, L.; Danielsson, J.; Oliveberg, M.; Marklund, S.L. Structural and kinetic analysis of protein-aggregate strains in vivo using binary epitope mapping. Proc. Natl. Acad. Sci. USA 2015, 112, 4489–4494. [Google Scholar] [CrossRef] [Green Version]
- Zetterström, P.; Graffmo, K.S.; Andersen, P.M.; Brännström, T.; Marklund, S.L. Proteins that bind to misfolded mutant superoxide dismutase-1 in spinal cords from transgenic amyotrophic lateral sclerosis (ALS) model mice. J. Biol. Chem. 2011, 286, 20130–20136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandelin, E.; Nordlund, A.; Andersen, P.M.; Marklund, S.S.; Oliveberg, M. Amyotrophic lateral sclerosis-associated copper/zinc superoxide dismutase mutations preferentially reduce the repulsive charge of the proteins. J. Biol. Chem. 2007, 282, 21230–21236. [Google Scholar] [CrossRef] [Green Version]
- Keskin, I.; Forsgren, E.; Lange, D.J.; Weber, M.; Birve, A.; Synofzik, M.; Gilthorpe, J.D.; Andersen, P.M.; Marklund, S.L. Effects of Cellular Pathway Disturbances on Misfolded Superoxide Dismutase-1 in Fibroblasts Derived from ALS Patients. PLoS ONE 2016, 11, e0150133. [Google Scholar] [CrossRef]
- Dash, B.P.; Naumann, M.; Sterneckert, J.; Hermann, A. Genome Wide Analysis Points towards Subtype-Specific Diseases in Different Genetic Forms of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 6938. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [Green Version]
- Datta, R.; Gillette, A.; Stefely, M.; Skala, M.C. Recent innovations in fluorescence lifetime imaging microscopy for biology and medicine. J. Biomed. Opt. 2021, 26, 70603. [Google Scholar] [CrossRef]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Walker, C.; El-Khamisy, S.F. Perturbed autophagy and DNA repair converge to promote neurodegeneration in amyotrophic lateral sclerosis and dementia. Brain J. Neurol. 2018, 141, 1247–1262. [Google Scholar] [CrossRef]
- Penndorf, D.; Tadic, V.; Witte, O.W.; Grosskreutz, J.; Kretz, A. DNA strand breaks and TDP-43 mislocation are absent in the murine hSOD1G93A model of amyotrophic lateral sclerosis in vivo and in vitro. PLoS ONE 2017, 12, e0183684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bystrom, R.; Andersen, P.M.; Grobner, G.; Oliveberg, M. SOD1 mutations targeting surface hydrogen bonds promote amyotrophic lateral sclerosis without reducing apo-state stability. J. Biol. Chem. 2010, 285, 19544–19552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Johnson, J.L.; Agar, N.Y.; Agar, J.N. Protein aggregation and protein instability govern familial amyotrophic lateral sclerosis patient survival. PLoS Biol. 2008, 6, e170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef]
- Redler, R.L.; Dokholyan, N.V. The complex molecular biology of amyotrophic lateral sclerosis (ALS). Prog. Mol. Biol. Transl. Sci. 2012, 107, 215–262. [Google Scholar] [CrossRef] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Models Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Beck, M.V.; Griffith, J.D.; Deshmukh, M.; Dokholyan, N.V. Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 4661–4665. [Google Scholar] [CrossRef] [Green Version]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Ait-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; De Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SOD1 Line | Sex | Age at Biopsy | Mutation | Primarily Characterized in | |
|---|---|---|---|---|---|
| Wt | Ctrl1 | Female | 48 | - | [39] |
| Wt | Ctrl2 | Female | 43 | - | [40] |
| Wt | Ctrl3 | Female | 49 | - | [40] |
| Wt | Ctrl4 | Male | 34 | - | [41] |
| IGC | SOD1 D90A igc | Female | 46 | - | [37] |
| Mt | SOD1 D90A | Female | 46 | D90A | [35] |
| Mt | SOD1 R115G | Male | 59 | R115G | [35] |
| Mt | SOD1 A4V | Female | 73 | A4V |
| Human Spinal Motor Neurons, DIV 30 (Endogenous SOD1 Expression Level) | |||
|---|---|---|---|
| R115G | D90A | 4AV | |
| Mitochondrial elongation (Figure 4D–F) | ↑ | ↑ | - |
| Mitochondrial moving fraction (Figure 5B) | ↑ | ↑ | - |
| Mitochondrial inner membrane potential (Figure 7B) | n.d. | ↓ | ↓ |
| Lysosomal size (Figure 6D) | - | - | - |
| Lysosomal moving fraction (Figure 6A) | - | - | ↓ |
| DNA damage (Figure 3) | |||
| Mock (DMSO) | - | - | - |
| Etoposide | - | - | - |
| X-ray irradiation | - | - | - |
| Total SOD1 in SG (Figure 1A,D *) | |||
| Untreated ** | - | - | - |
| Arsenite | - | - | - |
| Heat stress | - | - | - |
| Ver + Puro | |||
| Misfolded SOD1 in SG (Figure 1A,C *) | |||
| Untreated ** | - | - | - |
| Arsenite | - | - | - |
| Heat stress | - | - | - |
| Ver & Pur | - | - | - |
| Total SOD1 in aggresome (Figure 1B) | |||
| Mock (DMSO) | - | - | - |
| 0 | - | - | - |
| Misfolded SOD1 in aggresome (Figure 1B) | |||
| Mock (DMSO) | - | - | - |
| 0 | - | - | - |
| Soluble total SOD1 (ELISA, Figure 2A) | |||
| Untreated | ↓ | ↓ | ↓ |
| Additional Ver & Pur | - | - | - |
| Additional MG132 | - | - | - |
| Soluble misfolded SOD1 (ELISA, Figure 2B) | |||
| Untreated | ↑ | - | ↑ |
| Additional Ver & Pur | - | - | - |
| Additional MG132 | ↑ | - | ↑ |
| Detergent-resistant SOD1 (WB, Figure 2C,D) | |||
| Untreated | - | - | - |
| Additional Ver & Pur | - | - | - |
| Additional MG132 | ↑ | - | ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Günther, R.; Pal, A.; Williams, C.; Zimyanin, V.L.; Liehr, M.; von Neubeck, C.; Krause, M.; Parab, M.G.; Petri, S.; Kalmbach, N.; et al. Alteration of Mitochondrial Integrity as Upstream Event in the Pathophysiology of SOD1-ALS. Cells 2022, 11, 1246. https://doi.org/10.3390/cells11071246
Günther R, Pal A, Williams C, Zimyanin VL, Liehr M, von Neubeck C, Krause M, Parab MG, Petri S, Kalmbach N, et al. Alteration of Mitochondrial Integrity as Upstream Event in the Pathophysiology of SOD1-ALS. Cells. 2022; 11(7):1246. https://doi.org/10.3390/cells11071246
Chicago/Turabian StyleGünther, René, Arun Pal, Chloe Williams, Vitaly L. Zimyanin, Maria Liehr, Cläre von Neubeck, Mechthild Krause, Mrudula G. Parab, Susanne Petri, Norman Kalmbach, and et al. 2022. "Alteration of Mitochondrial Integrity as Upstream Event in the Pathophysiology of SOD1-ALS" Cells 11, no. 7: 1246. https://doi.org/10.3390/cells11071246
APA StyleGünther, R., Pal, A., Williams, C., Zimyanin, V. L., Liehr, M., von Neubeck, C., Krause, M., Parab, M. G., Petri, S., Kalmbach, N., Marklund, S. L., Sterneckert, J., Munch Andersen, P., Wegner, F., Gilthorpe, J. D., & Hermann, A. (2022). Alteration of Mitochondrial Integrity as Upstream Event in the Pathophysiology of SOD1-ALS. Cells, 11(7), 1246. https://doi.org/10.3390/cells11071246