Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease with dysfunction of memory, language and thinking. More than 55 million people were diagnosed with AD or other dementia around the world in 2020. The pathology of AD is still unclear and there are no applicable therapies for AD. MicroRNAs (miRNAs) play key roles in AD pathology and have great potential for the diagnosis and treatment of AD. Extracellular vesicles (EVs) widely exist in body fluids such as blood and cerebrospinal fluid (CSF) and contain miRNAs that are involved in cell-to-cell communication. We summarized the dysregulated miRNAs in EVs derived from the different body fluids of AD patients, as well as their potential function and application in AD. We also compared these dysregulated miRNAs in EVs to those in the brain tissues of AD patients aiming to provide a comprehensive view of miRNAs in AD. After careful comparisons, we found that miR-125b-5p and miR-132-3p were upregulated and downregulated in several different brain tissues of AD and EVs of AD, respectively, suggesting their value in AD diagnosis based on EV miRNAs. Furthermore, miR-9-5p was dysregulated in EVs and different brain tissues of AD patients and had also been tested as a potential therapy for AD in mice and human cell models, suggesting that miR-9-5p could be used to design new therapies for AD.
1. Introduction
As a degenerative neurological disease and the most common form of dementia, Alzheimer’s disease (AD) is marked with dysfunction of memory, language and thinking [1]. Based on the data of the World Health Organization in 2020, 55 million people were diagnosed with AD or other dementia, which was the seventh-largest cause of death [2]. Amyloid plaques and neurofibrillary tangles (NFTs) are the pathologic characteristics of AD and related to the most generally accepted hypotheses, i.e., the amyloid cascade hypothesis and the Tau hypothesis [3]. The amyloid cascade hypothesis posits that the overexpression and dysfunction of APP cause the overproduction of -amyloid (A) protein, combined with the dysfunction of A clearance resulting in misfolded insoluble protein and amyloid plaque formation, which are the major cause of neuronal dysfunction and death [4,5]. A recent study supplemented this hypothesis. The cellular responses to A in different types of brain cells determined whether the brain’s function could be performed normally [6]. Tau proteins are involved in microtubule formation, which is related to cell development, cell polarity and cellular transport [7]. In the Tau hypothesis of AD, Tau shows hyperphosphorylation and aggregation, leading to the breakdown of microtubule and cellular transport, aggregation of paired helical filaments and degeneration of neurons [8]. Thus, Tau protein is considered to play a key role in AD pathology. Moreover, many studies have suggested that A and Tau are acting collaboratively in AD pathology [9]. Based on their important roles in AD pathology, Tau and A are also important targets in AD treatment [10]. A review summarizing the treatment strategies for AD indicated that therapies targeting A did not yield the results expected in clinical trials [11]. However, therapies targeting Tau are in the early stage of development and have a great therapeutic potential in the future [11].
In addition to the amyloid cascade and Tau hypotheses of AD, three other hypotheses have been reported. Firstly, ApoE protein, an apolipoprotein, is thought to be a strong genetic risk factor for late-onset AD (LOAD) [12]. A longitudinal study of individuals from families with autosomal dominant AD suggested that the ApoE allele may promote amyloid deposition [13]. ApoE4, a variant protein product of the APOE gene, induced more Tau hyperphosphorylation and GABAergic neuron degeneration compared to ApoE3 [14]. In mice, the expression of ApoE4 also leads to the blood–brain barrier breakdown, promoting neuronal dysfunction [15]. Secondly, Efthymiou and Goate [16] propose that several genes, i.e., CR1, SPI1, the MS4As, TREM2, ABCA7, CD33 and INPP5D, are expressed by microglia and contribute to AD in a non-A-dependent fashion. In particular, Gratuze et al. [17] summarized the role of TREM2 in AD development. TREM2 is involved in the innate immune system and affects Tau and amyloid pathologies [17]. Based on the findings of the dysregulated immune response in AD development, a novel model for amyloidogenesis, the antimicrobial protection hypothesis, was proposed. A deposition is thought to be an early immune response in this hypothesis. However, the high level of A deposition induced microglial activation, which, combined with amyloid plaques and neurofibrillary tangle pathology, mediated neurodegeneration [18]. Thirdly, several studies uncovered that sleep impairment could play an important role in AD development. A study of isotope tracing A peptides in human cerebrospinal fluid (CSF) showed increased A production in sleep-deprived participants [19]. Another study showed a similar result for Tau production in both humans and mice [20].
MiRNAs are small non-coding RNAs, usually with 22 nucleotides [21]. MiRNAs are transcribed in the nucleus, and their primary transcripts are called pri-miRNAs [22]. Pri-miRNA is processed into a hairpin structure through the RNase III enzyme Drosha and other cofactors and then processed into a shorter hairpin, called pre-miRNA (precursor miRNA), under the participation of several proteins [23,24]. Then pre-miRNA is transported to the cytoplasm and the loop is cleaved to produce a double-stranded RNA called an miRNA:miRNA* duplex [21]. One strand of the miRNA:miRNA* duplex is loaded with AGO protein to form the silencing complex. Meanwhile, the other strand is discarded [25,26,27,28]. MiRNAs usually suppress the expression of their targets by complementary binding to the target mRNAs [29,30]. MiRNAs play key roles in many biological or pathological processes, including metabolism, proliferation, differentiation, development, apoptotic cell death, viral infection and molecular mechanisms of diseases [29].
Extracellular vesicles (EVs) are membrane-enclosed vesicles secreted by cells [31,32]. In consideration of differences in their size, biogenesis, cellular origin and biophysical properties, EVs can be divided into exosomes, microvesicles and apoptotic bodies [33]. Exosomes (30–100 nm in diameter) and microvesicles (100 nm to 1000 nm in diameter) are released from healthy living cells [34], while apoptotic bodies (800–5000 nm in diameter) are produced during programmed cell death [35]. A study of distinct RNA in extracellular vesicles from three different kinds of cell lines including a human erythroleukemia cell line (TF-1), a human mast cell line (HMC-1) and a mouse microglia cell line (BV-2) indicated that there was a little or no RNA in microvesicles except for those collected from TF-1 cells [35]. Meanwhile, Budnik et al. [34] thought that apoptotic bodies might not be involved in transcellular communication in the nervous system, since they were engulfed by phagocytic cells rapidly after being released. Only exosomes are involved in cell-to-cell communication in the central nervous system [36]. Exosomes from neurons regulate the integrity of the brain vascular system [37], and exosomes from astrocytes may impact neuronal morphology and function [38].
EVs contain different kinds of molecules, such as nucleic acids and proteins [39]. The miRNAs of exosomes keep similarities with the parent cells in cancer [40] and the proteins of EVs consist of membrane proteins and the lumen of EVs [39]. It is reasonable to expect that pathological miRNAs in EVs might be detected earlier and be more notable than those of proteins in EVs. Indeed, it has been reported that miRNAs in exosomes are dysregulated in AD patients when compared to healthy people [36,41]. To provide a comprehensive view of miRNAs in EVs of AD and their relations to AD, this review summarizes the miRNAs of extracellular vesicles in different samples of AD patients and their potential roles in AD pathology, diagnosis and treatment.
2. Relevant miRNAs in AD
Several studies revealed that miRNAs were involved in AD pathology, usually through targeting AD-related genes or signalling pathways, as summarized in Table 1. As a brain-rich miRNA, miR-9-5p has been investigated in many studies. As listed in Table 1, miR-9-5p targets several AD-related genes including BACE1, SIRT1, CAMKK2 [42] and TGFBIp [43]. A is formed during the cleavage of the amyloid precursor protein (APP) by BACE1 [44]. CAMKK2 has been inhibited by miR-9-5p in AD resulting in the aggravation of A-induced synaptotoxic impairment [45]. Interestingly, SIRT1 was studied as a protective factor in AD by inhibiting the accumulation of A and neuroinflammation [46]. Although the level of miR-9-5p was downregulated in AD brains and EVs from CSF [47,48,49], the expression of SIRT1 was decreased in AD patients [46]. This suggests that the downregulated miR-9-5p might be a kind of positive response in AD development, but the other regulators suppressed the production of SIRT1, except for miR-9-5p. Interestingly, Sethi and Lukiw found that miR-9-5p was upregulated in the temporal lobe neocortex of AD brains [50]. The mutation of TGFBIp accelerated A aggregation [51]. All these targets of miR-9-5p are related to the A accumulation in AD. Furthermore, miR-9-5p also decreased the Tau degradation by targeting UBE4B [52].
In addition to the contribution to the A accumulation and Tau hyperphosphorylation, miR-9-5p is also related to apoptosis [53]. Zhang et al. [54] reported that apoptosis might be associated with neuron loss in AD since A could induce neuronal apoptosis. In the forebrain, miR-9-5p regulates the expression of hairy1, which mediates neurogenesis, apoptosis and proliferation [53] (Table 1). Moreover, miR-9-5p was downregulated in AD cell models, which resulted in an increased expression of GSK-3[55]. Moreover, the overexpression of GSK-3 contributed to cell apoptosis, oxidative stress and mitochondrial dysfunction [55]. Therefore, miR-9-5p is also involved in AD development through other ways, such as apoptosis and oxidative stress.
Other miRNAs play important roles in AD pathology too. As listed in Table 1, these miRNAs can be generally divided into two categories. The first category includes miRNAs related to A accumulation. For example, miR-17-5p, miR-20a and miR-106b repressed the expression of APP [56] (as listed in Table 1). miR-153 [57] and miR-193b [58] also inhibited the level of APP (as listed in Table 1). miR-124 was involved in the splicing of APP mRNA [59] (see Table 1). miR-144 and miR-451 were reported as inhibitors of ADAM10 [60] (as listed in Table 1). The ADAM10 protein inhibits the generation of A[60]. miR-107 was downregulated in the brains of AD patients compared to the HC and played a positive role in the generation of A by decreasing the inhibition of BACE1 [61] (as listed in Table 1). Similarly, miR-9-5p, miR-29a, miR-29b-1 [47], miR-29c [62], miR-339-5p [63] and miR-485-5p [64] are also BACE1 inhibitors (as listed in Table 1).
The second category includes miRNAs involved in Tau hyperphosphorylation (see Table 1). For example, miR-15a might be a potential regulator of ERK1 [65]. miR-125b-5p upregulated the expression of Erk1/2, an activator of kinase cdk5, and was involved in Tau phosphorylation in AD development [66]. miR-125b-5p also decreases the production of phosphatases, including DUSP6 and PPP1CA [66] (see Table 1). miR-26b was up-regulated in AD and might induce cell apoptosis, aberrant cell cycle entry and increasing Tau-phosphorylation [67] (see Table 1). It was reported that miR-219 directly regulated the synthesis of Tau protein by binding the 3’-UTR of the Tau mRNA [68] (see Table 1).

Table 1.
The miRNAs with relevant functions in AD.
Table 1.
The miRNAs with relevant functions in AD.
| MicroRNAs | Pathological Roles | Reference |
|---|---|---|
| let-7e-5p | Let-7e in small neuron-derived extracellular vesicles can trigger inflammatory responses in microglia by increasing inflammatory cytokines, such as interleukin 6. | [69] |
| miR-9-5p | Accompanied by STUB1, decreases the clearance of Tau proteins by targeting UBE4B. | [52] |
| Alleviated A-induced synaptotoxic impairment by inhibiting CAMKK2. | [45] | |
| Related to the amyloid cascade hypothesis and memory loss by targeting BACE1, SIRT1 and CAMKK2. | [42] | |
| Repressed BACE1 and TGFBIp mRNA. | [43] | |
| [47] | ||
| Mediated neurogenesis, apoptosis and proliferation by regulating the expression of hairy1. | [53] | |
| Induced oxidative stress, mitochondrial dysfunction and cell apoptosis through targeting GSK-3. | [55] | |
| As the target of miR-9-5p, SIRT1 suppressed A accumulation and attenuated neuroinflammation. | [46] | |
| miR-15a | Might be a potential regulateor of ERK1, which is a candidate of kinase in Tau phosphorylation. | [65] |
| miR-17-5p, miR-20a, miR-106b | Repressed APP expression in neuronal cell lines. | [56] |
| miR-26b | Might induce aberrant cell cycle entry, increasing Tau-phosphorylation and cell apoptosis. | [67] |
| miR-29a, miR-29b-1 | Repressed BACE1 expression in vitro. | [47] |
| miR-29c | Repressed the expression of BACE1. | [62] |
| miR-34a | Related to the energy metabolism, synaptic plasticity and resting-state network activity by targeting several genes. | [70] |
| miR-107 | Contributed to the A generation by targeting BACE1. | [61] |
| miR-124 | Involved in APP mRNA alternative splicing. | [59] |
| miR-125b-5p | Might play a pro-inflammatory role. | [71] |
| Contributed to Tau hyperphosphorylation by upregulating the expression and activity of kinases, such as Erk1/2 and p35, an activator of kinase cdk5, and downregulating phosphatase production, including DUSP6 and PPP1CA. | [66] | |
| miR-132-3p, miR-212 | Inhibited apoptosis by regulating PTEN, FOXO3a and P300 and against oxidative stress. | [72] |
| miR-146a-5p | Contributed to the inflammatory response in AD by targeting CFH. | [73] |
| Mediated downregulation of the IRAK-1. It combined with NF-B induced upregulation of IRAK-2, resulting in the inflammatory response. | [74] | |
| miR-153 | Mediated the miRNA-induced suppression of APP. | [57] |
| miR-193b | Repressed the expression of APP. | [58] |
| miR-219 | Directly regulated the production of Tau protein by mediating the miRNA-induced suppression of Tau protein. | [68] |
| miR-339-5p | Repressed the expression of BACE1. | [63] |
| miR-144, miR-451 | Repressed the expression of ADAM10. | [60] |
| miR-485-5p | Mediated the miRNA-induced suppression of BACE1. | [64] |
Some miRNAs have other contributions to AD development. As listed in Table 1, miR-125b-5p (miR-125b) was upregulated in the CSF of AD [71]. miR-125b-5p was also specifically over-expressed in microglia compared to other immune cell types, suggesting its role in the innate immune response [71]. Similar to miR-125b-5p, let-7e-5p also triggered an increase in pro-inflammatory cytokines (such as interleukin 6) and inflammation in microglia [69]. miR-146a-5p was upregulated in AD brains and contributed to the inflammatory pathology in AD by targeting complement factor H (CFH) [73] and IRAK-1 [74] (see Table 1). The target genes of miR-34a were related to energy metabolism, synaptic plasticity and resting-state network activity [70], suggesting that miR-34a might be an important regulator in AD development (see Table 1). miR-132-3p (miR-132) and miR-212 inhibited apoptosis by regulating the expression of PTEN, FOXO3a and P300 and acted against oxidative stress in vitro [72] (as listed in Table 1).
3. Dysregulated miRNAs in Brains of AD Patients
In order to know whether there are common dysregulated miRNAs in the EVs and brain tissues of AD patients, we summarized the dysregulated miRNAs in different brain regions of AD patients in Table 2. In a study of the entorhinal cortex (EC) and superior temporal gyrus (STG), three miRNAs (miR-129-5p, miR-132-5p, miR-138-5p) were downregulated and miR-195-5p was upregulated in 99 AD brain samples compared to 91 healthy controls (HC) [75] (see Table 2). In another study of an unknown brain region, three upregulated miRNAs and 13 downregulated miRNAs were found in five AD brains compared to the five normal controls [47] (as listed in Table 2).
The Braak stage, also called the Tau Braak stage, reflects the changes in Tau hyperphosphorylation in AD development. It can be divided into six stages [76]. The Braak stage is usually used for the stage classification of AD with different criteria in different studies [48,61,63,66,67,70,77]. Since the disease changes happening in Braak III correspond to the early symptoms of AD and are defined as mild cognitive impairment (MCI) [78], some studies would regard the Braak stages I and II as a control group [48,66,67,70,77]. The Braak stages IV–VI are classified as a late stage or severe stage of AD [48,66,67,70,77]. The Braak stage was often used in AD studies and we summarized the information about the Braak stage in Table 2 when available.
miR-29a was downregulated in late-stage AD patients (Braak IV or VI) [77] (as listed in Table 2). miR-125b-5p was upregulated in AD (Braak V and VI) brains and miR-29a and miR-29b were downregulated compared to HC (Braak I and II) [66]. miR-26b was upregulated in both very early AD (Braak III) and severe AD (Braak VI) patients [67] (see Table 2). miR-34a and miR-146a-5p were upregulated in the temporal cortex of both mild (Braak III) and severe AD (Braak VI) patients [70] (as listed in Table 2). In a study by Cogswell et al. [48], Braak I was regarded as the normal control, and Braak III–VI were regarded as AD. They did not conduct the early- and late-stage classification of AD. In this study, 12 miRNAs were upregulated and six miRNAs were downregulated in AD brains [48] (see Table 2). Different from the previous studies, Long et al. took Braak I–III as the control group [63]. They found that miR-339-5p was downregulated in AD brains when compared to HC [63] (see Table 2). Wang et al. [61] separated all cases into four groups in their research, including nondemented with no/negligible AD-type pathology, nondemented with incipient AD pathology, MCI and AD. Among the four groups, miR-107 was downregulated in AD and MCI compared to nondemented with no/negligible AD-type pathology [61] (see Table 2).
We also found that the Braak stage combined with other pathological features were used to classify the stages of AD. As listed in Table 2, three dysregulated miRNAs (one upregulated miRNA and two downregulated miRNAs) were found in a 20-individual study with clear criteria of samples [79]. The authors combined the neurofibrillary tangles and neuritic plaques with clinical cognition evaluations to categorize the AD and HC groups [79]. Lau et al. [80] found 35 miRNAs dysregulated in the hippocampus (HP) and 41 miRNAs in the prefrontal cortex (PC) (see Table 2). For the HP cohort, Lau et al. [80] gave detailed information of the involved individuals but did not have a clear criterion for AD patients and normal controls. For the PC cohort, Lau et al. [80] found that 41 miRNAs were expressed differently in different Braak stages including the controls (Braak 0), early stages (Braak I and II), mid stages (Braak III and IV) and late stages (Braak V and VI). Santa-Maria et al. [68] found that miR-219 was downregulated in AD (Braak V and VI) and severe primary age-related tauopathy (Braak III and IV) compared to HC (see Table 2). Some studies did not use the Tau Braak stages in AD description. Lukiw et al. [73] used the criteria of the center to establish a registry for Alzheimer’s disease/National Institutes of Health to distinguish the AD group and HC group (as listed in Table 2). They found that miR-146a-5p was upregulated in AD brains when compared to HC [73].
Table 2.
Dysregulated miRNAs in brain tissues of AD patients.
Table 2.
Dysregulated miRNAs in brain tissues of AD patients.
| Sample (AD/Control) | Brain Region | Upregulated miRNAs | Downregulated miRNAs | Braak Stage | Reference |
|---|---|---|---|---|---|
| 190 (99/91) | EC and STG | miR-195-5p | miR-129-5p, miR-132-5p, miR-138-5p | / | [75] |
| 10 (5/5) | / | miR-197, miR-320, miR-511 | let-7i, miR-9-5p, miR-15a, miR-19b, miR-22, miR-26b, miR-29a, miR-29b-1, miR-93, miR-101, miR-106b, miR-181c, miR-210, miR-363 | / | [47] |
| 11 (7/4) | FC | / | miR-29a | Braak IV or VI | [77] |
| 15 (10/5) | FC | miR-125b-5p | miR-29a, miR-29b | Braak V and VI | [66] |
| 28 (20/8) | TC | miR-26b | / | Braak III and VI | [67] |
| 19 (10/9) | TC | miR-34a, miR-146a-5p | / | Braak III and VI | [70] |
| 27 (20/7) | HP; MFG; CB | miR-27a, miR-27b, miR-30e-5p, miR-34a, miR-92, miR-100, miR-125b-5p, miR-145, miR-148a, miR-381, miR-422a, miR-423 | miR-9-5p, miR-210, miR-212, miR-132-3p, miR-146b, miR-425 | Braak III-VI | [48] |
| 25 (20/5) | FC | / | miR-339-5p | Braak IV-VI | [63] |
| 22 (12/11) | TC | / | miR-107 | Braak III-V | [61] |
| 20 (11/9) | FC | miR-32-5p | miR-182-5p, miR-1304-5p | / | [79] |
| 64 (41/23) | HP | let-7f-5p, let-7i-5p, miR-23a-3p, miR-27a-3p, miR-92b-3p, miR-142-3p, miR-150-5p, miR-195-5p, miR-199a-3p, miR-199b-3p, miR-200a-3p, miR-223-3p, miR-362-3p, miR-363-3p, miR-455-5p | miR-124-3p, miR-127-3p, miR-128, miR-129-2-3p, miR-129-5p, miR-132-3p, miR-136-5p, miR-138-5p, miR-219-2-3p, miR-329, miR-370, miR-409-5p, miR-410, miR-425-5p, miR-433, miR-487a, miR-487b, miR-495-3p, miR-543, miR-769-5p | / | [80] |
| 41 (34/7) | PC | miR-27a-3p, miR-92b-3p, miR-190b, miR-200a-3p, miR-214-3p, miR-424-5p, miR-517c-3p, miR-519a-3p, miR-744-5p, miR-874, miR-1260a, miR-1275 | miR-127-5p, miR-129-2-3p, miR-10b-5p, miR-129-5p, miR-132-3p, miR-133b, miR-135b-5p, miR-136-5p, miR-210, miR-219-1-3p, miR-337-3p, miR-370, miR-382-5p, miR-409-5p, miR-421, miR-431-5p, miR-485-5p, miR-487a, miR-491-3p, miR-496, miR-508-3p, miR-520a-3p, miR-548j, miR-551b-3p, miR-633, miR-758-3p, miR-1178-3p, miR-1179, miR-1321 | Braak I-VI | |
| 27 (7/20) | / | / | miR-219 | Braak I-VI | [68] |
| 46 (23/23) | HP and TL | miR-146a-5p | / | / | [73] |
| 22 (11/11) | ATC | / | miR-124, miR-107 | / | [59] |
| 66 (36/30) | STLN | miR-146a-5p | / | / | [74] |
| 42 (21/21) | HP and CB | miR-132-3p, miR-152 | miR-138, miR-139-5p, miR-149, miR-191, miR-204, miR-328, miR-370 | / | [81] |
| 8 (4/4) | PLC | miR-134, miR-185, miR-188, miR-320, miR-382, miR-432, miR-486, miR-572, miR-575, miR-601, miR-617, miR-671, miR-765 | miR-15a, miR-20b, miR-29b, miR-29c, miR-30e-5p, miR-95, miR-101, miR-130a, miR-148b, miR-181c, miR-368, miR-374, miR-376a, miR-494, miR-582, miR-598 | / | [82] |
| 18 (8/10) | / | / | miR-15a | / | [65] |
| 60 (31/29) | / | / | miR-29c | / | [62] |
| 30 (19/11) | ATC or CB | / | miR-106b | / | [56] |
| 46 (30/16) | TC | / | miR-132-3p, miR-212 | / | [72] |
| 21 (10/11) | HP | miR-16, miR-34c, miR-146a-5p | / | Braak III and IV | [83] |
| 21 (10/11) | HP | / | miR-16, miR-107, miR-128a, miR-146a-5p | Braak VI | |
| 20 (10/10) | FC | / | miR-153 | Braak III-VI | [57] |
| 10–24 (5–12/5–12) | PL, CB, HP, EC | / | miR-485-5p | / | [64] |
| 12 (6/6) | TLN | miR-9-5p, miR-125b-5p, miR-146a-5p | / | / | [50] |
| 16 (8/8) | STG and MTG | / | miR-100, miR-132-3p | / | [84] |
* The abbreviations of brain regions are ATC: anterior temporal cortex; CB: cerebellum; EC: entorhinal cortex; FC: frontal cortex; HP: hippocampus; MTG: middle temporal gyrus; PL: parietal lobe; STG: superior temporal gyrus; STLN: superior temporal lobe neocortex; TL: temporal lobe; and TLN: temporal lobe neocortex.
Smith et al. [59] found that miR-124 and miR-107 were downregulated in AD brains with an unclear description of AD group (see Table 2). Cui et al. [74] found that miR-146a-5p was upregulated in AD brains (as listed in Table 2). A 42-brain-sample study found two upregulated miRNAs and seven downregulated miRNAs in AD compared to HC [81] (see Table 2). Although the Braak stage information was contained in the description of patients, a clear criterion for the AD and HC groups was not given in this study [81]. Some studies focused more on the function of dysregulated miRNAs and ignored the specific description of AD individuals. For example, Lei et al. [62] found that miR-29c was downregulated in AD brains (see Table 2). Nunez-Iglesias et al. [82] found 29 dysregulated miRNAs from the parietal lobe cortex (PLC) of AD patients. They focused more on the relationship of mRNA and miRNA [82] (see Table 2). miR-15a [65], miR-29c [62], miR-106b [56], miR-132-3p and miR-212 [72] were downregulated in AD brains and the authors paid more attention to the function of dysregulated miRNAs as well (as listed in Table 2).
The expression levels of miRNAs were altered not only at different stages of AD development but also in different brain regions. miR-16, miR-34c and miR-146a-5p were upregulated in early AD (Braak III and IV), while miR-16, miR-107, miR-128a and miR-146a-5p were downregulated in late AD (Braak VI) compared to HC (Braak I and II) [83] (see Table 2). The expression level of miR-153 did not have a significant difference between HC and Braak I and II, Braak III and IV or Braak V and VI [57] (see Table 2). However, miR-153 was significantly downregulated in Braak III–VI compared with HC and Braak I and II [57]. A similar condition also happened in a different brain region. Faghihi et al. [64] found that miR-485-5p was significantly downregulated in the parietal lobe, cerebellum, entorhinal cortex and hippocampus (as listed in Table 2), but was not significantly changed in the cerebellum and superior frontal gyrus.
Some studies compared dysregulated miRNAs in AD to those in other neurogenic diseases [50,84]. For instance, miR-9-5p, miR-125b-5p and miR-146a-5p were upregulated in AD brains, but not in other neurogenic diseases, such as amyotrophic lateral sclerosis, Parkinson’s disease and schizophrenia [50] (as listed in Table 2). Similarly, miR-100 was only downregulated in AD brains compared with HC, but had no statistical changes in frontotemporal lobar dementia (FTLD) and progressive supranuclear palsy (PSP) [84] (see Table 2). However, miR-132-3p was downregulated in AD, FTLD and PSP [84].
4. MiRNAs in EVs of Different Biological Fluids of AD Patients
4.1. Dysregulated miRNAs in Blood EVs
The miRNAs in EVs of blood samples from AD patients will be discussed first. There are three types of blood samples, comprising plasma, serum and whole blood. As a very common and easy-to-collect body fluid, plasma appears in many AD-related studies. Many miRNAs of EVs isolated from plasma are dysregulated in AD patients [79,85,86,87,88] (as listed in Table 3). A study of exosomes in 70 plasma samples (35 AD and 35 HC) showed that four miRNAs were upregulated, whereas 16 miNRAs were downregulated in AD compared to HC [85] (see Table 3). In another study, six miRNAs were downregulated in EVs derived from the plasma of AD patients compared with HC, but no miRNA was statistically upregulated in the same study [86] (see Table 3). In another study, 37 dysregulated miRNAs (15 upregulated miRNAs and 22 downregulated miRNAs) were found in the plasma EVs of AD patients compared with HC [87] (as listed in Table 3). miR-132-3p and miR-212-3p were downregulated in neural exosomes derived from the plasma of AD patients [79] (see Table 3). Serpente et al. also found several dysregulated miRNAs (two upregulated miRNAs and one downregulated miRNA) in neural EVs collected from the plasma of AD patients [88] (see Table 3). Durur et al. [69] found that let-7e-5p was upregulated in small neuron-derived EVs (sNDEV) of 23 AD patients’ plasma compared to 28 HC. The last three studies used neural EVs, which are not mentioned in other studies. Cheng et al. [89] indicated that the expression of miRNAs in EVs from peripheral blood had a lower correlation with that of the EVs from brain tissues because other organs also released EVs to the blood. The isolation method of brain-derived EVs in the blood should be emphasized to find more AD-related miRNAs [89].
Table 3.
Dysregulated EV miRNAs in blood of AD patients.
As another common peripheral biofluid, serum is also used frequently in AD studies [49,58,90,91,92,93]. A study found that 17 miRNAs were upregulated and three miRNAs were downregulated in the serum sample of 53 AD compared to 62 controls [49] (see Table 3). Another study found that 14 miRNAs were upregulated and three miRNAs were downregulated in the serum sample of 31 AD compared to 28 controls [90] (as listed in Table 3). miR-135a-5p and miR-384 were upregulated and miR-193b was downregulated in the serum of 208 AD compared to 228 controls [91] (see Table 3). However, Liu et al. found that miR-193b was upregulated in ABCA1-labeled exosomes of AD patients’ serum [92] (see Table 3). The reason for the opposite change of miR-193b from the last two studies might be the different methods of exosome isolation applied. Liu et al. [92] found that an exosome protein, ATP-binding cassette transporter A1 (ABCA1), was significantly upregulated in the CSF samples of AD patients than in those of controls. In [91], all exosomes in the serum of peripheral blood were analyzed, and ABCA1-labeled exosomes were captured and analyzed in [92].
Liu et al. found that ABCA1-labeled exosomal miR-135a-5p was upregulated in the serum of MCI and AD as well [94] (as listed Table 3). Dong et al. found 24 dysregulated miRNAs in the RNA sequence profile and validated five of them with quantitative reverse transcription PCR (qRT-PCR) [93] (see Table 3). Three out of five had significant differences but all five miRNAs had same tendency in RNA sequencing and qRT-PCR [93]. Similar to the results of Dong et al. [93], miR-193b was downregulated in AD and verified with quantitative PCR (qPCR) [58] (see Table 3).
The last type of blood sample is whole blood without any extra treatment after collection. Several dysregulated miRNAs were found in the whole-blood sample of AD patients [31] (see Table 3). miR-146a-5p was downregulated in EVs from the whole blood of nine moderate AD and 13 severe AD compared with 19 controls [31] (as listed in Table 3). Furthermore, let-7g and miR-142-3p were downregulated in EVs from the whole blood of nine moderate AD only compared to the 19 controls [31]. No significantly upregulated miRNAs were found in the EVs of AD patients’ whole blood [31].
4.2. Dysregulated miRNAs in Cerebrospinal Fluid EVs
The cerebrospinal fluid (CSF) contains many materials that are important to the brain function. Combined with the choroid plexus, the CSF forms the blood–CSF barrier (B-CSF-B). The B-CSF-B and the blood–brain barrier (BBB) form the barrier of the central nervous system, preventing substances in the blood from reaching the brain directly [95]. AD is divided into early-onset Alzheimer’s disease (EOAD) and late-onset Alzheimer’s disease (LOAD) [96]. A study about the miRNA expression of exosomes isolated from the CSF of EOAD patients found that miR-125b-5p was upregulated and miR-16-5p, miR-451a and miR-605-5p were downregulated compared to HC (see Table 4) [36]. The study also found the same altered tendency in LOAD patients and HC, except for miR-16-5p [36]. There is no significant difference in miR-16-5p expression between LOAD and HC [36], suggesting that the three common miRNAs might be potential candidates to distinguish AD from HC. Another study showed more dysregulated miRNAs in cell-free CSF [49] (see Table 4). Forty-one miRNAs were downregulated in AD [49]. Moreover, miR-193b was upregulated in ABCA1-labeled exosomes derived from the CSF in different stages of AD, including subjective cognitive decline, MCI and AD [92] (as listed in Table 4). The same condition happened in miR-135a-5p too [94] (see Table 4). miR-135a-5p in ABCA1-labeled exosomes derived from the CSF was also upregulated in 32 AD compared to seven controls [94]. Different from the ABCA1-labeled exosome, miR193b in exosomes isolated from the CSF was downregulated in AD [58] (as listed in Table 4).
Table 4.
Dysregulated EV miRNAs in CSF of AD patients.
5. Application of EV miRNAs in Diagnosis of AD
To figure out the possibility of exosomal miRNAs as AD biomarkers, McKeever et al. [36] performed logistic regression for the significantly dysregulated miRNAs in exosomes of AD patients’ CSF. According to ROC analysis, miR-451a has the best performance in distinguishing EOAD or LOAD from HC (AUC = 0.95 and 0.85, respectively, as listed in Table 5) [36]. Another study of exosomes in plasma obtained a similar performance using three different machine learning algorithms on seven miRNAs with AUC values of 0.83 to 0.92 [85] (see Table 5). miR-132-3p (AUC = 0.77) and miR-212-3p (AUC = 0.84) from exosomes in plasma were used as biomarkers to distinguish AD from HC, respectively [79] (as listed in Table 5). Durur et al. used let-7e-5p in sNDEV derived from the plasma to distinguish AD patients from HC, and the AUC was 0.92 [69]. miR-135a-5p, miR193b and miR-384 were used to distinguish AD from HC with a high sensitivity and specificity [91] (see Table 5). These three miRNAs were also used to distinguish MCI from HC separately and in combination [91]. The highest AUC (0.995) was obtained when the combination of miR-135a-5p, miR193b and miR-384 was used in the diagnosis of MCI [91]. Similarly, miR-22-3p, miR-30b-5p and miR-378a-3p were used to build a model for AD prediction as well [93] (see Table 5). However, the number of samples was smaller than that in Yang et al. [91] and the performance of the model was much worse than those in Yang et al. [91].
Table 5.
Dysregulated miRNAs as biomarkers of AD.
In addition to the machine learning methods, potential diagnostic methods based on cell conversion and differentiation have appeared [97,98]. Drouin-Ouellet et al. [97] successfully converted human dermal fibroblasts from patients with AD, Parkinson’s disease and Huntington’s disease into neurons by inhibiting RE1-silencing transcription factor expression. The conversion was partially regulated by miR-9-5p and miR-124 [97]. Ishikawa et al. [98] also achieved a rapid differentiation of neurons from human pluripotent stem cells (iPSCs) through the Neurogenin2 gene, miR-9-5p and miR-124. These neurons converted from iPSCs could be used for disease diagnosis, drug screening and in disease model studies [98].
6. Application of miRNAs in AD Treatment
Some miRNAs have been tested as therapeutic targets of AD in vitro or in animal model experiments (as listed in Table 6). For example, osthole, a natural coumarin derivative, has protective effects in neurons of both mice and humans by preventing cell death, improving cell viability and increasing the production of synaptic proteins [99] (see Table 6). Osthole decreases the expression of CAMKK2 and p-AMPK through upregulating miR-9-5p to achieve the therapeutic effects [99]. Osthole also stimulates the differentiation of neural stem cells into neurons by upregulating the expression of miR-9-5p, which inhibits the Notch signaling pathway [100]. miR-16-5p mimics induced a reduction in AD-related genes, including APP, BACE1 and Tau, in AD mice [101] (as listed in Table 6). The overexpression of miR-16-5p decreased the apoptosis of neurons in a cellular AD model [102] (see Table 6). miR-124 was downregulated in AD patients [103] (as listed in Table 6). Delivering miR-124 and rutin, a small molecular ancillary drug, to the brain of AD mice significantly increased miR-124 and inhibited the expression of BACE1 and APP [103].
Table 6.
MiRNAs as potential targets of AD treatment.
Based on the biological functions of miRNAs in gene regulation and the roles EVs play in cell communication [106], miRNAs were delivered using EVs to achieve the therapeutic purpose. MiRNAs or short interfering RNA (siRNA) can be delivered to inhibit the expression of pathological genes in AD. For example, siRNA can be delivered to neural cells specifically with exosomes, resulting BACE1 gene knockdown in AD mice [107]. miR-29b was contained in the exosomes and its target genes included BACE1 and BIM [104] (Table 6). The injection of miR-29b-containing exosomes reduced the pathological effects of A in a rat model of AD [104].
For the miRNAs to play a positive role in AD pathology, the miRNAs inhibitors must have therapeutic effects. For example, the EVs of astrocytes treated with aFGF or miR-206-3p inhibitor reduced the expression of miR-206-3p, improved synaptic plasticity and cognitive deficits and attenuated A accumulation in AD mice [105]. The repression of miRNAs that are upregulated in the EVs of AD patients can yield therapeutic effects.
7. Discussion
We first summarized the dysregulated miRNAs to find the miRNAs appearing in different brain regions. As shown in Figure 1A and Table A1, eight upregulated miRNAs and 16 downregulated miRNAs were found in at least two different brain regions. Among these miRNAs, miR-146a-5p [50,70,73,74,83] and miR-132-3p [48,72,80,81,84] were reported to be dysregulated in six independent AD studies, respectively. miR-146a-5p is related to the inflammatory response in AD [73,74]. Combined with miR-212, miR-132-3p is related to apoptosis [72]. Interestingly, miR-146a-5p was upregulated in the brains of AD patients compared to HC [50,70,73,74,83], but one study reported that miR-146a-5p was downregulated in the brain of AD patients compared to HC [83]. Similar to miR-146a-5p, miR-132-3p was reported to be downregulated in AD in five independent studies [48,72,80,84] but up-regulated in AD in only one independent study [81]. More studies are needed to verified the dysregulation of miRNAs in AD.
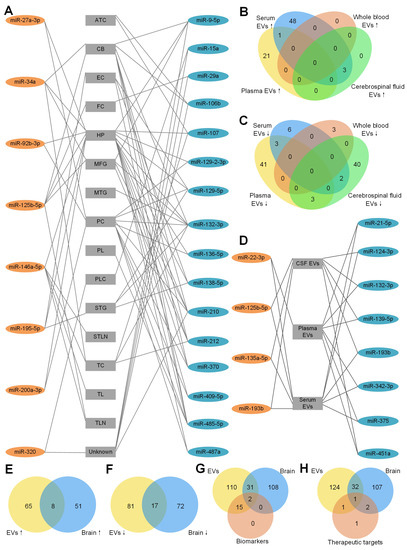
Figure 1.
Dysregulated miRNAs in different brain regions of AD patients. The detailed legend is given on the next page. (A) Dysregulated miRNAs in different brain regions of AD patients. A line between a miRNA and a brain region means the miRNA is dysregulated in the brain region. These miRNAs were reported by at least 2 studies. 8 miRNAs in orange ellipses of the left column were upregulated in AD patients compared to HC. 16 miRNAs in blue ellipses of the right column were downregulated in AD patients compared to HC. Abbreviations of the different brain regions in the central column: ATC: anterior temporal cortex; CB: cerebellum; CSF: cerebrospinal fluid; EC: entorhinal cortex; EVs: extracellular vesicles; FC: frontal cortex; HP: hippocampus; MFG: medial frontal gyrus; MTG: middle temporal gyrus; PC: prefrontal cortex; PL: parietal lobe; PLC: parietal lobe cortex; STG: superior temporal gyrus; STLN: superior temporal lobe neocortex; TC: temporal cortex; TL: temporal lobe; TLN: temporal lobe neocortex; and Unknown: unknown brain region. (B) Upregulated miRNAs of EVs derived from plasma, serum, whole blood and cerebrospinal fluid (CSF). There is no upregulated miRNA in exosomes derived from whole blood. Among the upregulated miRNAs from serum EVs, only miR-22-3p is common in serum EVs and plasma EVs and 3 miRNAs (miR-125b-5p, miR-135a-5p, miR-193b) are common in serum EVs and CSF EVs. (C) Downregulated miRNAs of EVs derived from plasma, serum, whole blood and CSF. There is no common miRNA in plasma EVs, serum EVs and whole-blood EVs. Among the upregulated miRNAs from serum EVs, 3 miRNAs (miR-21-5p, miR-342-3p, miR-375) are shared with plasma EVs and 2 miRNAs (miR-124-3p, miR-193b) are shared with CSF EVs. There are 3 (miR-132-3p, miR-139-5p, miR-451a) downregulated miRNAs in both plasma EVs and CSF EVs. (D) Dysregulated miRNAs in EVs of different body fluids of AD patients. These miRNAs are dysreguated in EVs from 2 different body fluids. 4 miRNAs in orange ellipse of the left column were upregulated in AD patients compared to HC. 8 miRNAs in blue ellipse of the right column were downregulated in AD patients compared to HC. (E) EVs and brain tissue have 8 upregulated miRNAs in common. (F) EVs and brain tissue have 17 downregulated miRNAs in common. (G) MiRNAs as AD biomarkers and all of the dysregulated miRNAs from EVs and brain tissue. Among all 17 miRNAs in EVs that could be an AD biomarker, only miR-125b-5p and miR-132-3p are dysregulated in EVs and brain tissue. Moreover, another 31 common miRNAs are dysregulated in EVs and brain tissue. (H) MiRNAs as AD therapeutic targets and all of the dysregulated miRNAs from EVs and brain tissue. For the 5 therapeutic miRNAs, miR-16-5p is dysregulated in EVs only. miR-29b and miR-124 are dysregulated in brain only. Only miR-9-5p was dysregulated in EVs and brain of AD patients and was tested as a therapeutic target of AD. miR-206-3p was not found to be dysregulated in either EVs or brain tissue.
To determine whether there are common miRNAs with the same tendency of EVs derived from different body fluids, the upregulated and downregulated miRNAs are summarized in Figure 1B,C respectively. As shown in Figure 1B, no upregulated miRNAs were found in EVs derived from whole blood. Twenty-two and 52 upregulated miRNAs were found in EVs derived from plasma and serum, respectively. Only miR-22-3p was in common. As a dysregulated miRNA, miR-22-3p was used as an AD biomarker to distinguish AD from HC [93]. miR-125b-5p, miR-135a-5p and miR-193b were common in EVs in serum and CSF, suggesting the exchange between the CSF and peripheral blood. miR-125-5p [85], miR-135a-5p and miR-193b [91] were used in AD identification too. miR-125-5p might be a positive factor in AD development by inducing Tau hyperphosphorylation [66] and being specifically expressed in microglia [71].
The downregulated miRNAs in EVs from different body fluids are shown in Figure 1C. Forty-seven, 11, three and 45 downregulated miRNAs in EVs were found from plasma, serum, whole blood and CSF, respectively. miR-21-5p, miR-342-3p and miR-375 were common in EVs of plasma and serum. miR-132-3p, miR-139-5p and miR-451a were common in EVs of plasma and CSF. miR-124-3p and miR-193b were common in EVs of serum and CSF. Interestingly, one common miRNA, miR-193b, was downregulated in exosomes from serum [58,91] and CSF [58], but upregulated in ABCA1-labeled exosomes from serum and CSF [92], indicating that the different methods of exosome isolation might influence the results. As shown in Figure 1D, four upregulated miRNAs and eight downregulated miRNAs were found in EVs of at least two different types of body fluid samples (details shown in Table A2).
To answer whether body fluids carry the deregulation patterns of miRNAs in the brain tissues of AD patients, we compared the upregulated miRNAs in EVs and brain tissues of AD patients (Figure 1E). Seventy-three and 59 upregulated miRNAs were found in EVs and brain tissues, respectively. Eight common miRNAs were found in both brain tissue and EVs of plasma or serum. They are miR-23a-3p, miR-27a-3p, miR-30e-5p, miR-92b-3p, miR-125b-5p, miR-199a-3p, miR-223-3p and miR-424-5p. No common miRNA was found in brain tissues and EVs derived from CSF, potentially because of the limited number of upregulated miRNAs in CSF studies. Seventeen downregulated miRNAs were common in EVs and brain tissues (Figure 1F). Among all the common miRNAs, 12 miRNAs (miR-9-5p, miR-95, miR-127-3p, miR-127-5p, miR-129-5p, miR-136-5p, miR-138-5p, miR-329, miR-410, miR-433, miR-598 and miR-769-5p) were common in brain tissues and EVs of CSF only. Two miRNAs (miR-132-3p, miR-139-5p) were common in brain tissues and EVs of plasma and CSF. miR-124-3p was common in brain tissues and EVs from serum and CSF. miR-182-5p was common in brain tissues and EVs from serum. miR-146a-5p was common in brain tissues and EVs from whole blood. In total, 15 out of 17 downregulated miRNAs were common in brain tissues and EVs from CSF, consistent with the expectation that the CSF should share more common miRNAs with brain tissues than with other body fluids.
In order to know whether the diagnostic miRNAs from EVs share the common miRNAs with brain tissues, an overlap of all of the dysregulated miRNAs from EVs, brain tissues and biomarkers was drawn. As shown in Figure 1G, only miR-132-3p (AUC = 0.77) and miR-125b-5p (combined with other miRNAs, AUC = 0.83∼0.92) were common in EVs and brain tissues. miR-132-3p was downregulated in AD brains [56,84] and EVs from plasma [79] and CSF [49]. miR-125b-5p was upregulated in brains [36,48,50] and EVs from CSF of AD patients compared to HC [49,66]. We did not find miR-135a-5p dysregulated in the brain of AD patients, but it has the best performance in single-miRNA biomarkers (Table 5). In addition, another 31 dysregulated miRNAs were common in EVs and brain tissues (Figure 1G). Some of them have the same alteration tendency, such as miR-199a-3p, which was upregulated in AD brains [80] and EVs from plasma [87]. However, some of them have the opposite tendency in different types of samples, such as miR-127-3p, which was downregulated in AD brains [80] and EVs from CSF, but upregulated in EVs from serum [49]. miR-146a-5p was upregulated in brains and EVs from plasma of AD patients compared to HC [50,70,73,74,83,87]. However, some studies reported that miR-146a-5p was downregulated in brains and EVs from whole blood [31,83].
Finally, we compared five therapeutic miRNAs in Table 6 to the dysregulated miRNAs in EVs and brain tissues of AD patients in Figure 1H. Only miR-9-5p was downregulated in EVs and brain tissues of AD patients and used as a therapeutic miRNA. miR-16-5p was downregulated in AD EVs only. miR-29b and miR-124 were downregulated in brain tissues only. miR-206-3p was not found to be dysregulated in either EVs or brain tissues. The other miRNAs that were dysregulated in EVs and brains of AD patients might be therapeutic targets in future studies. miR-9-5p [42,43], miR-16-5p [101], miR-29b [47], miR-124 [59] and miR-206-3p [105] were related to the generation of A. Delivering miR-9-5p, miR-16-5p, miR-29b and miR-124 and suppressing miR-206-3p have therapeutic effects (Table 6).
In summary, we compared the dysregulated miRNAs in EVs to those in brain tissues of AD patients, aiming to provide a comprehensive view of the relevant miRNAs in EVs of AD. By comparing Figure 1A,D, miR-125b-5p (miR-125) is upregulated in several different brain tissues of AD (Figure 1A) and EVs of AD (Figure 1D), and miR-132-3p (miR-132) is downregulated in several different brain tissues of AD (Figure 1A) and EVs of AD (Figure 1D). These results suggest that miR-125-5p and miR-132-3p are good markers for the diagnosis of AD based on EV miRNAs because they carry the miRNA dereglation patterns of brain tissues. We also found that miR-9-5p was normally downregulated in both EVs and brain tissues of AD patients and was tested as a therapy for AD in both mice and human cell models of AD. Therefore, miR-9-5p is a promising candidate for designing potential therapies of AD.
Author Contributions
Y.Z.: conceived and designed this work; W.L.: writing and revision, Y.Z.: revision. All authors have read and agreed to the published version of the manuscript.
Funding
The research was supported in part by a grant (No. 31460295) accessed on 17 August 2017 from the National Natural Science Foundation of China (http://www.nsfc.gov.cn/, accessed on 3 May 2023) and an open research fund (No. SKLGE-2107) of the State Key Laboratory of Genetic Engineering, Fudan University, China, to Y.Z. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| A | -amyloid |
| ABCA1 | ATP-binding cassette transporter A1 |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| ATC | Anterior temporal cortex |
| BBB | Blood–brain barrier |
| B-CSF-B | Blood–CSF barrier |
| CB | Cerebellum |
| CFH | Complement factor H |
| CSF | Cerebrospinal fluid |
| EC | Entorhinal cortex |
| EOAD | Early-onset Alzheimer’s disease |
| EVs | Extracellular vesicles |
| FC | Frontal cortex |
| FTLD | Frontotemporal lobar dementia |
| HC | Healthy controls |
| HP | Hippocampus |
| LOAD | Late-onset Alzheimer’s disease |
| MCI | Mild cognitive impairment |
| MFG | Medial frontal gyrus |
| miRNAs | MicroRNAs |
| MTG | Middle temporal gyrus |
| NFTs | Neurofibrillary tangles |
| PC | Prefrontal cortex |
| PL | Parietal lobe |
| PLC | Parietal lobe cortex |
| PSP | Progressive supranuclear palsy |
| qPCR | Quantitative PCR |
| qRT-PCR | Quantitative reverse transcription PCR |
| siRNA | Short interfering RNA |
| sNDEV | Small neuron-derived EVs |
| STG | Superior temporal gyrus |
| STLN | Superior temporal lobe neocortex |
| TC | Temporal cortex |
| TL | Temporal lobe |
| TLN | Temporal lobe neocortex |
Appendix A
Table A1.
Dysregulated miRNAs in at least two different brain regions of AD patients.
Table A1.
Dysregulated miRNAs in at least two different brain regions of AD patients.
| MiRNAs | Brain Region | Reference |
|---|---|---|
| Upregulated miRNAs | ||
| miR-27a-3p | HP | [80] |
| PC | [80] | |
| miR-34a | TC | [70] |
| HP; MFG; CB | [48] | |
| miR-92b-3p | HP | [80] |
| PC | [80] | |
| miR-125b-5p | FC | [66] |
| HP; MFG; CB | [48] | |
| TLN | [50] | |
| Upregulated miRNAs | ||
| miR-146a-5p | TC | [70] |
| HP, TL | [73] | |
| STLN | [74] | |
| HP | [83] | |
| TLN | [50] | |
| miR-195-5p | EC, STG | [75] |
| HP | [80] | |
| miR-200a-3p | HP | [80] |
| PC | [80] | |
| miR-320 | Unknown | [47] |
| PLC | [82] | |
| Downregulated miRNAs | ||
| miR-9-5p | Unknown | [47] |
| HP; MFG; CB | [48] | |
| miR-15a | PLC | [82] |
| Unknown | [47] | |
| Unknown | [65] | |
| miR-29a | Unknown | [47] |
| FC | [77] | |
| FC | [66] | |
| miR-106b | Unknown | [47] |
| ATC; CB | [56] | |
| miR-107 | TC | [61] |
| ATC | [59] | |
| HP | [83] | |
| miR-129-2-3p | HP | [80] |
| PC | [80] | |
| miR-129-5p | EC, STG | [75] |
| HP | [80] | |
| PC | [80] | |
| miR-132-3p | HP; MFG; CB | [48] |
| TC | [72] | |
| HP | [80] | |
| PC | [80] | |
| STG, MTG | [84] | |
| miR-136-5p | HP | [80] |
| PC | [80] | |
| miR-138-5p | EC, STG | [75] |
| HP | [80] | |
| miR-210 | Unknown | [47] |
| HP; MFG; CB | [48] | |
| PC | [80] | |
| miR-212 | HP; MFG; CB | [48] |
| TC | [72] | |
| miR-370 | HP | [80] |
| PC | [80] | |
| HP, CB | [81] | |
| miR-409-5p | HP | [80] |
| PC | [80] | |
| miR-485-5p | PC | [80] |
| PL, CB, HP, EC | [64] | |
| miR-487a | HP | [80] |
| PC | [80] |
Table A2.
Dysregulated miRNAs in EVs of at least two different types of body fluid samples of AD patients.
Table A2.
Dysregulated miRNAs in EVs of at least two different types of body fluid samples of AD patients.
| MiRNAs | Sample Source | Reference |
|---|---|---|
| Upregulated miRNAs | ||
| miR-22-3p | Plasma | [87] |
| Serum | [93,94] | |
| miR-125b-5p | Serum | [93] |
| CSF | [36] | |
| miR-135a-5p | Serum | [91,94] |
| CSF | [94] | |
| miR-193b | Serum | [92] |
| CSF | [92] | |
| Downregulated miRNAs | ||
| miR-21-5p | Plasma | [86] |
| Serum | [49] | |
| miR-342-3p | Plasma | [85] |
| Serum | [90] | |
| miR-375 | Plasma | [87] |
| Serum | [49] | |
| miR-132-3p | Plasma | [85] |
| CSF | [49] | |
| miR-139-5p | Plasma | [85] |
| CSF | [49] | |
| miR-451a | Plasma | [86] |
| CSF | [36] | |
| miR-124-3p | Serum | [93] |
| CSF | [49] | |
| miR-193b | Serum | [58,91] |
| CSF | [58] |
References
- Association, A. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 327–406. [Google Scholar] [CrossRef]
- Webster, C. What is dementia, why make a diagnosis and what are the current roadblocks? In World Alzheimer Report 2021; CARDI., Ed.; Alzheimer’s Disease International: London, UK, 2021; pp. 2–7. [Google Scholar]
- Mengli, W.; Lixia, Q.; Beisha, T. MicroRNAs in Alzheimer’s Disease. Front. Genet. 2019, 10, 153. [Google Scholar] [CrossRef]
- Clive, B.; Serge, G.; Anne, C.; Carol, B.; Dag, A.; Emma, J. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Miya Shaik, M.; Tamargo, I.A.; Abubakar, M.B.; Kamal, M.A.; Greig, N.H.; Gan, S.H. The role of microRNAs in Alzheimer’s disease and their therapeutic potentials. Genes 2018, 9, 174. [Google Scholar] [CrossRef]
- Philip, S.; Bart De, S.; Miia, K.; Henne, H.; Gael, C.; Charlotte E, T.; Jeffrey, C.; Wiesje M van der, F. MicroRNAs in Alzheimer’s Disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Drewes, G.; Ebneth, A.; Mandelkow, E.M. MAPs, MARKs and microtubule dynamics. Trends Biochem. Sci. 1998, 23, 307–311. [Google Scholar] [CrossRef]
- Mandelkow, E.M.; Mandelkow, E. Tau in Alzheimer’s disease. Trends Cell Biol. 1998, 8, 425–427. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement. Transl. Res. Clin. Interv. 2021, 7, e12179. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Bussy, A.; Snider, B.J.; Coble, D.; Xiong, C.; Fagan, A.M.; Cruchaga, C.; Benzinger, T.L.S.; Gordon, B.A.; Hassenstab, J.; Bateman, R.J. Effect of Apolipoprotein E4 on Clinical, Neuroimaging, and Biomarker Measures in Noncarrier Participants in the Dominantly Inherited Alzheimer Network. Neurobiol Aging 2019, 75, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Lucey, B.P.; Hicks, T.J.; McLeland, J.S.; Toedebusch, C.D.; Boyd, J.; Elbert, D.L.; Patterson, B.W.; Baty, J.; Morris, J.C.; Ovod, V.; et al. Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann. Neurol. 2018, 83, 197–204. [Google Scholar] [CrossRef]
- Holth, J.K.; Fritschi, S.K.; Wang, C.; Pedersen, N.P.; Cirrito, J.R.; Mahan, T.E.; Finn, M.B.; Manis, M.; Geerling, J.C.; Fuller, P.M.; et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 2019, 363, 880–884. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Denli, A.M.; Tops, B.B.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Zamore, P.D. microPrimer: The biogenesis and function of microRNA. Development 2005, 132, 4645–4652. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef]
- Kawamata, T.; Tomari, Y. Making risc. Trends Biochem. Sci. 2010, 35, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Shen, X.J.; Zou, Q.; Wang, S.P.; Tang, S.M.; Zhang, G.Z. Biological functions of microRNAs: A review. J. Physiol. Biochem. 2011, 67, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Aharon, A.; Spector, P.; Ahmad, R.S.; Horrany, N.; Sabbach, A.; Brenner, B.; Aharon-Peretz, J. Extracellular Vesicles of Alzheimer’s Disease Patients as a Biomarker for Disease Progression. Mol. Neurobiol. 2020, 57, 4156–4169. [Google Scholar] [CrossRef]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; d’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Budnik, V.; Ruiz-Cañada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Crescitelli, R.; Lässer, C.; Szabó, T.G.; Kittel, A.; Eldh, M.; Dianzani, I.; Buzás, E.I.; Lötvall, J. Distinct RNA profiles in subpopulations of extracellular vesicles: Apoptotic bodies, microvesicles and exosomes. J. Extracell. Vesicles 2013, 2, 20677. [Google Scholar] [CrossRef]
- McKeever, P.M.; Schneider, R.; Taghdiri, F.; Weichert, A.; Multani, N.; Brown, R.A.; Boxer, A.L.; Karydas, A.; Miller, B.; Robertson, J.; et al. MicroRNA expression levels are altered in the cerebrospinal fluid of patients with young-onset Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 8826–8841. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, Y.; Du, X.F.; Li, J.; Zi, H.X.; Bu, J.W.; Yan, Y.; Han, H.; Du, J.L. Neurons secrete miR-132-containing exosomes to regulate brain vascular integrity. Cell Res. 2017, 27, 882–897. [Google Scholar] [CrossRef]
- Lafourcade, C.; Ramírez, J.P.; Luarte, A.; Fernández, A.; Wyneken, U. MIRNAS in astrocyte-derived exosomes as possible mediators of neuronal plasticity: Supplementary issue: Brain plasticity and repair. J. Exp. Neurosci. 2016, 10, JEN-S39916. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef]
- Thind, A.; Wilson, C. Exosomal miRNAs as cancer biomarkers and therapeutic targets. J. Extracell. Vesicles 2016, 5, 31292. [Google Scholar] [CrossRef]
- Iranifar, E.; Seresht, B.M.; Momeni, F.; Fadaei, E.; Mehr, M.H.; Ebrahimi, Z.; Rahmati, M.; Kharazinejad, E.; Mirzaei, H. Exosomes and microRNAs: New potential therapeutic candidates in Alzheimer disease therapy. J. Cell. Physiol. 2019, 234, 2296–2305. [Google Scholar] [CrossRef]
- Turk, A.; Kunej, T.; Peterlin, B. MicroRNA-Target Interaction Regulatory Network in Alzheimer’s Disease. J. Pers. Med. 2021, 11, 1275. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Lee, B.; Woo, J.H.; Jeong, J.B.; Jun, I.; Kim, E.K. APP processing and metabolism in corneal fibroblasts and epithelium as a potential biomarker for Alzheimer’s disease. Exp. Eye Res. 2019, 182, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Yan, R. Role of BACE1 in Alzheimer’s synaptic function. Transl. Neurodegener. 2017, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Zhang, L.H.; Xu, W.P.; Jing, P.; Zhan, P.Y. microRNA-9 attenuates amyloidβ-induced synaptotoxicity by targeting calcium/calmodulin-dependent protein kinase kinase 2. Mol. Med. Rep. 2014, 9, 1917–1922. [Google Scholar] [CrossRef]
- Watroba, M.; Szukiewicz, D. Sirtuins promote brain homeostasis, preventing Alzheimer’s disease through targeting neuroinflammation. Front. Physiol. 2022, 13, 962769. [Google Scholar] [CrossRef]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef]
- Burgos, K.; Malenica, I.; Metpally, R.; Courtright, A.; Rakela, B.; Beach, T.; Shill, H.; Adler, C.; Sabbagh, M.; Villa, S.; et al. Profiles of extracellular miRNA in cerebrospinal fluid and serum from patients with Alzheimer’s and Parkinson’s diseases correlate with disease status and features of pathology. PLoS ONE 2014, 9, e94839. [Google Scholar] [CrossRef]
- Sethi, P.; Lukiw, W.J. Micro-RNA abundance and stability in human brain: Specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 2009, 459, 100–104. [Google Scholar] [CrossRef]
- Yam, G.H.; Wang, K.; Jhanji, V.; Choy, K.W.; Baum, L.; Pang, C.P. In vitro amyloid aggregate forming ability of TGFBI mutants that cause corneal dystrophies. Invest. Ophthalmol. Vis. Sci. 2012, 53, 5890–5898. [Google Scholar] [CrossRef]
- Subramanian, M.; Hyeon, S.J.; Das, T.; Suh, Y.S.; Kim, Y.K.; Lee, J.S.; Song, E.J.; Ryu, H.; Yu, K. UBE4B, a microRNA-9 target gene, promotes autophagy-mediated Tau degradation. Nat. Commun. 2021, 12, 3291. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Pisco, A.; Papalopulu, N. MicroRNA-9 reveals regional diversity of neural progenitors along the anterior-posterior axis. Dev. Cell 2011, 20, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qian, Y.; Li, J.; Zhou, X.; Xu, H.; Yan, J.; Xiang, J.; Yuan, X.; Sun, B.; Sisodia, S.S.; et al. BAD-mediated neuronal apoptosis and neuroinflammation contribute to Alzheimer’s disease pathology. iScience 2021, 24, 102942. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zuo, X.; Han, J.; Dai, Q.; Xu, H.; Liu, Y.; Cui, S. MiR-9-5p inhibits mitochondrial damage and oxidative stress in AD cell models by targeting GSK-3β. Biosci. Biotechnol. Biochem. 2020, 84, 2273–2280. [Google Scholar] [CrossRef]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Bergmans, B.; Papadopoulou, A.S.; Delacourte, A.; De Strooper, B. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis. 2009, 33, 422–428. [Google Scholar] [CrossRef]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-153 physiologically inhibits expression of amyloid-β precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J. Biol. Chem. 2012, 287, 31298–31310. [Google Scholar] [CrossRef]
- Liu, C.G.; Song, J.; Zhang, Y.Q.; Wang, P.C. MicroRNA-193b is a regulator of amyloid precursor protein in the blood and cerebrospinal fluid derived exosomal microRNA-193b is a biomarker of Alzheimer’s disease. Mol. Med. Rep. 2014, 10, 2395–2400. [Google Scholar] [CrossRef]
- Smith, P.; Al Hashimi, A.; Girard, J.; Delay, C.; Hébert, S.S. In vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J. Neurochem. 2011, 116, 240–247. [Google Scholar] [CrossRef]
- Cheng, C.; Li, W.; Zhang, Z.; Yoshimura, S.; Hao, Q.; Zhang, C.; Wang, Z. MicroRNA-144 is regulated by activator protein-1 (AP-1) and decreases expression of Alzheimer disease-related a disintegrin and metalloprotease 10 (ADAM10). J. Biol. Chem. 2013, 288, 13748–13761. [Google Scholar] [CrossRef]
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef]
- Lei, X.; Lei, L.; Zhang, Z.; Zhang, Z.; Cheng, Y. Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 1565–1574. [Google Scholar] [PubMed]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-339-5p down-regulates protein expression of β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J. Biol. Chem. 2014, 289, 5184–5198. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Zhang, M.; Huang, J.; Modarresi, F.; Van der Brug, M.P.; Nalls, M.A.; Cookson, M.R.; St-Laurent, G.; Wahlestedt, C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010, 11, R56. [Google Scholar] [CrossRef] [PubMed]
- Hébert, S.S.; Papadopoulou, A.S.; Smith, P.; Galas, M.C.; Planel, E.; Silahtaroglu, A.N.; Sergeant, N.; Buée, L.; De Strooper, B. Genetic ablation of Dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum. Mol. Genet. 2010, 19, 3959–3969. [Google Scholar] [CrossRef]
- Banzhaf-Strathmann, J.; Benito, E.; May, S.; Arzberger, T.; Tahirovic, S.; Kretzschmar, H.; Fischer, A.; Edbauer, D. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014, 33, 1667–1680. [Google Scholar] [CrossRef]
- Absalon, S.; Kochanek, D.M.; Raghavan, V.; Krichevsky, A.M. MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J. Neurosci. 2013, 33, 14645–14659. [Google Scholar] [CrossRef]
- Santa-Maria, I.; Alaniz, M.E.; Renwick, N.; Cela, C.; Fulga, T.A.; Van Vactor, D.; Tuschl, T.; Clark, L.N.; Shelanski, M.L.; McCabe, B.D.; et al. Dysregulation of microRNA-219 promotes neurodegeneration through post-transcriptional regulation of tau. J. Clin. Investig. 2015, 125, 681–686. [Google Scholar] [CrossRef]
- Durur, D.Y.; Tastan, B.; Ugur Tufekci, K.; Olcum, M.; Uzuner, H.; Karakülah, G.; Yener, G.; Genc, S. Alteration of miRNAs in Small Neuron-Derived Extracellular Vesicles of Alzheimer’s Disease Patients and the Effect of Extracellular Vesicles on Microglial Immune Responses. J. Mol. Neurosci. 2022, 72, 1182–1194. [Google Scholar] [CrossRef]
- Sarkar, S.; Jun, S.; Rellick, S.; Quintana, D.D.; Cavendish, J.Z.; Simpkins, J.W. Expression of microRNA-34a in Alzheimer’s disease brain targets genes linked to synaptic plasticity, energy metabolism, and resting state network activity. Brain Res. 2016, 1646, 139–151. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef]
- Wong, H.K.; Veremeyko, T.; Patel, N.; Lemere, C.A.; Walsh, D.M.; Esau, C.; Vanderburg, C.; Krichevsky, A.M. De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer’s disease. Hum. Mol. Genet. 2013, 22, 3077–3092. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Zhao, Y.; Cui, J.G. An NF-κB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 2008, 283, 31315–31322. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.G.; Li, Y.Y.; Zhao, Y.; Bhattacharjee, S.; Lukiw, W.J. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NF-κB in stressed human astroglial cells and in Alzheimer disease. J. Biol. Chem. 2010, 285, 38951–38960. [Google Scholar] [CrossRef] [PubMed]
- Dobricic, V.; Schilling, M.; Schulz, J.; Zhu, L.S.; Zhou, C.W.; Fuß, J.; Franzenburg, S.; Zhu, L.Q.; Parkkinen, L.; Lill, C.M.; et al. Differential microRNA expression analyses across two brain regions in Alzheimer’s disease. Transl. Psychiatry 2022, 12, 352. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J. Aberrant microRNA expression in the brains of neurodegenerative diseases: MiR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 2010, 36, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Weldon Furr, J.; Morales-Scheihing, D.; Manwani, B.; Lee, J.; McCullough, L.D. Cerebral Amyloid Angiopathy, Alzheimer’s Disease and MicroRNA: MiRNA as Diagnostic Biomarkers and Potential Therapeutic Targets. Neuromol. Med. 2019, 21, 369–390. [Google Scholar] [CrossRef]
- Cha, D.J.; Mengel, D.; Mustapic, M.; Liu, W.; Selkoe, D.J.; Kapogiannis, D.; Galasko, D.; Rissman, R.A.; Bennett, D.A.; Walsh, D.M. miR-212 and miR-132 Are Downregulated in Neurally Derived Plasma Exosomes of Alzheimer’s Patients. Front. Neurosci. 2019, 13, 1208. [Google Scholar] [CrossRef]
- Lau, P.; Bossers, K.; Janky, R.; Salta, E.; Frigerio, C.S.; Barbash, S.; Rothman, R.; Sierksma, A.S.; Thathiah, A.; Greenberg, D.; et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 1613–1634. [Google Scholar] [CrossRef]
- Bekris, L.M.; Lutz, F.; Montine, T.J.; Yu, C.E.; Tsuang, D.; Peskind, E.R.; Leverenz, J.B. MicroRNA in Alzheimer’s disease: An exploratory study in brain, cerebrospinal fluid and plasma. Biomarkers 2013, 18, 455–466. [Google Scholar] [CrossRef]
- Nunez-Iglesias, J.; Liu, C.C.; Morgan, T.E.; Finch, C.E.; Zhou, X.J. Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS ONE 2010, 5, e8898. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Kuiperij, H.B.; Claassen, J.A.; Küsters, B.; Verbeek, M.M. MicroRNAs in Alzheimer’s disease: Differential expression in hippocampus and cell-free cerebrospinal fluid. Neurobiol. Aging 2014, 35, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Hébert, S.S.; Wang, W.X.; Zhu, Q.; Nelson, P.T. A study of small RNAs from cerebral neocortex of pathology-verified Alzheimer’s disease, dementia with lewy bodies, hippocampal sclerosis, frontotemporal lobar dementia, and non-demented human controls. J Alzheimers Dis 2013, 35, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma Exosomal miRNAs in Persons with and without Alzheimer Disease: Altered Expression and Prospects for Biomarkers. PLoS ONE 2015, 10, e0139233. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Valero, A.; Campdelacreu, J.; Vilas, D.; Ispierto, L.; Reñé, R.; Álvarez, R.; Armengol, M.P.; Borràs, F.E.; Beyer, K. Exploratory study on microRNA profiles from plasma-derived extracellular vesicles in Alzheimer’s disease and dementia with Lewy bodies. Transl. Neurodegener. 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Sun, Y.; Zhen, H.; Guo, M.; Ye, J.; Liu, Z.; Yang, Y.; Zhang, X. Differential Expression of Plasma Exo-miRNA in Neurodegenerative Diseases by Next-Generation Sequencing. Front. Neurosci. 2020, 14, 438. [Google Scholar] [CrossRef]
- Serpente, M.; Fenoglio, C.; D’Anca, M.; Arcaro, M.; Sorrentino, F.; Visconte, C.; Arighi, A.; Fumagalli, G.G.; Porretti, L.; Cattaneo, A.; et al. MiRNA Profiling in Plasma Neural-Derived Small Extracellular Vesicles from Patients with Alzheimer’s Disease. Cells 2020, 9, 1443. [Google Scholar] [CrossRef]
- Cheng, L.; Vella, L.J.; Barnham, K.J.; McLean, C.; Masters, C.L.; Hill, A.F. Small RNA fingerprinting of Alzheimer’s disease frontal cortex extracellular vesicles and their comparison with peripheral extracellular vesicles. J. Extracell. Vesicles 2020, 9, 1766822. [Google Scholar] [CrossRef]
- Li, F.; Xie, X.Y.; Sui, X.F.; Wang, P.; Chen, Z.; Zhang, J.B. Profile of Pathogenic Proteins and MicroRNAs in Plasma-derived Extracellular Vesicles in Alzheimer’s Disease: A Pilot Study. Neuroscience 2020, 432, 240–246. [Google Scholar] [CrossRef]
- Yang, T.T.; Liu, C.G.; Gao, S.C.; Zhang, Y.; Wang, P.C. The Serum Exosome Derived MicroRNA-135a, -193b, and -384 Were Potential Alzheimer’s Disease Biomarkers. Biomed. Environ. Sci. 2018, 31, 87–96. [Google Scholar] [CrossRef]
- Liu, C.G.; Zhao, Y.; Lu, Y.; Wang, P.C. ABCA1-Labeled Exosomes in Serum Contain Higher MicroRNA-193b Levels in Alzheimer’s Disease. BioMed Res. Int. 2021, 2021, 5450397. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Gu, H.; Guo, Q.; Liang, S.; Xue, J.; Yao, F.; Liu, X.; Li, F.; Liu, H.; Sun, L.; et al. Profiling of Serum Exosome MiRNA Reveals the Potential of a MiRNA Panel as Diagnostic Biomarker for Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3084–3094. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.G.; Meng, S.; Li, Y.; Lu, Y.; Zhao, Y.; Wang, P.C. MicroRNA-135a in ABCA1-labeled Exosome is a Serum Biomarker Candidate for Alzheimer’s Disease. Biomed. Environ. Sci. 2021, 34, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Bonfio, C.; Chadwick, J.; Begum, F.; Skehel, M.; Lancaster, M.A. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science 2020, 369, eaaz5626. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, H.; Fereshtehnejad, S.M.; Falahati, F.; Farahmand, B.; Religa, D.; Eriksdotter, M. Differences in routine clinical practice between early and late onset Alzheimer’s disease: Data from the Swedish Dementia Registry (SveDem). J. Alzheimers Dis. 2014, 41, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Drouin-Ouellet, J.; Lau, S.; Brattås, P.L.; Rylander Ottosson, D.; Pircs, K.; Grassi, D.A.; Collins, L.M.; Vuono, R.; Andersson Sjöland, A.; Westergren-Thorsson, G.; et al. REST suppression mediates neural conversion of adult human fibroblasts via microRNA-dependent and -independent pathways. EMBO Mol. Med. 2017, 9, 1117–1131. [Google Scholar] [CrossRef]
- Ishikawa, M.; Aoyama, T.; Shibata, S.; Sone, T.; Miyoshi, H.; Watanabe, H.; Nakamura, M.; Morota, S.; Uchino, H.; Yoo, A.S.; et al. miRNA-Based Rapid Differentiation of Purified Neurons from hPSCs Advancestowards Quick Screening for Neuronal Disease Phenotypes In Vitro. Cells 2020, 9, 532. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Gao, P.; Wang, L.T.; Yan, Y.H.; Xia, Y.; Song, J.; Li, H.Y.; Yang, J.X. Osthole Stimulated Neural Stem Cells Differentiation into Neurons in an Alzheimer’s Disease Cell Model via Upregulation of MicroRNA-9 and Rescued the Functional Impairment of Hippocampal Neurons in APP/PS1 Transgenic Mice. Front. Neurosci. 2017, 11, 340. [Google Scholar] [CrossRef]
- Li, S.; Yan, Y.; Jiao, Y.; Gao, Z.; Xia, Y.; Kong, L.; Yao, Y.; Tao, Z.; Song, J.; Yan, Y.; et al. Neuroprotective Effect of Osthole on Neuron Synapses in an Alzheimer’s Disease Cell Model via Upregulation of MicroRNA-9. J. Mol. Neurosci. 2016, 60, 71–81. [Google Scholar] [CrossRef]
- Parsi, S.; Smith, P.Y.; Goupil, C.; Dorval, V.; Hébert, S.S. Preclinical evaluation of miR-15/107 family members as multifactorial drug targets for Alzheimer’s disease. Mol. Ther.-Nucleic Acids 2015, 4, e256. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, C.; Wang, A.; Lin, Q. MiR-16 regulates cell death in Alzheimer’s disease by targeting amyloid precursor protein. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4020–4027. [Google Scholar] [PubMed]
- Ouyang, Q.; Liu, K.; Zhu, Q.; Deng, H.; Le, Y.; Ouyang, W.; Yan, X.; Zhou, W.; Tong, J. Brain-Penetration and Neuron-Targeting DNA Nanoflowers Co-Delivering miR-124 and Rutin for Synergistic Therapy of Alzheimer’s Disease. Small 2022, 18, e2107534. [Google Scholar] [CrossRef] [PubMed]
- Jahangard, Y.; Monfared, H.; Moradi, A.; Zare, M.; Mirnajafi-Zadeh, J.; Mowla, S.J. Therapeutic Effects of Transplanted Exosomes Containing miR-29b to a Rat Model of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 564. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Wang, Y.; Xiao, Y.; Peng, M.; Mai, W.; Hu, B.; Jia, Y.; Chen, H.; Yang, Y.; Xiang, Q.; et al. Extracellular vesicles derived from astrocyte-treated with haFGF(14-154) attenuate Alzheimer phenotype in AD mice. Theranostics 2022, 12, 3862–3881. [Google Scholar] [CrossRef]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Krämer-Albers, E.M. Extracellular vesicles as mediators of neuron-glia communication. Front. Cell. Neurosci. 2013, 7, 182. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).