

Desmosomes in Cell Fate Determination: From Cardiogenesis to Cardiomyopathy



Abstract
:1. Cardiogenesis and Mechanical Cues
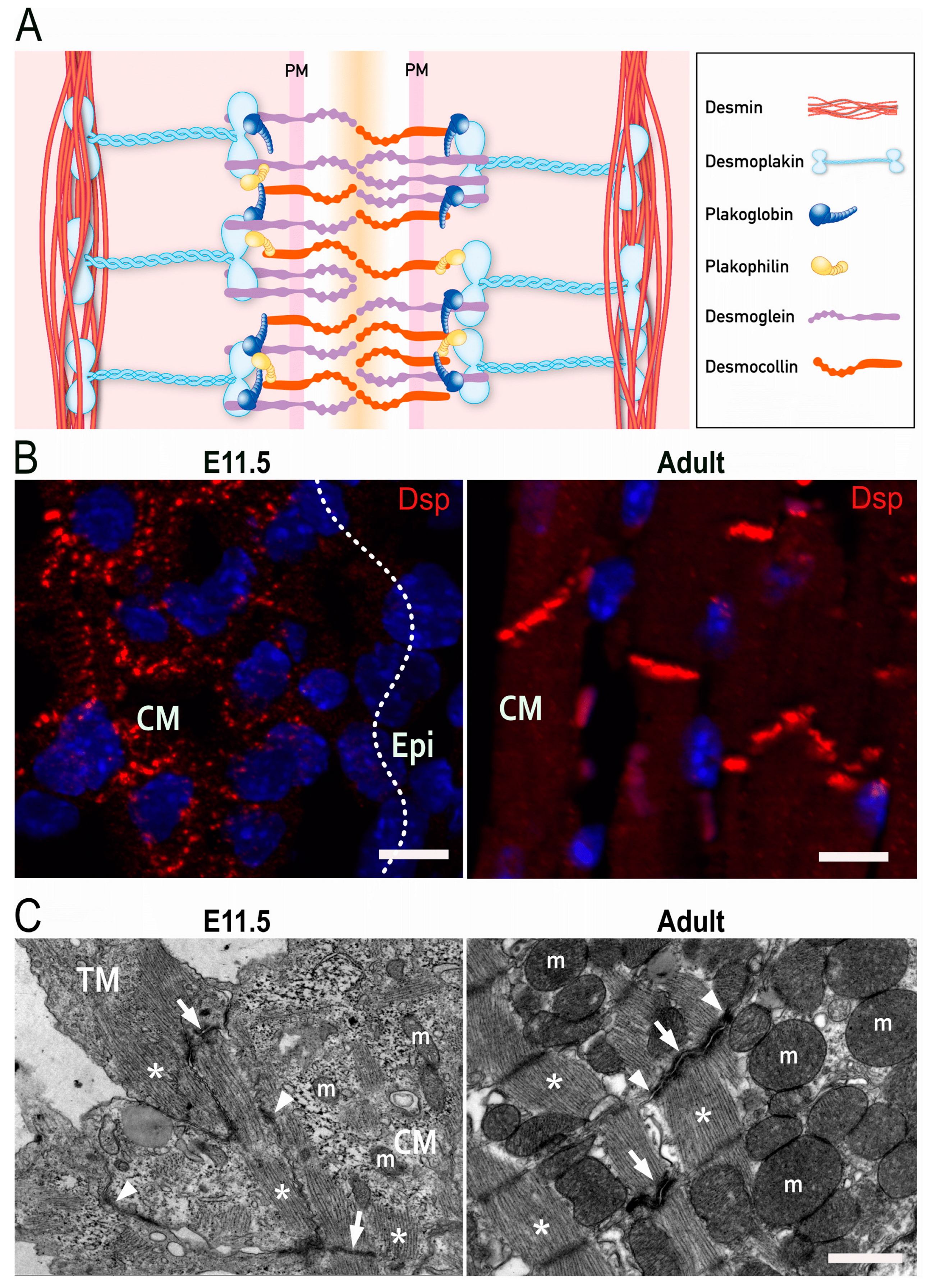
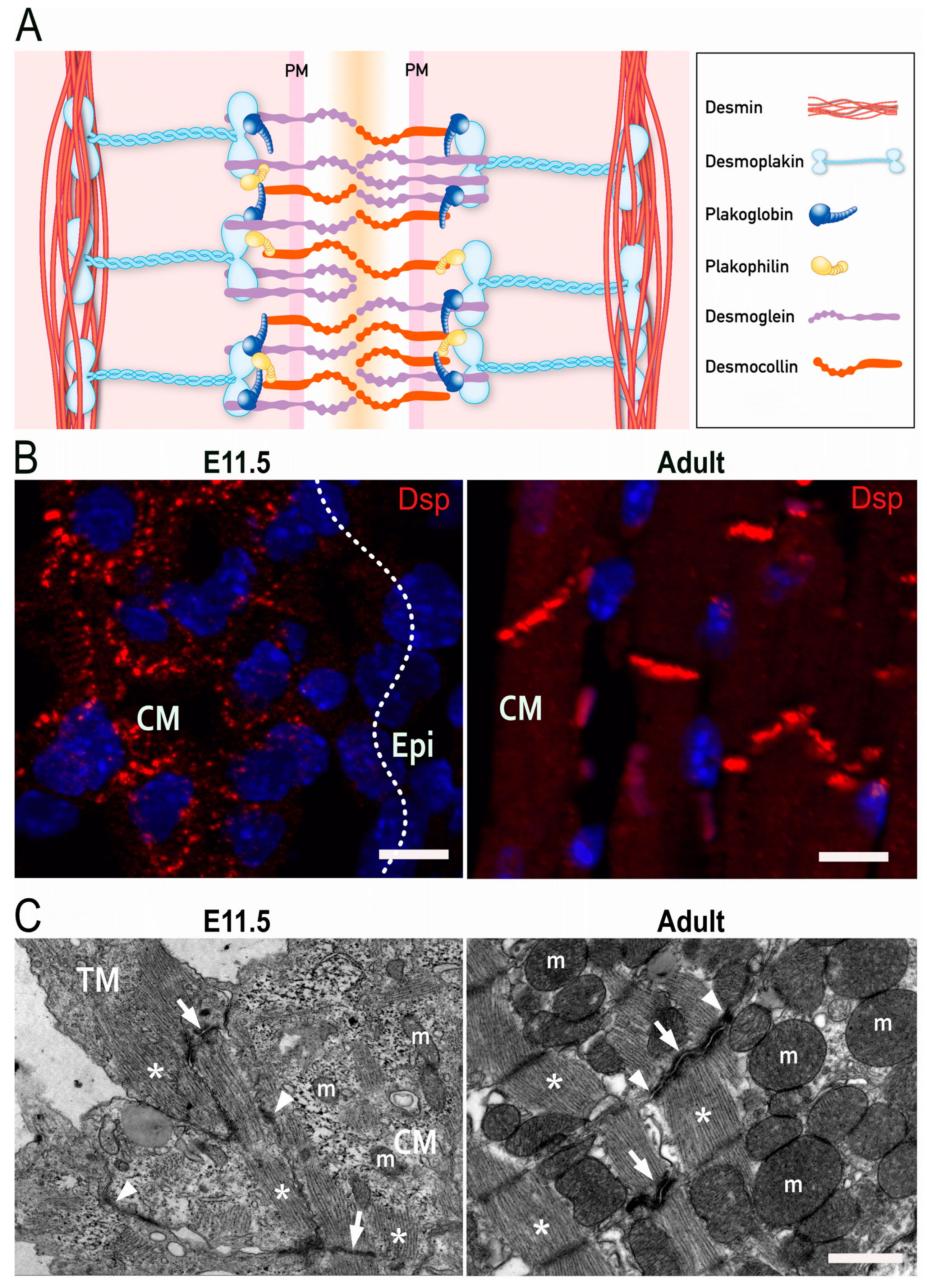
2. Molecular Structure of Desmosomes
3. Cardiogenesis
3.1. Contribution of Different Heart Fields
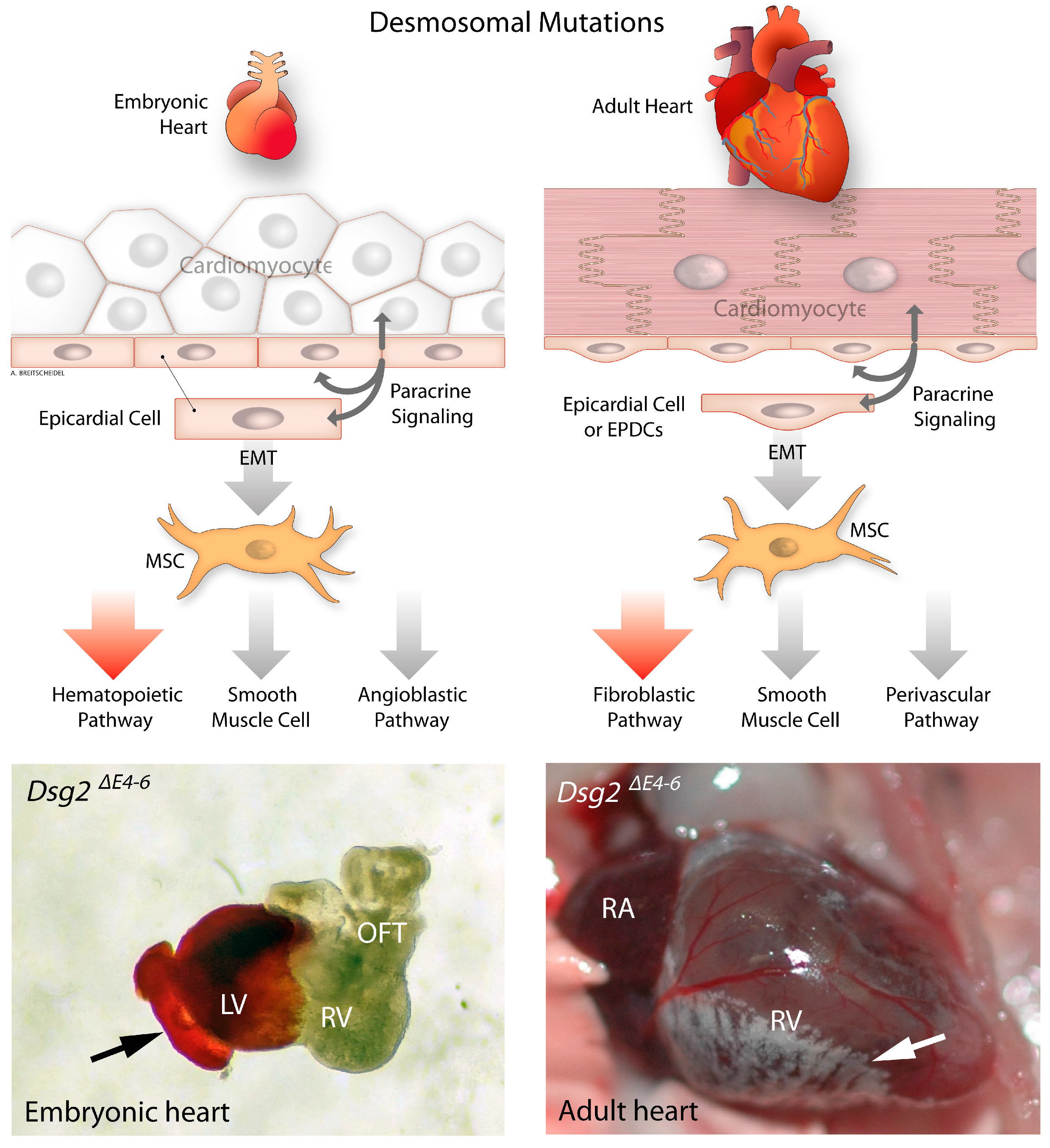
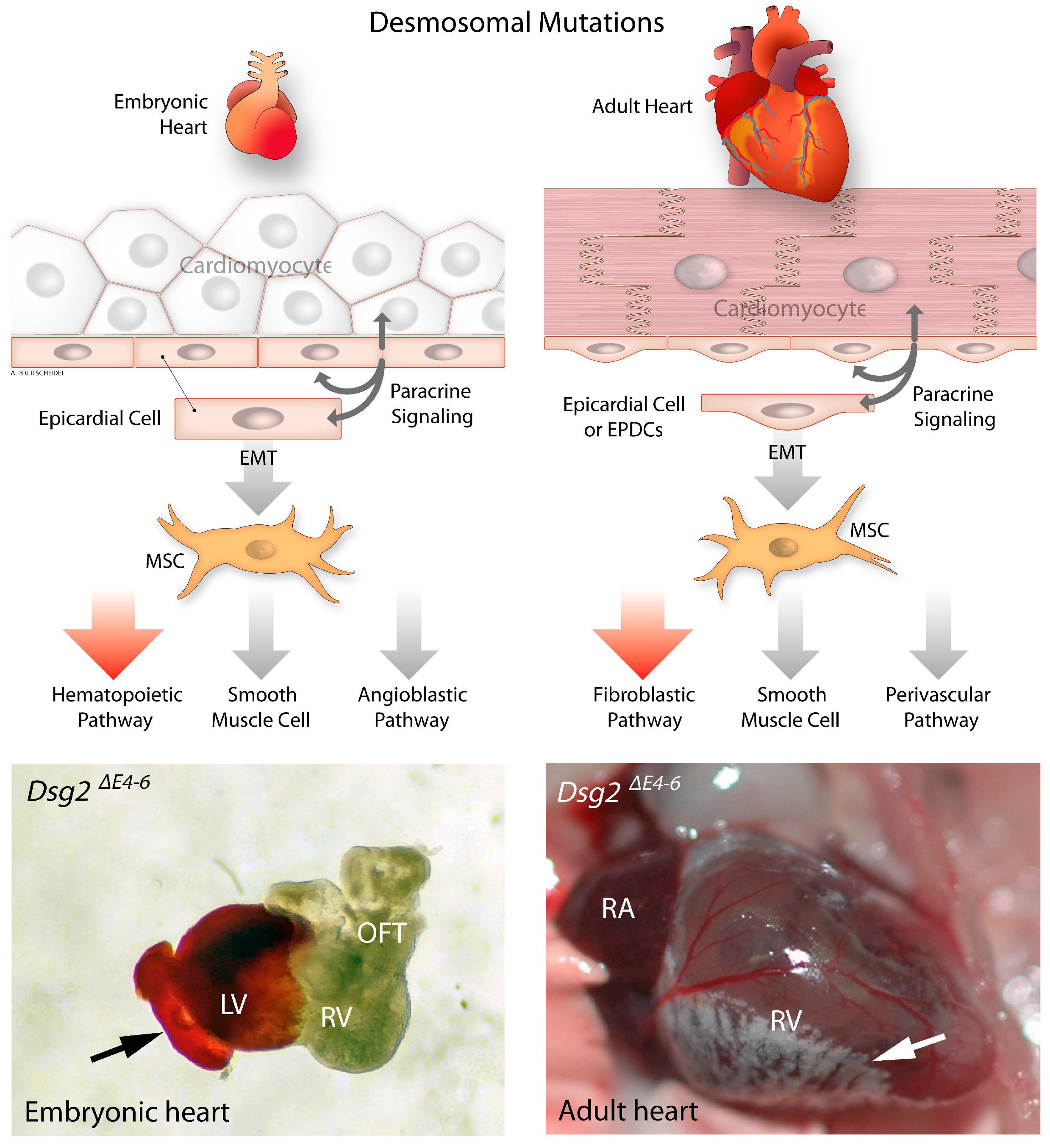
3.2. Development of Epicardium and Epicardial-Derived Cells
4. Development of Intercellular Junctions in Embryonic Cardiomyocytes
5. The Impact of Desmosomal Proteins on Cardiac Morphogenesis
6. Epithelial to Mesenchymal Transition in Desmosome Deficient Models
6.1. Desmosomes Communicate with Gap Junctions in the Regulation of Epicardial EMT
6.2. Desmosome-TGF-β Cross-Talk in the Regulation of Epicardium Development
6.3. Desmosomes and Modulation of YAP/TAZ Signaling
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paige, S.L.; Plonowska, K.; Xu, A.; Wu, S.M. Molecular regulation of cardiomyocyte differentiation. Circ. Res. 2015, 116, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Bartman, T.; Hove, J. Mechanics and function in heart morphogenesis. Dev. Dyn. 2005, 233, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, R.; Zizzi, E.A.; Deriu, M.A.; Morbiducci, U.; Pesce, M.; Messina, E. When Stiffness Matters: Mechanosensing in Heart Development and Disease. Front. Cell Dev. Biol. 2020, 8, 334. [Google Scholar] [CrossRef] [PubMed]
- Munch, J.; Abdelilah-Seyfried, S. Sensing and Responding of Cardiomyocytes to Changes of Tissue Stiffness in the Diseased Heart. Front. Cell Dev. Biol. 2021, 9, 642840. [Google Scholar] [CrossRef]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pilichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Brodehl, A. Genetic Animal Models for Arrhythmogenic Cardiomyopathy. Front. Physiol. 2020, 11, 624. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef]
- Gao, S.; Puthenvedu, D.; Lombardi, R.; Chen, S.N. Established and Emerging Mechanisms in the Pathogenesis of Arrhythmogenic Cardiomyopathy: A Multifaceted Disease. Int. J. Mol. Sci. 2020, 21, 6320. [Google Scholar] [CrossRef]
- Coscarella, I.L.; Landim-Vieira, M.; Pinto, J.R.; Chelko, S.P. Arrhythmogenic Cardiomyopathy: Exercise Pitfalls, Role of Connexin-43, and Moving beyond Antiarrhythmics. Int. J. Mol. Sci. 2022, 23, 8753. [Google Scholar] [CrossRef]
- Thiene, G.; Basso, C.; Pilichou, K.; Bueno Marinas, M. Desmosomal Arrhythmogenic Cardiomyopathy: The Story Telling of a Genetically Determined Heart Muscle Disease. Biomedicines 2023, 11, 2018. [Google Scholar] [CrossRef]
- Reisqs, J.B.; Moreau, A.; Sleiman, Y.; Boutjdir, M.; Richard, S.; Chevalier, P. Arrhythmogenic cardiomyopathy as a myogenic disease: Highlights from cardiomyocytes derived from human induced pluripotent stem cells. Front. Physiol. 2023, 14, 1191965. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Sommariva, E.; Di Resta, C.; Rabino, M.; Villatore, A.; Lazzeroni, D.; Sala, S.; Pompilio, G.; Cooper, L.T. Myocardial Inflammation as a Manifestation of Genetic Cardiomyopathies: From Bedside to the Bench. Biomolecules 2023, 13, 646. [Google Scholar] [CrossRef] [PubMed]
- Eshkind, L.; Tian, Q.; Schmidt, A.; Franke, W.W.; Windoffer, R.; Leube, R.E. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur. J. Cell Biol. 2002, 81, 592–598. [Google Scholar] [CrossRef]
- Den, Z.; Cheng, X.; Merched-Sauvage, M.; Koch, P.J. Desmocollin 3 is required for pre-implantation development of the mouse embryo. J. Cell Sci. 2006, 119, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Son, Y.; Lee, N.G.; Lee, K.; Lee, D.G.; Song, J.; Lee, J.; Kim, S.; Cho, M.J.; Jang, J.H.; et al. DSG2 Is a Functional Cell Surface Marker for Identification and Isolation of Human Pluripotent Stem Cells. Stem. Cell Rep. 2018, 11, 115–127. [Google Scholar] [CrossRef]
- Gallicano, G.I.; Bauer, C.; Fuchs, E. Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development 2001, 128, 929–941. [Google Scholar] [CrossRef]
- Grossmann, K.S.; Grund, C.; Huelsken, J.; Behrend, M.; Erdmann, B.; Franke, W.W.; Birchmeier, W. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J. Cell Biol. 2004, 167, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Viragh, S.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; Kalman, F. Early development of quail heart epicardium and associated vascular and glandular structures. Anat. Embryol. 1993, 188, 381–393. [Google Scholar] [CrossRef]
- Eroglu, E.; Yen, C.Y.T.; Tsoi, Y.L.; Witman, N.; Elewa, A.; Joven Araus, A.; Wang, H.; Szattler, T.; Umeano, C.H.; Sohlmer, J.; et al. Epicardium-derived cells organize through tight junctions to replenish cardiac muscle in salamanders. Nat. Cell Biol. 2022, 24, 645–658. [Google Scholar] [CrossRef]
- Matthes, S.A.; Taffet, S.; Delmar, M. Plakophilin-2 and the migration, differentiation and transformation of cells derived from the epicardium of neonatal rat hearts. Cell Commun. Adhes. 2011, 18, 73–84. [Google Scholar] [CrossRef]
- Yuan, P.; Cheedipudi, S.M.; Rouhi, L.; Fan, S.; Simon, L.; Zhao, Z.; Hong, K.; Gurha, P.; Marian, A.J. Single-Cell RNA Sequencing Uncovers Paracrine Functions of the Epicardial-Derived Cells in Arrhythmogenic Cardiomyopathy. Circulation 2021, 143, 2169–2187. [Google Scholar] [CrossRef] [PubMed]
- Kohela, A.; van Kampen, S.J.; Moens, T.; Wehrens, M.; Molenaar, B.; Boogerd, C.J.; Monshouwer-Kloots, J.; Perini, I.; Goumans, M.J.; Smits, A.M.; et al. Epicardial differentiation drives fibro-fatty remodeling in arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2021, 13, eabf2750. [Google Scholar] [CrossRef] [PubMed]
- Reant, P.; Hauer, A.D.; Castelletti, S.; Pantazis, A.; Rosmini, S.; Cheang, M.H.; Peyrou, J.; Tome-Esteban, M.; Syrris, P.; Lafitte, S.; et al. Epicardial myocardial strain abnormalities may identify the earliest stages of arrhythmogenic cardiomyopathy. Int. J. Cardiovasc. Imaging 2016, 32, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Chen, S.N.; Ruggiero, A.; Gurha, P.; Czernuszewicz, G.Z.; Willerson, J.T.; Marian, A.J. Cardiac Fibro-Adipocyte Progenitors Express Desmosome Proteins and Preferentially Differentiate to Adipocytes Upon Deletion of the Desmoplakin Gene. Circ. Res. 2016, 119, 41–54. [Google Scholar] [CrossRef]
- Maione, A.S.; Faris, P.; Iengo, L.; Catto, V.; Bisonni, L.; Lodola, F.; Negri, S.; Casella, M.; Guarino, A.; Polvani, G.; et al. Ca(2+) dysregulation in cardiac stromal cells sustains fibro-adipose remodeling in Arrhythmogenic Cardiomyopathy and can be modulated by flecainide. J. Transl. Med. 2022, 20, 522. [Google Scholar] [CrossRef]
- Gerull, B.; Brodehl, A. Insights Into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef]
- Quijada, P.; Trembley, M.A.; Small, E.M. The Role of the Epicardium During Heart Development and Repair. Circ. Res. 2020, 126, 377–394. [Google Scholar] [CrossRef]
- Holthofer, B.; Windoffer, R.; Troyanovsky, S.; Leube, R.E. Structure and function of desmosomes. Int. Rev. Cytol. 2007, 264, 65–163. [Google Scholar] [CrossRef]
- Hegazy, M.; Perl, A.L.; Svoboda, S.A.; Green, K.J. Desmosomal Cadherins in Health and Disease. Annu. Rev. Pathol. 2022, 17, 47–72. [Google Scholar] [CrossRef]
- Hirschy, A.; Schatzmann, F.; Ehler, E.; Perriard, J.C. Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev. Biol. 2006, 289, 430–441. [Google Scholar] [CrossRef]
- Franke, W.W.; Borrmann, C.M.; Grund, C.; Pieperhoff, S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur. J. Cell Biol. 2006, 85, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Leo-Macias, A.; Liang, F.X.; Delmar, M. Ultrastructure of the intercellular space in adult murine ventricle revealed by quantitative tomographic electron microscopy. Cardiovasc. Res. 2015, 107, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Pieperhoff, S.; Franke, W.W. The area composita of adhering junctions connecting heart muscle cells of vertebrates - IV: Coalescence and amalgamation of desmosomal and adhaerens junction components - late processes in mammalian heart development. Eur. J. Cell Biol. 2007, 86, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Martin-Puig, S.; Wang, Z.; Chien, K.R. Lives of a heart cell: Tracing the origins of cardiac progenitors. Cell Stem Cell 2008, 2, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sanchez-Danes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef]
- Ma, Q.; Zhou, B.; Pu, W.T. Reassessment of Isl1 and Nkx2-5 cardiac fate maps using a Gata4-based reporter of Cre activity. Dev. Biol. 2008, 323, 98–104. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Garcia-Martinez, V. Molecular determinants of cardiac specification. Cardiovasc. Res. 2011, 91, 185–195. [Google Scholar] [CrossRef]
- Bruneau, B.G.; Logan, M.; Davis, N.; Levi, T.; Tabin, C.J.; Seidman, J.G.; Seidman, C.E. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev. Biol. 1999, 211, 100–108. [Google Scholar] [CrossRef]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Galdos, F.X.; Wu, S.M. Single-Cell Delineation of Who's on First and Second Heart Fields During Development. Circ. Res. 2019, 125, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Luo, Y.; Yue, Y.; Zhang, J.; Ai, S.; Li, X.; Wang, X.; Zhang, Y.L.; Wei, Y.; Li, H.H.; et al. Single-Cell Transcriptomics Reveals Chemotaxis-Mediated Intraorgan Crosstalk During Cardiogenesis. Circ. Res. 2019, 125, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Misfeldt, A.M.; Boyle, S.C.; Tompkins, K.L.; Bautch, V.L.; Labosky, P.A.; Baldwin, H.S. Endocardial cells are a distinct endothelial lineage derived from Flk1+ multipotent cardiovascular progenitors. Dev. Biol. 2009, 333, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Dye, B.; Lincoln, J. The Endocardium and Heart Valves. Cold Spring Harb. Perspect. Biol. 2020, 12, a036723. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.C.; Pereira, C.; Fonseca, A.; Pinto-do, O.P.; Nascimento, D.S. Bearing My Heart: The Role of Extracellular Matrix on Cardiac Development, Homeostasis, and Injury Response. Front. Cell Dev. Biol. 2020, 8, 621644. [Google Scholar] [CrossRef]
- Rienks, M.; Papageorgiou, A.P.; Frangogiannis, N.G.; Heymans, S. Myocardial extracellular matrix: An ever-changing and diverse entity. Circ. Res. 2014, 114, 872–888. [Google Scholar] [CrossRef]
- Jallerat, Q.; Feinberg, A.W. Extracellular Matrix Structure and Composition in the Early Four-Chambered Embryonic Heart. Cells 2020, 9, 285. [Google Scholar] [CrossRef]
- Kalman, F.; Viragh, S.; Modis, L. Cell surface glycoconjugates and the extracellular matrix of the developing mouse embryo epicardium. Anat. Embryol. 1995, 191, 451–464. [Google Scholar] [CrossRef]
- Zhang, H.; Lui, K.O.; Zhou, B. Endocardial Cell Plasticity in Cardiac Development, Diseases and Regeneration. Circ. Res. 2018, 122, 774–789. [Google Scholar] [CrossRef]
- Zhang, F.; Pasumarthi, K.B. Ultrastructural and immunocharacterization of undifferentiated myocardial cells in the developing mouse heart. J. Cell Mol. Med. 2007, 11, 552–560. [Google Scholar] [CrossRef]
- Wu, M. Mechanisms of Trabecular Formation and Specification During Cardiogenesis. Pediatr. Cardiol. 2018, 39, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Männer, J.; Pérez-Pomares, J.M.; Macías, D.; Muñoz-Chápuli, R. The origin, formation and developmental significance of the epicardium: A review. Cells Tissues Organs 2001, 169, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Nahirney, P.C.; Mikawa, T.; Fischman, D.A. Evidence for an extracellular matrix bridge guiding proepicardial cell migration to the myocardium of chick embryos. Dev. Dyn. 2003, 227, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.T.; Rayburn, H.; Hynes, R.O. Cell adhesion events mediated by alpha 4 integrins are essential in placental and cardiac development. Development 1995, 121, 549–560. [Google Scholar] [CrossRef]
- Cao, Y.; Duca, S.; Cao, J. Epicardium in Heart Development. Cold Spring Harb. Perspect. Biol. 2020, 12, a037192. [Google Scholar] [CrossRef]
- Dettman, R.W.; Denetclaw, W., Jr.; Ordahl, C.P.; Bristow, J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 1998, 193, 169–181. [Google Scholar] [CrossRef]
- Chong, J.J.; Chandrakanthan, V.; Xaymardan, M.; Asli, N.S.; Li, J.; Ahmed, I.; Heffernan, C.; Menon, M.K.; Scarlett, C.J.; Rashidianfar, A.; et al. Adult cardiac-resident MSC-like stem cells with a proepicardial origin. Cell Stem Cell 2011, 9, 527–540. [Google Scholar] [CrossRef]
- Boezio, G.L.M.; Zhao, S.; Gollin, J.; Priya, R.; Mansingh, S.; Guenther, S.; Fukuda, N.; Gunawan, F.; Stainier, D.Y.R. The developing epicardium regulates cardiac chamber morphogenesis by promoting cardiomyocyte growth. Dis. Model Mech. 2023, 16, dmm049571. [Google Scholar] [CrossRef]
- Weeke-Klimp, A.; Bax, N.A.; Bellu, A.R.; Winter, E.M.; Vrolijk, J.; Plantinga, J.; Maas, S.; Brinker, M.; Mahtab, E.A.; Gittenberger-de Groot, A.C.; et al. Epicardium-derived cells enhance proliferation, cellular maturation and alignment of cardiomyocytes. J. Mol. Cell Cardiol. 2010, 49, 606–616. [Google Scholar] [CrossRef]
- Giacomelli, E.; Meraviglia, V.; Campostrini, G.; Cochrane, A.; Cao, X.; van Helden, R.W.J.; Krotenberg Garcia, A.; Mircea, M.; Kostidis, S.; Davis, R.P.; et al. Human-iPSC-Derived Cardiac Stromal Cells Enhance Maturation in 3D Cardiac Microtissues and Reveal Non-cardiomyocyte Contributions to Heart Disease. Cell Stem Cell 2020, 26, 862–879 e811. [Google Scholar] [CrossRef]
- Linask, K.K. N-cadherin localization in early heart development and polar expression of Na+,K(+)-ATPase, and integrin during pericardial coelom formation and epithelialization of the differentiating myocardium. Dev. Biol. 1992, 151, 213–224. [Google Scholar] [CrossRef]
- Navaratnam, V.; Kaufman, M.H.; Skepper, J.N.; Barton, S.; Guttridge, K.M. Differentiation of the myocardial rudiment of mouse embryos: An ultrastructural study including freeze-fracture replication. J. Anat. 1986, 146, 65–85. [Google Scholar]
- Linask, K.K.; Knudsen, K.A.; Gui, Y.H. N-cadherin-catenin interaction: Necessary component of cardiac cell compartmentalization during early vertebrate heart development. Dev. Biol. 1997, 185, 148–164. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K.; Knudsen, K.A.; Linask, K.K. N-cadherin is required for the differentiation and initial myofibrillogenesis of chick cardiomyocytes. Cell Motil. Cytoskelet. 1998, 39, 52–62. [Google Scholar] [CrossRef]
- Radice, G.L.; Rayburn, H.; Matsunami, H.; Knudsen, K.A.; Takeichi, M.; Hynes, R.O. Developmental defects in mouse embryos lacking N-cadherin. Dev. Biol. 1997, 181, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Vreeker, A.; van Stuijvenberg, L.; Hund, T.J.; Mohler, P.J.; Nikkels, P.G.; van Veen, T.A. Assembly of the cardiac intercalated disk during pre- and postnatal development of the human heart. PLoS ONE 2014, 9, e94722. [Google Scholar] [CrossRef] [PubMed]
- Kostetskii, I.; Li, J.; Xiong, Y.; Zhou, R.; Ferrari, V.A.; Patel, V.V.; Molkentin, J.D.; Radice, G.L. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ. Res. 2005, 96, 346–354. [Google Scholar] [CrossRef]
- Piven, O.O.; Kostetskii, I.E.; Macewicz, L.L.; Kolomiets, Y.M.; Radice, G.L.; Lukash, L.L. Requirement for N-cadherin-catenin complex in heart development. Exp. Biol. Med. 2011, 236, 816–822. [Google Scholar] [CrossRef]
- Gallicano, G.I.; Kouklis, P.; Bauer, C.; Yin, M.; Vasioukhin, V.; Degenstein, L.; Fuchs, E. Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J. Cell Biol. 1998, 143, 2009–2022. [Google Scholar] [CrossRef]
- Ha, J.; Kim, B.S.; Min, B.; Nam, J.; Lee, J.G.; Lee, M.; Yoon, B.H.; Choi, Y.H.; Im, I.; Park, J.S.; et al. Intermediate cells of in vitro cellular reprogramming and in vivo tissue regeneration require desmoplakin. Sci. Adv. 2022, 8, eabk1239. [Google Scholar] [CrossRef]
- Heuser, A.; Plovie, E.R.; Ellinor, P.T.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Moazzen, H.; Venger, K.; Kant, S.; Leube, R.E.; Krusche, C.A. Desmoglein 2 regulates cardiogenesis by restricting hematopoiesis in the developing murine heart. Sci. Rep. 2021, 11, 21687. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.D.; Moriarty, M.A.; Byrnes, L.; Grealy, M. Plakoglobin has both structural and signalling roles in zebrafish development. Dev. Biol. 2009, 327, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Bierkamp, C.; McLaughlin, K.J.; Schwarz, H.; Huber, O.; Kemler, R. Embryonic heart and skin defects in mice lacking plakoglobin. Dev. Biol. 1996, 180, 780–785. [Google Scholar] [CrossRef]
- Schinner, C.; Xu, L.; Franz, H.; Zimmermann, A.; Wanuske, M.T.; Rathod, M.; Hanns, P.; Geier, F.; Pelczar, P.; Liang, Y.; et al. Defective Desmosomal Adhesion Causes Arrhythmogenic Cardiomyopathy by Involving an Integrin-alphaVbeta6/TGF-beta Signaling Cascade. Circulation 2022, 146, 1610–1626. [Google Scholar] [CrossRef]
- Ruiz, P.; Brinkmann, V.; Ledermann, B.; Behrend, M.; Grund, C.; Thalhammer, C.; Vogel, F.; Birchmeier, C.; Gunthert, U.; Franke, W.W.; et al. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J. Cell Biol. 1996, 135, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.D.; Grealy, M. Plakoglobin expression and localization in zebrafish embryo development. Biochem. Soc. Trans. 2004, 32, 797–798. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, M.A.; Ryan, R.; Lalor, P.; Dockery, P.; Byrnes, L.; Grealy, M. Loss of plakophilin 2 disrupts heart development in zebrafish. Int. J. Dev. Biol. 2012, 56, 711–718. [Google Scholar] [CrossRef]
- Moriarty, M.A.; Martin, E.D.; Byrnes, L.; Grealy, M. Molecular cloning and developmental expression of plakophilin 2 in zebrafish. Biochem. Biophys. Res. Commun. 2008, 367, 124–129. [Google Scholar] [CrossRef]
- Sanchez-Fernandez, C.; Rodriguez-Outeirino, L.; Matias-Valiente, L.; Ramirez de Acuna, F.; Hernandez-Torres, F.; Lozano-Velasco, E.; Dominguez, J.N.; Franco, D.; Aranega, A.E. Regulation of Epicardial Cell Fate during Cardiac Development and Disease: An Overview. Int. J. Mol. Sci. 2022, 23, 3220. [Google Scholar] [CrossRef]
- Bannerman, D.; Pascual-Gil, S.; Floryan, M.; Radisic, M. Bioengineering strategies to control epithelial-to-mesenchymal transition for studies of cardiac development and disease. APL Bioeng. 2021, 5, 021504. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Freytag, B.; Herzog, A.; Reich, A.; Merkel, R.; Hoffmann, B.; Krusche, C.A.; Leube, R.E. Desmoglein 2 mutation provokes skeletal muscle actin expression and accumulation at intercalated discs in murine hearts. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed]
- Oxford, E.M.; Musa, H.; Maass, K.; Coombs, W.; Taffet, S.M.; Delmar, M. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ. Res. 2007, 101, 703–711. [Google Scholar] [CrossRef]
- Dubash, A.D.; Kam, C.Y.; Aguado, B.A.; Patel, D.M.; Delmar, M.; Shea, L.D.; Green, K.J. Plakophilin-2 loss promotes TGF-beta1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes. J. Cell Biol. 2016, 212, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Kam, C.Y.; Dubash, A.D.; Magistrati, E.; Polo, S.; Satchell, K.J.F.; Sheikh, F.; Lampe, P.D.; Green, K.J. Desmoplakin maintains gap junctions by inhibiting Ras/MAPK and lysosomal degradation of connexin-43. J. Cell Biol. 2018, 217, 3219–3235. [Google Scholar] [CrossRef] [PubMed]
- Agullo-Pascual, E.; Reid, D.A.; Keegan, S.; Sidhu, M.; Fenyo, D.; Rothenberg, E.; Delmar, M. Super-resolution fluorescence microscopy of the cardiac connexome reveals plakophilin-2 inside the connexin43 plaque. Cardiovasc. Res. 2013, 100, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Rhee, D.Y.; Zhao, X.Q.; Francis, R.J.; Huang, G.Y.; Mably, J.D.; Lo, C.W. Connexin 43 regulates epicardial cell polarity and migration in coronary vascular development. Development 2009, 136, 3185–3193. [Google Scholar] [CrossRef] [PubMed]
- Li, W.E.; Waldo, K.; Linask, K.L.; Chen, T.; Wessels, A.; Parmacek, M.S.; Kirby, M.L.; Lo, C.W. An essential role for connexin43 gap junctions in mouse coronary artery development. Development 2002, 129, 2031–2042. [Google Scholar] [CrossRef]
- Dai, P.; Nakagami, T.; Tanaka, H.; Hitomi, T.; Takamatsu, T. Cx43 mediates TGF-beta signaling through competitive Smads binding to microtubules. Mol. Biol. Cell 2007, 18, 2264–2273. [Google Scholar] [CrossRef]
- Fukuda, S.; Akiyama, M.; Harada, H.; Nakahama, K.I. Effect of gap junction-mediated intercellular communication on TGF-beta induced epithelial-to-mesenchymal transition. Biochem. Biophys. Res. Commun. 2019, 508, 928–933. [Google Scholar] [CrossRef]
- Lim, M.C.; Maubach, G.; Zhuo, L. TGF-beta1 down-regulates connexin 43 expression and gap junction intercellular communication in rat hepatic stellate cells. Eur. J. Cell Biol. 2009, 88, 719–730. [Google Scholar] [CrossRef] [PubMed]
- de Boer, T.P.; van Veen, T.A.; Bierhuizen, M.F.; Kok, B.; Rook, M.B.; Boonen, K.J.; Vos, M.A.; Doevendans, P.A.; de Bakker, J.M.; van der Heyden, M.A. Connexin43 repression following epithelium-to-mesenchyme transition in embryonal carcinoma cells requires Snail1 transcription factor. Differentiation 2007, 75, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Lyon, H.; Yin, N.; Rupenthal, I.D.; Green, C.R.; Mugisho, O.O. Blocking connexin43 hemichannels prevents TGF-beta2 upregulation and epithelial-mesenchymal transition in retinal pigment epithelial cells. Cell Biol. Int. 2022, 46, 323–330. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, W.; Wei, J.; Cui, Y.; Zhang, D.; Xie, J. Transforming growth factor-beta1-induced N-cadherin drives cell-cell communication through connexin43 in osteoblast lineage. Int. J. Oral Sci. 2021, 13, 15. [Google Scholar] [CrossRef]
- Kahata, K.; Dadras, M.S.; Moustakas, A. TGF-beta Family Signaling in Epithelial Differentiation and Epithelial-Mesenchymal Transition. Cold Spring Harb. Perspect. Biol. 2018, 10, a022194. [Google Scholar] [CrossRef] [PubMed]
- Kwee, L.; Baldwin, H.S.; Shen, H.M.; Stewart, C.L.; Buck, C.; Buck, C.A.; Labow, M.A. Defective development of the embryonic and extraembryonic circulatory systems in vascular cell adhesion molecule (VCAM-1) deficient mice. Development 1995, 121, 489–503. [Google Scholar] [CrossRef]
- Dokic, D.; Dettman, R.W. VCAM-1 inhibits TGFbeta stimulated epithelial-mesenchymal transformation by modulating Rho activity and stabilizing intercellular adhesion in epicardial mesothelial cells. Dev. Biol. 2006, 299, 489–504. [Google Scholar] [CrossRef]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S.; et al. Interactions between beta-catenin and transforming growth factor-beta signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038. [Google Scholar] [CrossRef]
- Cho, H.J.; Yoo, J. Rho activation is required for transforming growth factor-beta-induced epithelial-mesenchymal transition in lens epithelial cells. Cell Biol. Int. 2007, 31, 1225–1230. [Google Scholar] [CrossRef]
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.H.; Moustakas, A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell 2005, 16, 1987–2002. [Google Scholar] [CrossRef]
- Zavadil, J.; Cermak, L.; Soto-Nieves, N.; Bottinger, E.P. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004, 23, 1155–1165. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertran, E.; Perez-Pomares, J.M.; Diez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisua-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef]
- del Monte, G.; Casanova, J.C.; Guadix, J.A.; MacGrogan, D.; Burch, J.B.; Perez-Pomares, J.M.; de la Pompa, J.L. Differential Notch signaling in the epicardium is required for cardiac inflow development and coronary vessel morphogenesis. Circ. Res. 2011, 108, 824–836. [Google Scholar] [CrossRef]
- Dimitrova, Y.; Gruber, A.J.; Mittal, N.; Ghosh, S.; Dimitriades, B.; Mathow, D.; Grandy, W.A.; Christofori, G.; Zavolan, M. TFAP2A is a component of the ZEB1/2 network that regulates TGFB1-induced epithelial to mesenchymal transition. Biol. Direct. 2017, 12, 8. [Google Scholar] [CrossRef]
- Xiong, Y.; Feng, Y.; Zhao, J.; Lei, J.; Qiao, T.; Zhou, Y.; Lu, Q.; Jiang, T.; Jia, L.; Han, Y. TFAP2A potentiates lung adenocarcinoma metastasis by a novel miR-16 family/TFAP2A/PSG9/TGF-beta signaling pathway. Cell Death Dis. 2021, 12, 352. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Luo, J.; Hou, N. Molecular Mechanism of Hippo-YAP1/TAZ Pathway in Heart Development, Disease, and Regeneration. Front. Physiol. 2020, 11, 389. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef]
- Hu, Y.; Pu, W.T. Hippo activation in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 402–405. [Google Scholar] [CrossRef]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef]
- Yang, Y.; Ren, J.; Sun, Y.; Xue, Y.; Zhang, Z.; Gong, A.; Wang, B.; Zhong, Z.; Cui, Z.; Xi, Z.; et al. A connexin43/YAP axis regulates astroglial-mesenchymal transition in hemoglobin induced astrocyte activation. Cell Death Differ. 2018, 25, 1870–1884. [Google Scholar] [CrossRef]
- Narimatsu, M.; Samavarchi-Tehrani, P.; Varelas, X.; Wrana, J.L. Distinct polarity cues direct Taz/Yap and TGFbeta receptor localization to differentially control TGFbeta-induced Smad signaling. Dev. Cell 2015, 32, 652–656. [Google Scholar] [CrossRef]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef]
- Kant, S.; Holthofer, B.; Magin, T.M.; Krusche, C.A.; Leube, R.E. Desmoglein 2-Dependent Arrhythmogenic Cardiomyopathy Is Caused by a Loss of Adhesive Function. Circ. Cardiovasc. Genet. 2015, 8, 553–563. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Cardiogenesis Phenotype | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | Organism | Edema | Pericardial Blood | Hypoplastic Myocard | Defective Contraction | Disrupted Myocardial Patterning | Defective Intercellular Adhesion | Rupture | Perturbed Endocardial Differentiation | Reduced Desmosomal Plaque | Refs. |
| Dsc2morpholino | Zebrafish | × | × | × | [71] | ||||||
| Dsg2ΔE4–E6 | Mouse | × | × | × | × | × | × | × | [72] | ||
| Dsg2W2A | Mouse | × | × | [75] | |||||||
| Pkp2morpholino | Zebrafish | × | × | [73] | |||||||
| Pkp2−/− | Mouse | × | × | × | × | [17] | |||||
| Jup−/− | Mouse | × | × | × | × | × | × | [74,76] | |||
| Dsp−/− extraembryonal rescue | Mouse | × | × | × | × | [16] | |||||
| Mutation | Cell/Animal Model | Pathway | Refs. |
|---|---|---|---|
| Dsg2RNAi | Human pluripotent stem cells (hPSCs) | Inhibition of E-cadherin and elevation of Slug | [15] |
| Dsg2ΔE4–E6 Dsg2ΔE4–E5 myocardial induction | Adult murine heart | TGFβ and SRF signaling | [82] |
| Dsg2W2A | Adult murine heart | Integrin-αVβ6/TGF-β signaling | [75] |
| Pkp2RNAi | Neonatal rat ventricular myocytes (NRVMs) Epcardial-mesenchymal cells (EPDCs) | Reduction and redistribution of Cx43 | [83] |
| Pkp2RNAi | Neonatal rat ventricular myocytes (NRVMs) | TGF-β1/p38 MAPK kinase signaling | [84] |
| Pkp2c.del2013c | Human-induced pluripotent stem cell- derived (hiPSC)-epicardial cells | Activation of TFAP2A | [22] |
| DspRNAi | Neonatal rat ventricular myocytes (NRVMs) Murine HL-1 atrial cardiomyocytes | ERK1/2-MAPK signaling Phosphorylation and degradation of Cx43 | [85] |
| DspW/F epicardial induction | Adult Mouse Heart | FGF2 and TGF-β1 signaling | [21] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moazzen, H.; Bolaji, M.D.; Leube, R.E. Desmosomes in Cell Fate Determination: From Cardiogenesis to Cardiomyopathy. Cells 2023, 12, 2122. https://doi.org/10.3390/cells12172122
Moazzen H, Bolaji MD, Leube RE. Desmosomes in Cell Fate Determination: From Cardiogenesis to Cardiomyopathy. Cells. 2023; 12(17):2122. https://doi.org/10.3390/cells12172122
Chicago/Turabian StyleMoazzen, Hoda, Mistura Dolapo Bolaji, and Rudolf E. Leube. 2023. "Desmosomes in Cell Fate Determination: From Cardiogenesis to Cardiomyopathy" Cells 12, no. 17: 2122. https://doi.org/10.3390/cells12172122
APA StyleMoazzen, H., Bolaji, M. D., & Leube, R. E. (2023). Desmosomes in Cell Fate Determination: From Cardiogenesis to Cardiomyopathy. Cells, 12(17), 2122. https://doi.org/10.3390/cells12172122