
Copy Number Variation That Influences the Ionizing Radiation Sensitivity of Oral Squamous Cell Carcinoma
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical and RNAseq Data of TCGA
2.2. Associating Gene Coordinate to TCGA RNAseq Data
2.3. Genomic Copy Number Assessment of TCGA SNP Array
2.4. Cell Lines
2.5. Transient RNA Interference with siRNA
2.6. Stable Cell Lines with Lentiviral shRNA Vectors
2.7. Immunoblot Assay
2.8. Comet Assay
2.9. X-ray Survival Assay
2.10. Determination of Copy Number Variation of OSCC Tissues
2.11. Tumor Growth Assay in Mouse Xenograft
2.12. Statistical Methods
3. Results
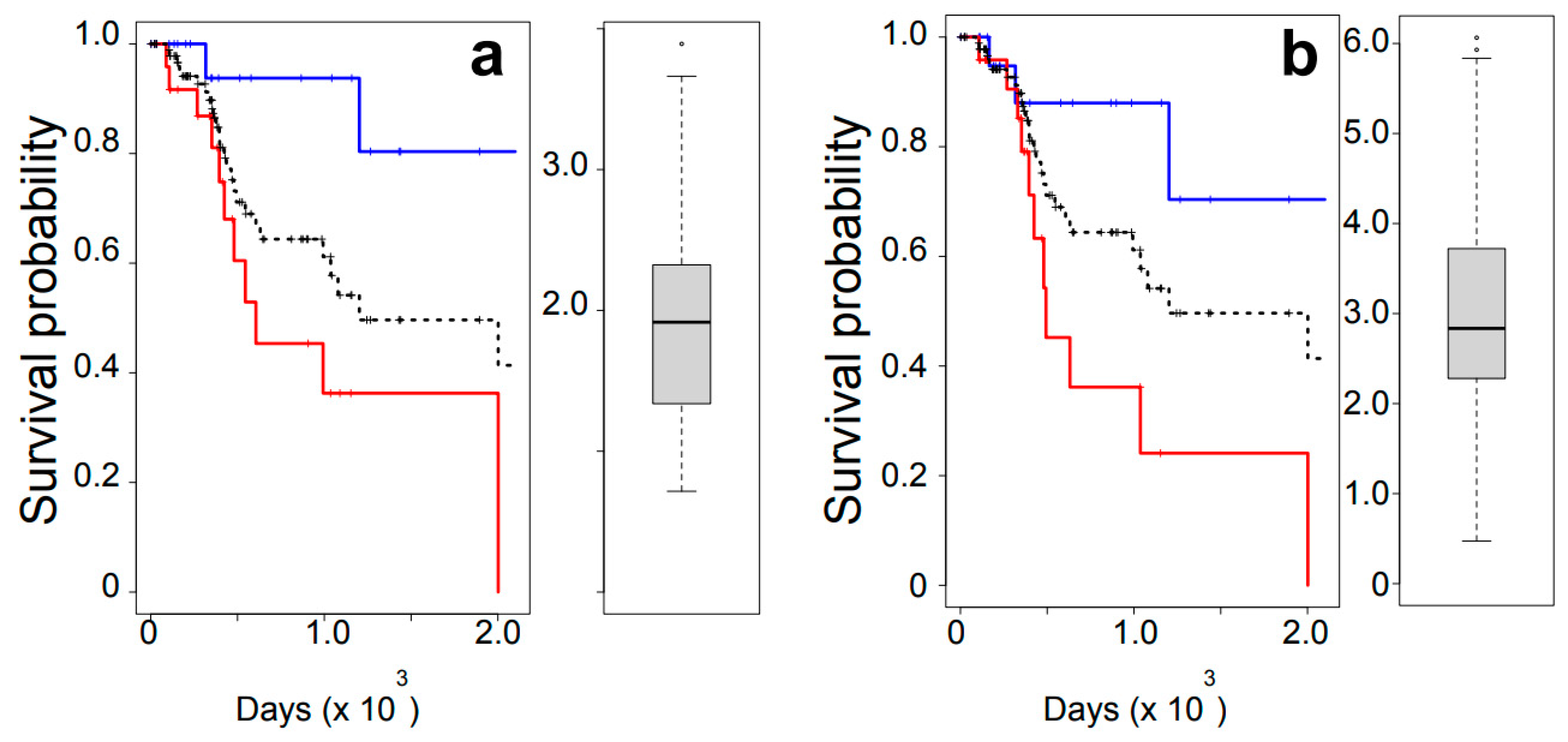
3.1. Overall Survival Analysis
3.2. Association of High Expression of APE1 and PRMT5 with Outcomes of Radiation Therapy
3.3. Tumor-Specific Co-Regulation of Genes with PRMT5
3.4. Copy Number Variation as the Main Driver of the High Correlation of PRMT5-APE1 Expression
3.5. Synergistic Sensitization of Cells to IR via Simultaneous Down-Regulation of PRMT5 and APE1
3.6. Synergistic Sensitization of Tumors in Xenograft
3.7. CNV at the APE1-PRMT5 Gene Region in OSCC Tumors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; McKenna, W.G.; Weber, C.N.; Feldman, M.D.; Goldsmith, J.D.; Mick, R.; Machtay, M.; Rosenthal, D.I.; Bakanauskas, V.J.; Cerniglia, G.J.; et al. Local recurrence in head and neck cancer: Relationship to radiation resistance and signal transduction. Clin. Cancer Res. 2002, 8, 885–892. [Google Scholar] [PubMed]
- Murakami, N.; Matsumoto, F.; Yoshimoto, S.; Ito, Y.; Mori, T.; Ueno, T.; Tuchida, K.; Kashihara, T.; Kobayashi, K.; Harada, K.; et al. Patterns of recurrence after selective postoperative radiation therapy for patients with head and neck squamous cell carcinoma. BMC Cancer 2016, 16, 192. [Google Scholar] [CrossRef]
- Blatt, S.; Voelxen, N.; Sagheb, K.; Pabst, A.M.; Walenta, S.; Schroeder, T.; Mueller-Klieser, W.; Ziebart, T. Lactate as a predictive marker for tumor recurrence in patients with head and neck squamous cell carcinoma (HNSCC) post radiation: A prospective study over 15 years. Clin. Oral. Investig. 2016, 20, 2097–2104. [Google Scholar] [CrossRef]
- Michna, A.; Schotz, U.; Selmansberger, M.; Zitzelsberger, H.; Lauber, K.; Unger, K.; Hess, J. Transcriptomic analyses of the radiation response in head and neck squamous cell carcinoma subclones with different radiation sensitivity: Time-course gene expression profiles and gene association networks. Radiat. Oncol. 2016, 11, 94. [Google Scholar] [CrossRef]
- Echarri, M.J.; Lopez-Martin, A.; Hitt, R. Targeted Therapy in Locally Advanced and Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma (LA-R/M HNSCC). Cancers 2016, 8, 27. [Google Scholar] [CrossRef]
- Su, Z.; Li, G.; Liu, C.; Ren, S.; Tian, Y.; Liu, Y.; Qiu, Y. Ionizing radiation promotes advanced malignant traits in nasopharyngeal carcinoma via activation of epithelial-mesenchymal transition and the cancer stem cell phenotype. Oncol. Rep. 2016, 36, 72–78. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Moncharmont, C.; Levy, A.; Gilormini, M.; Bertrand, G.; Chargari, C.; Alphonse, G.; Ardail, D.; Rodriguez-Lafrasse, C.; Magne, N. Targeting a cornerstone of radiation resistance: Cancer stem cell. Cancer Lett. 2012, 322, 139–147. [Google Scholar] [CrossRef]
- Breen, A.P.; Murphy, J.A. Reactions of oxyl radicals with DNA. Free Radic. Biol. Med. 1995, 18, 1033–1077. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog. Nucleic Acid. Res. Mol. Biol. 1988, 35, 95–125. [Google Scholar] [PubMed]
- Kievit, F.M.; Stephen, Z.R.; Wang, K.; Dayringer, C.J.; Sham, J.G.; Ellenbogen, R.G.; Silber, J.R.; Zhang, M. Nanoparticle mediated silencing of DNA repair sensitizes pediatric brain tumor cells to gamma-irradiation. Mol. Oncol. 2015, 9, 1071–1080. [Google Scholar] [CrossRef]
- Bansal, N.; Mims, J.; Kuremsky, J.G.; Olex, A.L.; Zhao, W.; Yin, L.; Wani, R.; Qian, J.; Center, B.; Marrs, G.S.; et al. Broad phenotypic changes associated with gain of radiation resistance in head and neck squamous cell cancer. Antioxid. Redox Signal. 2014, 21, 221–236. [Google Scholar] [CrossRef]
- Naidu, M.D.; Mason, J.M.; Pica, R.V.; Fung, H.; Pena, L.A. Radiation resistance in glioma cells determined by DNA damage repair activity of Ape1/Ref-1. J. Radiat. Res. 2010, 51, 393–404. [Google Scholar] [CrossRef]
- Liu, X.; Han, E.K.; Anderson, M.; Shi, Y.; Semizarov, D.; Wang, G.; McGonigal, T.; Roberts, L.; Lasko, L.; Palma, J.; et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol. Cancer Res. 2009, 7, 1686–1692. [Google Scholar] [CrossRef]
- Essers, J.; Hendriks, R.W.; Swagemakers, S.M.; Troelstra, C.; de Wit, J.; Bootsma, D.; Hoeijmakers, J.H.; Kanaar, R. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell 1997, 89, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Owens, J.L.; Beketova, E.; Liu, S.; Tinsley, S.L.; Asberry, A.M.; Deng, X.; Huang, J.; Li, C.; Wan, J.; Hu, C.D. PRMT5 Cooperates with pICln to Function as a Master Epigenetic Activator of DNA Double-Strand Break Repair Genes. iScience 2019, 23, 100750. [Google Scholar] [CrossRef]
- Clarke, T.L.; Sanchez-Bailon, M.P.; Chiang, K.; Reynolds, J.J.; Herrero-Ruiz, J.; Bandeiras, T.M.; Matias, P.M.; Maslen, S.L.; Skehel, J.M.; Stewart, G.S.; et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol. Cell 2017, 65, 900–916.e7. [Google Scholar] [CrossRef]
- Scott, T.L.; Rangaswamy, S.; Wicker, C.A.; Izumi, T. Repair of oxidative DNA damage and cancer: Recent progress in DNA base excision repair. Antioxid. Redox Signal. 2014, 20, 708–726. [Google Scholar] [CrossRef]
- Izumi, T.; Hazra, T.K.; Boldogh, I.; Tomkinson, A.E.; Park, M.S.; Ikeda, S.; Mitra, S. Requirement for human AP endonuclease 1 for repair of 3′-blocking damage at DNA single-strand breaks induced by reactive oxygen species. Carcinogenesis 2000, 21, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Fung, H.; Demple, B. Distinct roles of Ape1 protein in the repair of DNA damage induced by ionizing radiation or bleomycin. J. Biol. Chem. 2011, 286, 4968–4977. [Google Scholar] [CrossRef] [PubMed]
- Wicker, C.A.; Takiar, V.; Suganya, R.; Arnold, S.M.; Brill, Y.M.; Chen, L.; Horbinski, C.M.; Napier, D.; Valentino, J.; Kudrimoti, M.R.; et al. Evaluation of antioxidant network proteins as novel prognostic biomarkers for head and neck cancer patients. Oral Oncol. 2020, 111, 104949. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ronai, Z.A. PRMT5 function and targeting in cancer. Cell Stress 2020, 4, 199–215. [Google Scholar] [CrossRef]
- Shih, J.; Sarmashghi, S.; Zhakula-Kostadinova, N.; Zhang, S.; Georgis, Y.; Hoyt, S.H.; Cuoco, M.S.; Gao, G.F.; Spurr, L.F.; Berger, A.C.; et al. Cancer aneuploidies are shaped primarily by effects on tumour fitness. Nature 2023, 619, 793–800. [Google Scholar] [CrossRef]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000. [Google Scholar]
- Rangan, S.R. A new human cell line (FaDu) from a hypopharyngeal carcinoma. Cancer 1972, 29, 117–121. [Google Scholar] [CrossRef]
- Scott, T.L.; Wicker, C.A.; Suganya, R.; Dhar, B.; Pittman, T.; Horbinski, C.; Izumi, T. Polyubiquitination of apurinic/apyrimidinic endonuclease 1 by Parkin. Mol. Carcinog. 2017, 56, 325–336. [Google Scholar] [CrossRef]
- Tice, R.R.; Agurell, E.; Anderson, D.; Burlinson, B.; Hartmann, A.; Kobayashi, H.; Miyamae, Y.; Rojas, E.; Ryu, J.C.; Sasaki, Y.F. Single cell gel/comet assay: Guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 2000, 35, 206–221. [Google Scholar] [CrossRef]
- Chen, Q.; Molloy, J.; Izumi, T.; Sterpin, E. Impact of backscatter material thickness on the depth dose of orthovoltage irradiators for radiobiology research. Phys. Med. Biol. 2019, 64, 055001. [Google Scholar] [CrossRef]
- Chow, Z.; Johnson, J.; Chauhan, A.; Izumi, T.; Cavnar, M.; Weiss, H.; Townsend, C.M., Jr.; Anthony, L.; Wasilchenko, C.; Melton, M.L.; et al. PI3K/mTOR Dual Inhibitor PF-04691502 Is a Schedule-Dependent Radiosensitizer for Gastroenteropancreatic Neuroendocrine Tumors. Cells 2021, 10, 1261. [Google Scholar] [CrossRef]
- Hamada, N.; Funayama, T.; Wada, S.; Sakashita, T.; Kakizaki, T.; Ni, M.; Kobayashi, Y. LET-dependent survival of irradiated normal human fibroblasts and their descendents. Radiat. Res. 2006, 166, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T. Analysis of Copy Number Variation of DNA Repair/Damage Response Genes in Tumor Tissues. Methods Mol. Biol. 2023, 2701, 231–242. [Google Scholar] [PubMed]
- Sukati, S.; Ho, J.; Chaiswing, L.; Sompol, P.; Pandit, H.; Wei, W.; Izumi, T.; Chen, Q.; Weiss, H.; Noel, T.; et al. Extracellular vesicles released after cranial radiation: An insight into an early mechanism of brain injury. Brain Res. 2022, 1782, 147840. [Google Scholar] [CrossRef]
- Enot, D.P.; Vacchelli, E.; Jacquelot, N.; Zitvogel, L.; Kroemer, G. TumGrowth: An open-access web tool for the statistical analysis of tumor growth curves. Oncoimmunology 2018, 7, e1462431. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.; Hatahet, Z.; Wallace, S.S. In vitro repair of synthetic ionizing radiation-induced multiply damaged DNA sites. J. Mol. Biol. 1999, 290, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Dumitrache, L.C.; Shimada, M.; Downing, S.M.; Kwak, Y.D.; Li, Y.; Illuzzi, J.L.; Russell, H.R.; Wilson, D.M., 3rd; McKinnon, P.J. Apurinic endonuclease-1 preserves neural genome integrity to maintain homeostasis and thermoregulation and prevent brain tumors. Proc. Natl. Acad. Sci. USA 2018, 115, E12285–E12294. [Google Scholar] [CrossRef]
- Silber, J.R.; Bobola, M.S.; Blank, A.; Schoeler, K.D.; Haroldson, P.D.; Huynh, M.B.; Kolstoe, D.D. The apurinic/apyrimidinic endonuclease activity of Ape1/Ref-1 contributes to human glioma cell resistance to alkylating agents and is elevated by oxidative stress. Clin. Cancer Res. 2002, 8, 3008–3018. [Google Scholar]
- Koukourakis, M.I.; Giatromanolaki, A.; Kakolyris, S.; Sivridis, E.; Georgoulias, V.; Funtzilas, G.; Hickson, I.D.; Gatter, K.C.; Harris, A.L. Nuclear expression of human apurinic/apyrimidinic endonuclease (HAP1/Ref-1) in head-and-neck cancer is associated with resistance to chemoradiotherapy and poor outcome. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 27–36. [Google Scholar] [CrossRef]
- Bobola, M.S.; Blank, A.; Berger, M.S.; Stevens, B.A.; Silber, J.R. Apurinic/apyrimidinic endonuclease activity is elevated in human adult gliomas. Clin. Cancer Res. 2001, 7, 3510–3518. [Google Scholar]
- Bobola, M.S.; Emond, M.J.; Blank, A.; Meade, E.H.; Kolstoe, D.D.; Berger, M.S.; Rostomily, R.C.; Silbergeld, D.L.; Spence, A.M.; Silber, J.R. Apurinic endonuclease activity in adult gliomas and time to tumor progression after alkylating agent-based chemotherapy and after radiotherapy. Clin. Cancer Res. 2004, 10, 7875–7883. [Google Scholar] [CrossRef]
- Oliveira, D.V.; Kato, A.; Nakamura, K.; Ikura, T.; Okada, M.; Kobayashi, J.; Yanagihara, H.; Saito, Y.; Tauchi, H.; Komatsu, K. Histone chaperone FACT regulates homologous recombination by chromatin remodeling through interaction with RNF20. J. Cell Sci. 2014, 127, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Bilokapic, S.; Suskiewicz, M.J.; Ahel, I.; Halic, M. Bridging of DNA breaks activates PARP2-HPF1 to modify chromatin. Nature 2020, 585, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Bounaix Morand du Puch, C.; Barbier, E.; Kraut, A.; Coute, Y.; Fuchs, J.; Buhot, A.; Livache, T.; Seve, M.; Favier, A.; Douki, T.; et al. TOX4 and its binding partners recognize DNA adducts generated by platinum anticancer drugs. Arch. Biochem. Biophys. 2011, 507, 296–303. [Google Scholar] [CrossRef]
- Xiang, Y.; Laurent, B.; Hsu, C.H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Correlation Coefficient (vs. PRMT5) | ||
|---|---|---|---|
| Matched (N = 43) | All Tumors (N = 522) | ||
| Normal | Tumor | Tumor | |
| PRMT5 | 1 | 1 | 1 |
| BRCA1 | 0.367 * | 0.18 | 0.124 * |
| BRCA2 | 0.306 * | 0.265 | −0.024 |
| NHEJ1 | −0.057 | −0.037 | 0.098 * |
| PRKDC | 0.289 | 0.4 * | 0.147 ** |
| RAD51AP1 | 0.566 ** | 0.251 | 0.115 ** |
| RAD51 | 0.708 ** | 0.239 | 0.17 ** |
| XRCC4 | 0.446 ** | 0.112 | 0.045 |
| XRCC5 | 0.625 ** | −0.096 | 0.01 |
| XRCC6 | 0.726 ** | 0.348 * | 0.274 ** |
| Gene Symbol | r | p | Cytoband |
|---|---|---|---|
| APEX1 | 0.667 | 2.07 × 10−64 | 14q11.2 |
| NGDN | 0.664 | 1.93 × 10−63 | 14q11.2 |
| IPO4 | 0.663 | 2.49 × 10−63 | 14q12 |
| OXA1L | 0.618 | 4.43 × 10−52 | 14q11.2 |
| SUPT16H | 0.615 | 1.71 × 10−51 | 14q11.2 |
| C14orf119 | 0.612 | 1.14 × 10−50 | 14q11.2 |
| PSMB5 | 0.601 | 2.62 × 10−48 | 14q11.2 |
| GMPR2 | 0.538 | 2.85 × 10−36 | 14q12 |
| PARP2 | 0.530 | 5.60 × 10−35 | 14q11.2 |
| METTL3 | 0.529 | 8.76 × 10−35 | 14q11.1 |
| TOX4 | 0.527 | 1.64 × 10−34 | 14q11.2 |
| AP4S1 | 0.527 | 3.34 × 10−32 | 14q12 |
| X-ray | Post Incubation | Number of Cells Analyzed | Tail Moment | ||
|---|---|---|---|---|---|
| Mean ± sd | t-Test (vs. shBoth) | ||||
| WT | - | - | 277 | 1.46 ± 1.29 | |
| 4.5 Gy | 24 h | 152 | 0.77 ± 0.74 | <0.001 | |
| shBoth | - | - | 161 | 2.23 ± 2.64 | |
| 4.5 Gy | 24 h | 125 | 5.62 ± 6.25 | - | |
| shAPE1 | - | - | 165 | 0.58 ± 2.70 | |
| 4.5 Gy | 24 h | 106 | 0.27 ± 0.69 | <0.001 | |
| shPRMT5 | - | - | 89 | 0.09 ± 0.21 | |
| 4.5 Gy | 24 h | 101 | 0.49 ± 2.28 | <0.001 | |
| X-ray | Pairs | p-Value |
|---|---|---|
| - | WT vs. shPRMT5 | 1 |
| WT vs. shAPE1 | 0.247 | |
| WT vs. shBoth | 1 | |
| + | WT vs. shPRMT5 | 0.151 |
| WT vs. shAPE1 | 0.762 | |
| WT vs. shBoth | <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izumi, T.; Rychahou, P.; Chen, L.; Smith, M.H.; Valentino, J. Copy Number Variation That Influences the Ionizing Radiation Sensitivity of Oral Squamous Cell Carcinoma. Cells 2023, 12, 2425. https://doi.org/10.3390/cells12202425
Izumi T, Rychahou P, Chen L, Smith MH, Valentino J. Copy Number Variation That Influences the Ionizing Radiation Sensitivity of Oral Squamous Cell Carcinoma. Cells. 2023; 12(20):2425. https://doi.org/10.3390/cells12202425
Chicago/Turabian StyleIzumi, Tadahide, Piotr Rychahou, Li Chen, Molly H. Smith, and Joseph Valentino. 2023. "Copy Number Variation That Influences the Ionizing Radiation Sensitivity of Oral Squamous Cell Carcinoma" Cells 12, no. 20: 2425. https://doi.org/10.3390/cells12202425