
Assessment of Fallopian Tube Epithelium Features Derived from Induced Pluripotent Stem Cells of Both Fallopian Tube and Skin Origins

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Establishing iPSCs from FTECs
2.1.1. Ethics
2.1.2. FTEC Culture
2.1.3. Induction Protocol
2.1.4. iPSC Pluripotency Characterization
2.2. qPCR
2.3. iPSC IHC and Three Germ Layer Differentiation
2.4. Chromosomal Analysis
2.5. Animal Teratoma Formation Experiments
2.6. Histological Examination
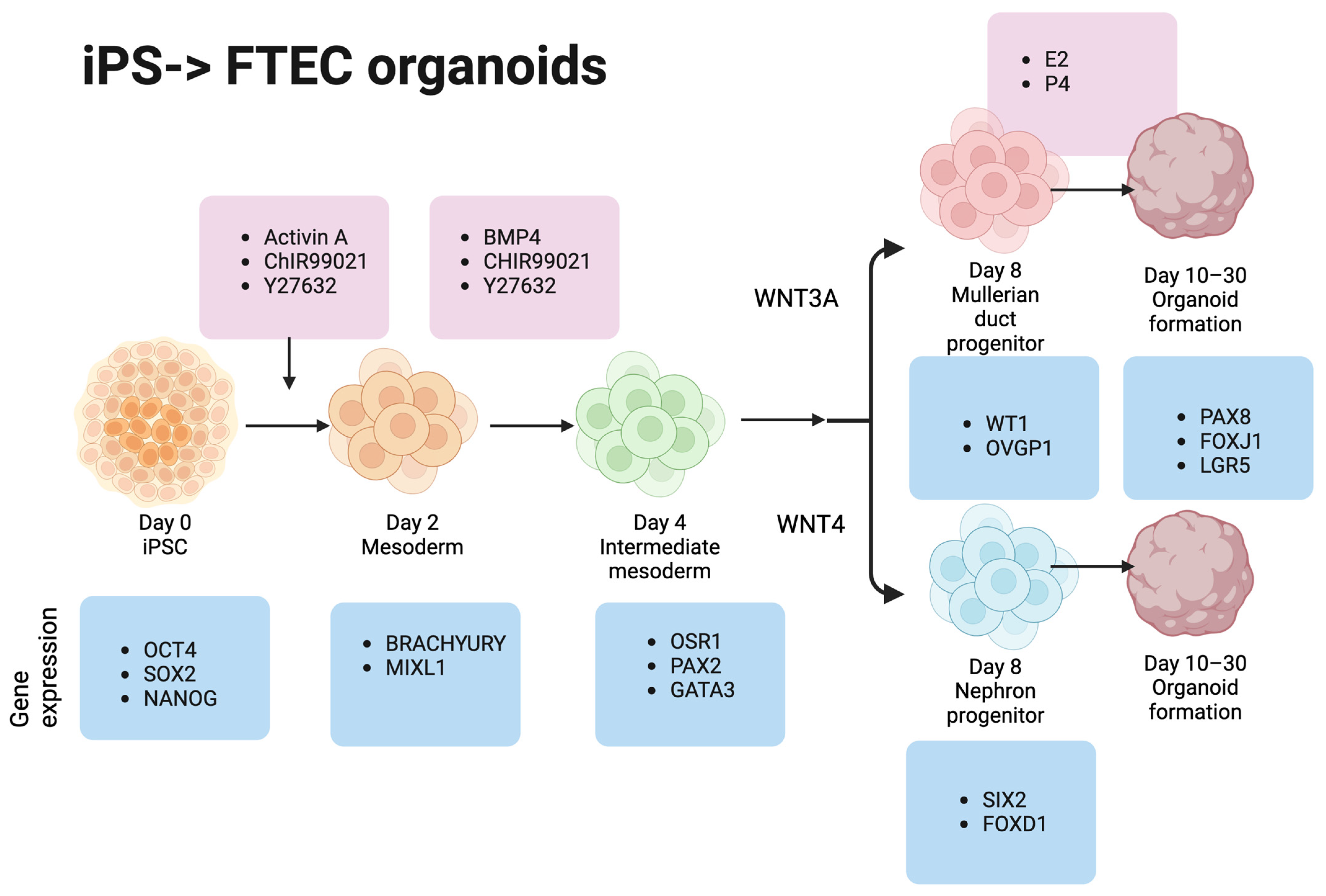
2.7. iPSC Differentiation to FTE
2.8. iPSCs to Mesoderm Differentiation (Day 0–2)
2.9. Mesoderm to Intermediate Mesoderm Differentiation (Days 2–4)
2.10. Spheroid to Mullerian Duct Epithelium Differentiation (Days 4–6)
2.11. FTE Differentiation (Days 6–8)
2.12. Growing FTE Organoids from Spheroids in the Matrigel
2.13. Organoid IHC
2.14. Statistical Analysis
3. Results
3.1. FTEC-iPSCs and Xenograft Tumor Characteristics
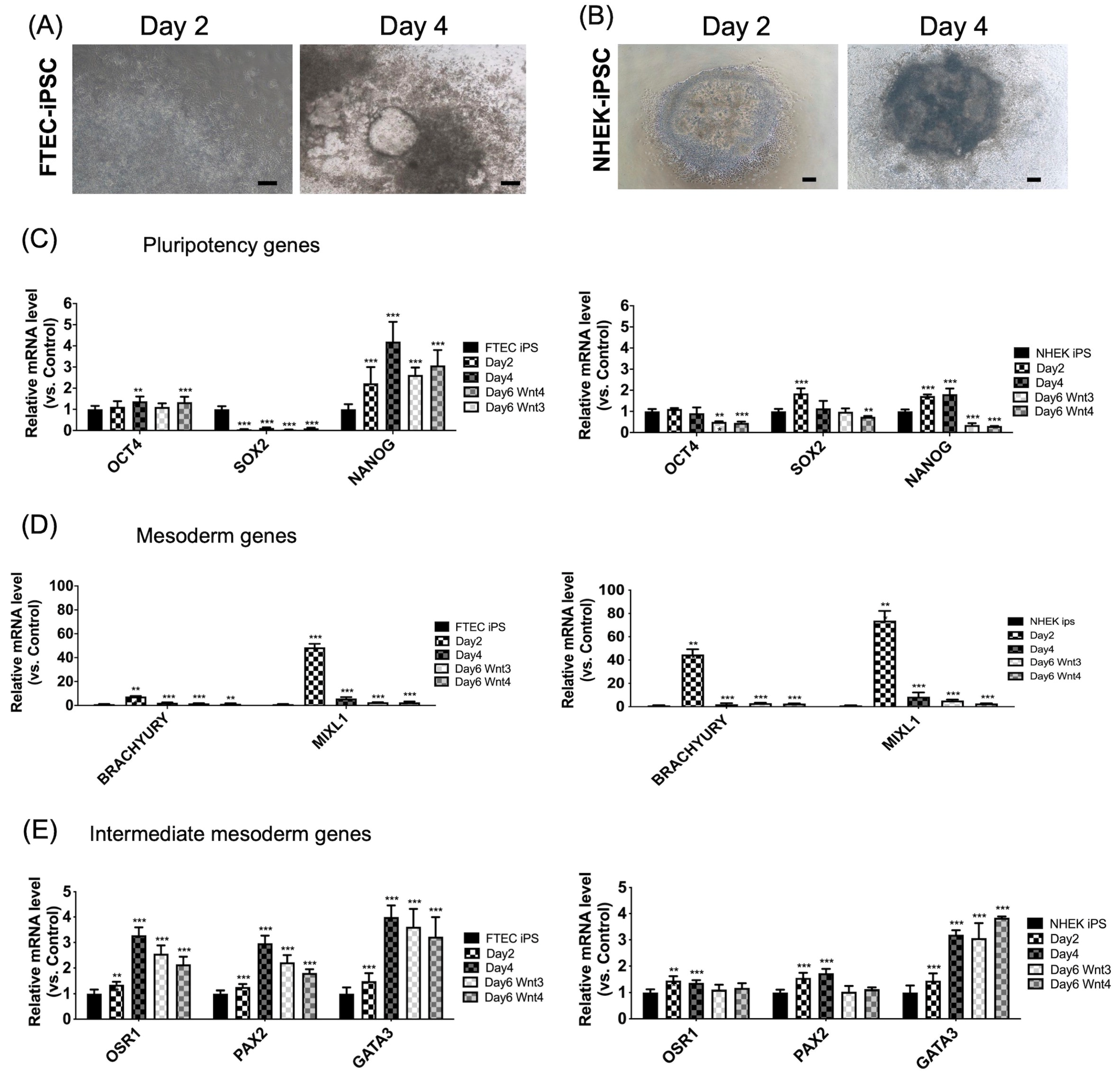
3.2. Sequential Differentiation of FTEC-iPSCs from iPSCs to Intermediate Mesoderm
3.3. Sequential Differentiation of Both iPSC Lines from Intermediate Mesoderm to FTEC
3.4. The FTE Organoid Generation from FTE-iPSC-FTE and NHEK-iPSC-FTE
3.5. Increased FTE-Related Gene Expression in FTE-iPSC-FTE and NHEK-iPSC-FTE
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
List of Abbreviations
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s Modified Eagle Medium |
| EB | Embryoid body |
| ES | Embryonic stem |
| FBS | Fetal bovine serum |
| FTE | Fallopian tube epithelial |
| FTEC | Fallopian tube epithelial cell |
| H&E | Hematoxylin and eosin |
| HGSOC | High-grade serous ovarian cancer |
| IHC | Immunohistochemistry |
| iPSC | Induced pluripotent stem cell |
| MD | Müllerian duct |
| MOI | Multiplicity of infection |
| NHEK | Normal human keratinocytes |
| NOD-SCID | Nonobese diabetic/severe combined immunodeficiency |
| OCT | Optimal cutting temperature |
| PBS | Phosphate buffered saline |
| PBST | Phosphate buffered saline triton X-100 |
| qPCR | Quantitative polymerase chain reaction |
| RT-qPCR | Real-time quantitative polymerase chain reaction |
References
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of Ovarian Cancer: A Review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [PubMed]
- Zhang, Y.; Luo, G.; Li, M.; Guo, P.; Xiao, Y.; Ji, H.; Hao, Y. Global Patterns and Trends in Ovarian Cancer Incidence: Age, Period and Birth Cohort Analysis. BMC Cancer 2019, 19, 984. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.-M. The Origin and Pathogenesis of Epithelial Ovarian Cancer: A Proposed Unifying Theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Eddie, S.L.; Quartuccio, S.M.; Zhu, J.; Shepherd, J.A.; Kothari, R.; Kim, J.J.; Woodruff, T.K.; Burdette, J.E. Three-Dimensional Modeling of the Human Fallopian Tube Fimbriae. Gynecol. Oncol. 2015, 136, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Lawrenson, K.; Notaridou, M.; Lee, N.; Benjamin, E.; Jacobs, I.J.; Jones, C.; Gayther, S.A. In Vitro Three-Dimensional Modeling of Fallopian Tube Secretory Epithelial Cells. BMC Cell Biol. 2013, 14, 43. [Google Scholar] [CrossRef]
- Levanon, K.; Ng, V.; Piao, H.Y.; Zhang, Y.; Chang, M.C.; Roh, M.H.; Kindelberger, D.W.; Hirsch, M.S.; Crum, C.P.; Marto, J.A.; et al. Primary Ex Vivo Cultures of Human Fallopian Tube Epithelium as a Model for Serous Ovarian Carcinogenesis. Oncogene 2010, 29, 1103–1113. [Google Scholar] [CrossRef]
- Elkas, J.C.; Baldwin, R.L.; Pegram, M.; Tseng, Y.; Slamon, D.; Karlan, B.Y. A Human Ovarian Carcinoma Murine Xenograft Model Useful for Preclinical Trials. Gynecol. Oncol. 2002, 87, 200–206. [Google Scholar] [CrossRef]
- Kim, J.; Coffey, D.M.; Ma, L.; Matzuk, M.M. The Ovary Is an Alternative Site of Origin for High-Grade Serous Ovarian Cancer in Mice. Endocrinology 2015, 156, 1975–1981. [Google Scholar] [CrossRef]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.H.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the Fallopian Tube Secretory Epithelium Leads to High-Grade Serous Ovarian Cancer in Brca;Tp53;Pten Models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef]
- Xu, Y.; Silver, D.F.; Yang, N.P.; Oflazoglu, E.; Hempling, R.E.; Piver, M.S.; Repasky, E.A. Characterization of Human Ovarian Carcinomas in a SCID Mouse Model. Gynecol. Oncol. 1999, 72, 161–170. [Google Scholar] [CrossRef]
- Scott, C.L.; Becker, M.A.; Haluska, P.; Samimi, G. Patient-Derived Xenograft Models to Improve Targeted Therapy in Epithelial Ovarian Cancer Treatment. Front. Oncol. 2013, 3, 295. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Drapkin, R. Primary Culture and Immortalization of Human Fallopian Tube Secretory Epithelial Cells. Nat. Protoc. 2012, 7, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-H.; Chu, T.-Y.; Ding, D.-C. Human Fallopian Tube Epithelial Cells Exhibit Stemness Features, Self-Renewal Capacity, and Wnt-Related Organoid Formation. J. Biomed. Sci. 2020, 27, 32. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Jones, P.M.; Vena, N.; Ligon, A.H.; Liu, J.F.; Hirsch, M.S.; Etemadmoghadam, D.; Bowtell, D.D.L.; Drapkin, R. Cyclin E1 Deregulation Occurs Early in Secretory Cell Transformation to Promote Formation of Fallopian Tube-Derived High-Grade Serous Ovarian Cancers. Cancer Res. 2014, 74, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Yucer, N.; Holzapfel, M.; Jenkins Vogel, T.; Lenaeus, L.; Ornelas, L.; Laury, A.; Sareen, D.; Barrett, R.; Karlan, B.Y.; Svendsen, C.N. Directed Differentiation of Human Induced Pluripotent Stem Cells into Fallopian Tube Epithelium. Sci. Rep. 2017, 7, 10741. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Yu, J.; Rose, F.F., Jr.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced Pluripotent Stem Cells from a Spinal Muscular Atrophy Patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-F.; Su, J.; Kim, H.S.; Chang, B.; Papatsenko, D.; Zhao, R.; Yuan, Y.; Gingold, J.; Xia, W.; Darr, H.; et al. Modeling Familial Cancer with Induced Pluripotent Stem Cells. Cell 2015, 161, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic Memory in Induced Pluripotent Stem Cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell Type of Origin Influences the Molecular and Functional Properties of Mouse Induced Pluripotent Stem Cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef]
- Shrestha, R.; Wen, Y.-T.; Ding, D.-C.; Tsai, R.-K. Aberrant hiPSCs-Derived from Human Keratinocytes Differentiates into 3D Retinal Organoids That Acquire Mature Photoreceptors. Cells 2019, 8, 36. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.M.; Diaz-Hernandez, M.E.; Chihab, S.; Priyadarshani, P.; Bhattaram, P.; Mortensen, L.J.; Guzzo, R.M.; Drissi, H. Differential Chondrogenic Differentiation between iPSC Derived from Healthy and OA Cartilage Is Associated with Changes in Epigenetic Regulation and Metabolic Transcriptomic Signatures. eLife 2023, 12, e83138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guo, J.; Zhang, P.; Xiong, Q.; Wu, S.C.; Xia, L.; Roy, S.S.; Tolar, J.; O’Connell, T.D.; Kyba, M.; et al. Derivation and High Engraftment of Patient-Specific Cardiomyocyte Sheet Using Induced Pluripotent Stem Cells Generated from Adult Cardiac Fibroblast. Circ. Heart Fail. 2015, 8, 156–166. [Google Scholar] [CrossRef]
- Kessler, M.; Hoffmann, K.; Brinkmann, V.; Thieck, O.; Jackisch, S.; Toelle, B.; Berger, H.; Mollenkopf, H.-J.; Mangler, M.; Sehouli, J.; et al. The Notch and Wnt Pathways Regulate Stemness and Differentiation in Human Fallopian Tube Organoids. Nat. Commun. 2015, 6, 8989. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-H.; Ding, D.-C.; Chu, T.-Y. Estradiol and Progesterone Induced Differentiation and Increased Stemness Gene Expression of Human Fallopian Tube Epithelial Cells. J. Cancer 2019, 10, 3028–3036. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Primers 2022, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Corrò, C.; Novellasdemunt, L.; Li, V.S.W. A Brief History of Organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165. [Google Scholar] [CrossRef]
- Yang, S.; Hu, H.; Kung, H.; Zou, R.; Dai, Y.; Hu, Y.; Wang, T.; Lv, T.; Yu, J.; Li, F. Organoids: The Current Status and Biomedical Applications. MedComm (2020) 2023, 4, e274. [Google Scholar] [CrossRef]
- Zhang, S.; Shen, J.; Wang, X.; Sun, X.; Wu, Y.; Zhang, M.-R.; Wang, R.; Hu, K. Integration of Organoids in Peptide Drug Discovery: Rise of the High-throughput Screening. View (Beijing) 2023, 20230010. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Sequence | Reverse Sequence | Product Size (bp) |
|---|---|---|---|
| Oct4 | CAG TGC CCG AAA CCC ACA C | CAG TGC CCG AAA CCC ACA C | 161 |
| Nanog | AGT CCC AAA GGC AAA CAA CCC ACT TC | TGC TGG AGG CTG AGG TAT TTC TGT CTC | 161 |
| Sox2 | GGG AAA TGG GAG GGG TGC AAA AGA GG | TTG CGT GAG TGT GGA TGG GAT TGG TG | 151 |
| Lin28 | TGCACCAGAGTAAGCTGCAC | CTCCTTTTGATCTGCGCTTC | 189 |
| Klf4 | CCCACACAGGTGAGAAACCT | ATGTGTAAGGCGAGGTGGTC | 169 |
| MAP2 | GCA TGA GCT CTT GGC AGG | CCA ATT GAA CCC ATG TAA AGC C | 194 |
| GFAP | AGG GCT GAC ACG TCC AC | GCC TTA GAG GGG AGA GGA G | 132 |
| GATA4 | TCC CTC TTC CCT CCT CAA AT | TCA GCG TGT AAA GGC ATC TG | 194 |
| Tubb3 | CAG AGC AAG AAC AGC AGC TAC TT | GTG AAC TCC ATC TCG TCC ATG CCC TC | 227 |
| Hand1 | TGC CTG AGA AAG AGA ACC AG | ATG GCA GGA TGA ACA AAC AC | 274 |
| GATA6 | CCT CAC TCC ACT CGT GTC TGC | GTC CTG GCT TCT GGA AGT GG | 225 |
| MIXL1 | GGTACCCCGACATCCACTT | GCCTGTTCTGGAACCATACCT | 87 |
| BRACHYURY | GCTGTGACAGGTACCCAACC | CATGCAGGTGAGTTGTCAGAA | 106 |
| PAX2 | GAAGTGCCCCCTTGTGTG | TCGTTGTAGGCCGTGTACTG | 82 |
| OSR1 | GGACCTCTGCGGAACAAG | TGCAGGGAAGGGTGGATA | 100 |
| GATA3 | CTCATTAAGCCCAAGCGAAG | GTCTGACAGTTCGCACAGGA | 68 |
| SIX2 | CAGGTCAGCAACTGGTTCAA | AGCTGCCTAACACCGACTTG | 136 |
| FOXD1 | GACTCTGCACCAAGGGACTG | CAATTGGAAATCCTAGCAGTAAAGT | 63 |
| FOXJ1 | GGGGTGGGAGCAACTTCT | CCTCCTCCGAATAAGTATGTGGT | 83 |
| PAX8 | CAACAGCACCCTGGACGAC | AGGGTGAGTGAGGATCTGCC | 113 |
| WT1 | GAATGCATGACCTGGAATCA | TCTGCCCTTCTGTCCATTTC | 94 |
| OVGP1 | AAGCTGTTGCTGTGGGTTG | TGTGCCCAGTTGGTGAAAT | 92 |
| GAPDH | GGTCTCCTCTGACTTGAACA | GTGAGGGTCTCTCTCTTCCT | 221 |
| RT-PCR primer | |||
| SeV | GGA TCA CTA GGT GAT ATC GAG C | ACC AGA CAA GAG TTT AAG AGA TAT GTA TC | 181 |
| SOX2 | ATG CAC CGC TAC GAC GTG AGC GC | ACC TTG ACA ATC CTG ATG TGG | 528 |
| KLF4 | TTC CTG CAT GCC AGA GGA GCC C | AAT GTA TCG AAG GTG CTC AA | 410 |
| C-MYC | TAA CTG ACT AGC AGG CTT GTC G | TCC ACA TAC AGT CCT GGA TGA TGA TG | 532 |
| OCT3/4 | CCC GAA AGA GAA AGC GAA CCA G | AAT GTA TCG AAG GTG CTC AA | 483 |
| GAPDH | CCA TCT TCC AGG AGC GAG | GCA GGA GGC ATT GCT GAT | 233 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.-H.; Wu, K.-C.; Wang, K.-H.; Ding, D.-C. Assessment of Fallopian Tube Epithelium Features Derived from Induced Pluripotent Stem Cells of Both Fallopian Tube and Skin Origins. Cells 2023, 12, 2635. https://doi.org/10.3390/cells12222635
Chang Y-H, Wu K-C, Wang K-H, Ding D-C. Assessment of Fallopian Tube Epithelium Features Derived from Induced Pluripotent Stem Cells of Both Fallopian Tube and Skin Origins. Cells. 2023; 12(22):2635. https://doi.org/10.3390/cells12222635
Chicago/Turabian StyleChang, Yu-Hsun, Kun-Chi Wu, Kai-Hung Wang, and Dah-Ching Ding. 2023. "Assessment of Fallopian Tube Epithelium Features Derived from Induced Pluripotent Stem Cells of Both Fallopian Tube and Skin Origins" Cells 12, no. 22: 2635. https://doi.org/10.3390/cells12222635