
Engineering of Salmonella Phages into Novel Antimicrobial Tailocins

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacteriophage, Bacterial Strain, and Culturing Media
2.2. From Phage DNA to Cloned Plasmids
2.3. Genetic Engineering
2.4. CRISPR-Cas gRNA Efficiency against S117
2.5. Construction of pEcgRNA Plasmid to Engineer S117
2.6. S117 Portal Gene Deletion
2.7. Fels-1, -2, Gifsy-1, -2 Major Capsid Gene Deletion
2.8. Tailocin Production: S117 Tailocin
2.9. Tailocin Production: Fels-1, -2, Gifsy-1, -2 Tailocins
2.10. Tailocin Killing Assay
3. Results
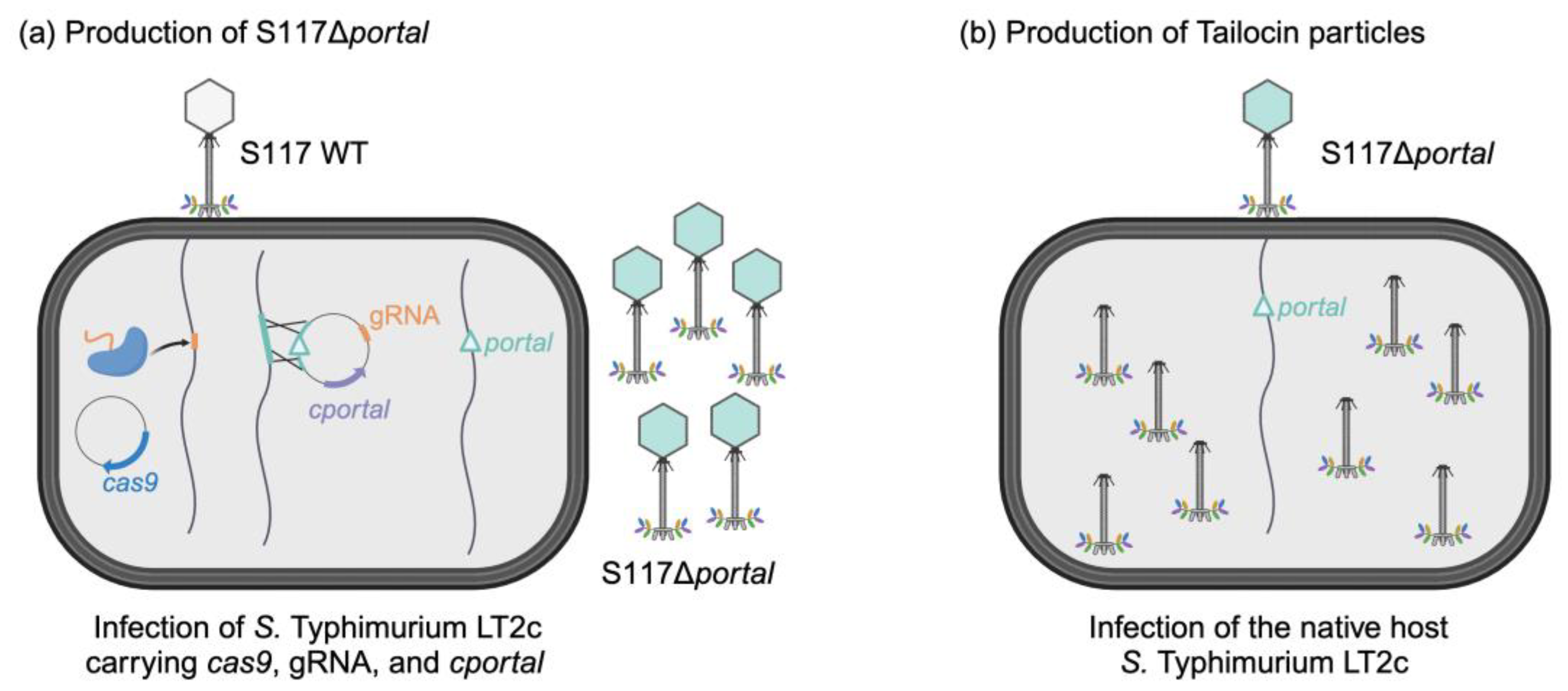
3.1. Overall Approach for Engineering of Phage S117 into a Tailocin
3.2. Engineering of Phage S117 Lacking the Portal Gene
3.2.1. CRISPR-Cas9 Guides Efficiencies
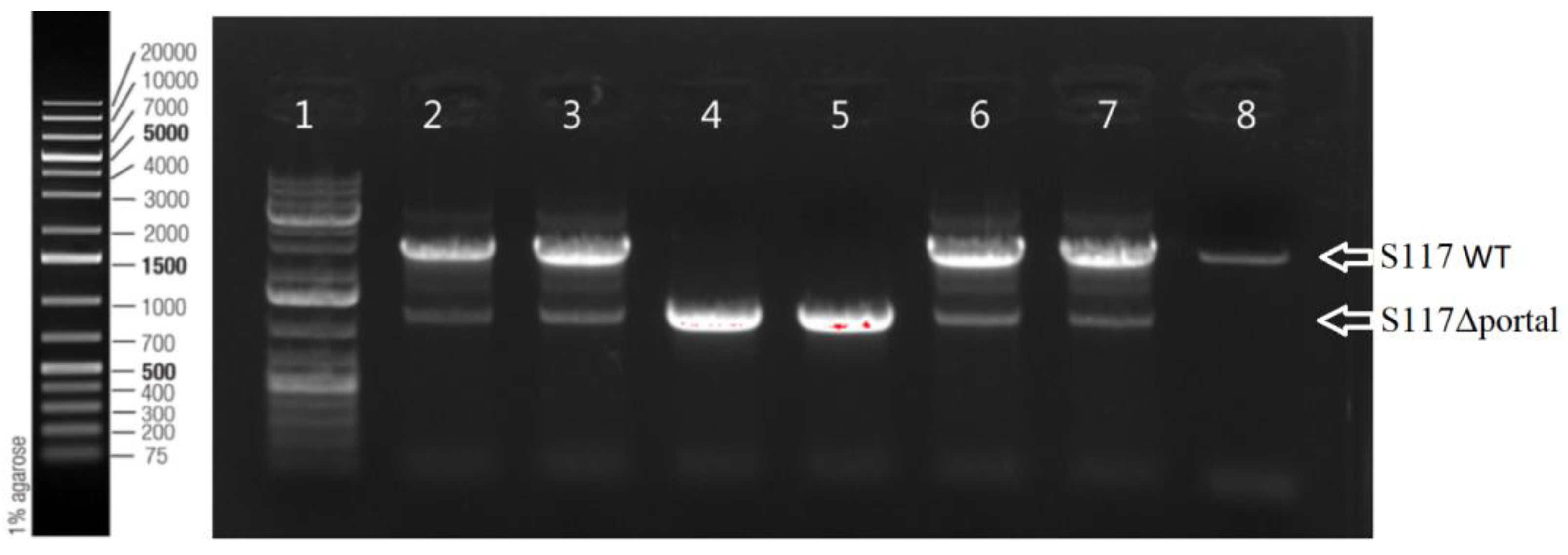
3.2.2. Production of Engineered S117Δportal
3.3. Characterization of the Engineered Phages S117Δportal
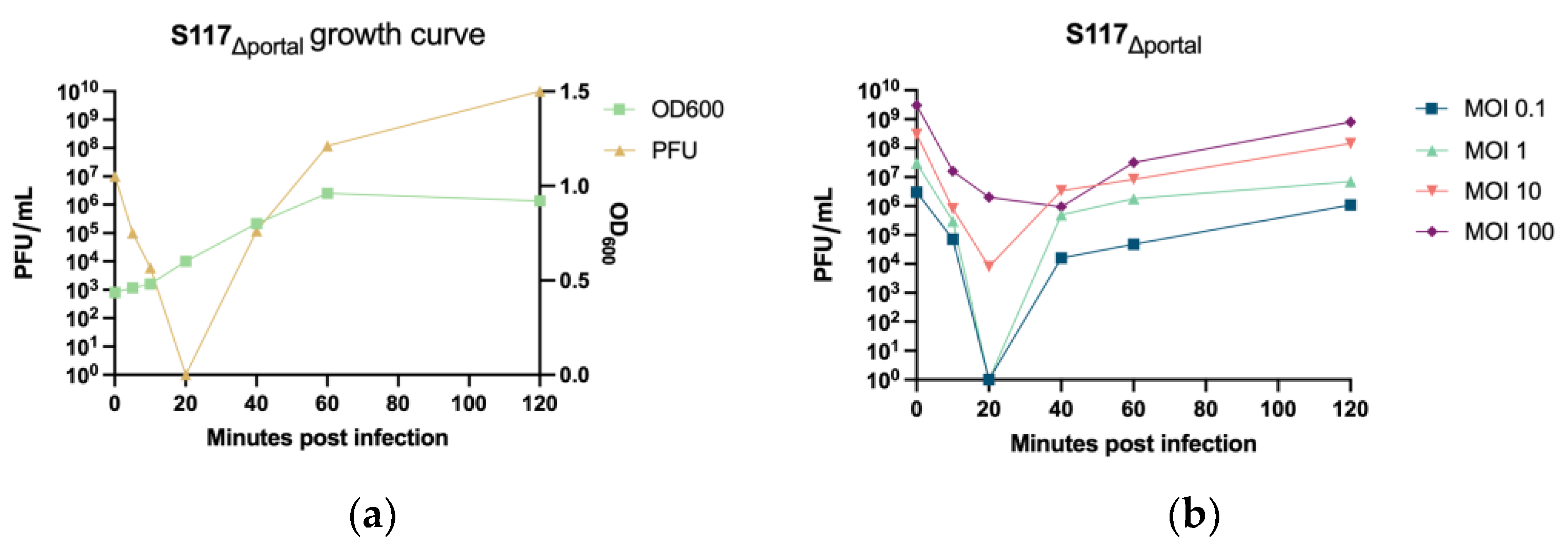
3.3.1. S117Δportal Replication Characterization
3.3.2. Production of S117Δportal Particles
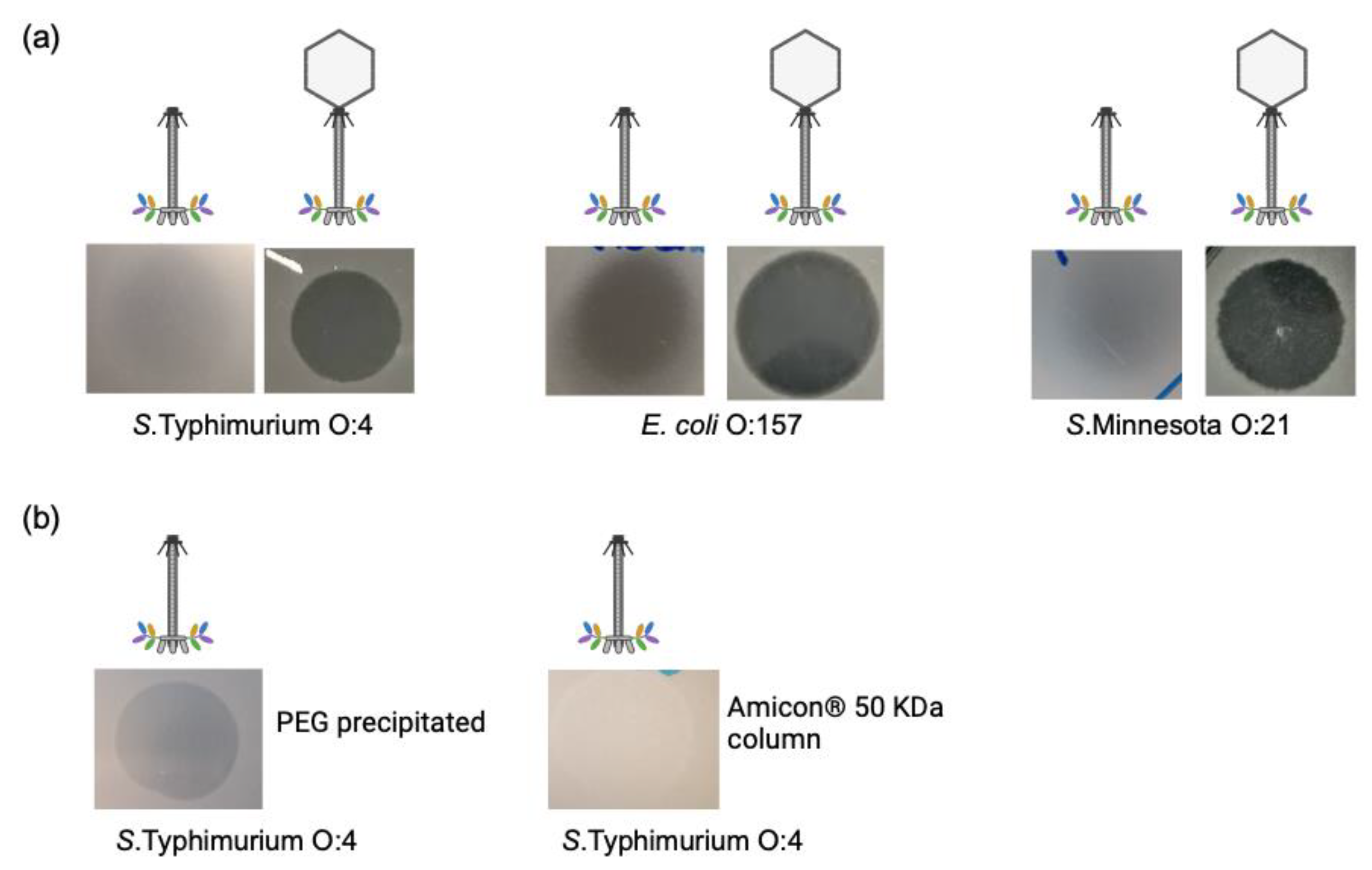
3.4. Tailocin Particles Produced from Phage S117Δportal Form Inhibition Zones
3.5. Tailocin Particles Produced by Engineering Temperate Phages of S. Typhimurium
3.5.1. Bioinformatic Identification of Fels-1 and Fels-2, Putative Receptors
3.5.2. Genetic Engineering of Temperate Phages Fels-1, Fels-2, Gifsy-1, and Gifsy-2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hatfull, G.F.; Dedrick, R.M.; Schooley, R.T. Phage Therapy for Antibiotic-Resistant Bacterial Infections. Annu. Rev. Med. 2022, 73, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Chanishvili, N.; Myelnikov, D.; Blauvelt, T.K. Professor Giorgi Eliava and the Eliava Institute of Bacteriophage. Ther. Appl. Res. 2022, 3, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, M.; Adamia, R. Phage therapy experience at the Eliava Institute. Med. Mal. Infect. 2008, 38, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, A.; Clark, J.; Karris, M.; Hernandez-Santos, H.; Green, S.; Aslam, S.; Maresso, A. Phage therapy related microbial succession associated with successful clinical outcome for a recurrent urinary tract infection. Viruses 2021, 13, 2049. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; A Woodworth, B.; Horne, B.; Fackler, J.; Brownstein, M.J. Case Report: Successful use of phage therapy in refractory MRSA chronic rhinosinusitis. Int. J. Infect. Dis. 2022, 121, 14–16. [Google Scholar] [CrossRef]
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Zolnikov, T.R. Global Health in Action Against a Superbug. Am. J. Public Health 2019, 109, 523–524. [Google Scholar] [CrossRef]
- Ghequire, M.G.K.; De Mot, R. The Tailocin Tale: Peeling off Phage Tails. Trends Microbiol. 2015, 23, 587–590. [Google Scholar] [CrossRef]
- Michel-Briand, Y.; Baysse, C. The pyocins of Pseudomonas aeruginosa. Biochimie 2002, 84, 499–510. [Google Scholar] [CrossRef]
- Kageyama, M.; Ikeda, K.; Egami, F. Studies of a pyocin: III. biological properties of the pyocin. J. Biochem. 1964, 55, 59–64. [Google Scholar] [CrossRef]
- Bali, V.; Panesar, P.S.; Bera, M.B.; Kennedy, J.F. Bacteriocins: Recent Trends and Potential Applications. Crit. Rev. Food Sci. Nutr. 2016, 56, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Behrens, H.M.; Six, A.; Walker, D.; Kleanthous, C. The therapeutic potential of bacteriocins as protein antibiotics. Emerg. Top. Life Sci. 2017, 1, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.W.; Duarte, I.; Le, T.T.; Carmody, L.; LiPuma, J.J.; Young, R.; Gonzalez, C.F. A broadhost-range tailocin from Burkholderia cenocepacia. Appl. Environ. Microbiol. 2017, 83, e03414-16. [Google Scholar] [CrossRef] [PubMed]
- Grinter, R.; Milner, J.; Walker, D. Ferredoxin Containing Bacteriocins Suggest a Novel Mechanism of Iron Uptake in Pectobacterium spp. PLoS ONE 2012, 7, e33033. [Google Scholar] [CrossRef] [PubMed]
- Príncipe, A.; Fernandez, M.; Torasso, M.; Godino, A.; Fischer, S. Effectiveness of tailocins produced by Pseudomonas fluorescens SF4c in controlling the bacterial-spot disease in tomatoes caused by Xanthomonas vesicatoria. Microbiol. Res. 2018, 212–213, 94–102. [Google Scholar] [CrossRef]
- Williams, S.R.; Gebhart, D.; Martin, D.W.; Scholl, D. Retargeting R-type pyocins to generate novel bactericidal protein complexes. Appl. Environ. Microbiol. 2008, 74, 3868–3876. [Google Scholar] [CrossRef]
- Benítez-Chao, D.F.; León-Buitimea, A.; Lerma-Escalera, J.A.; Morones-Ramírez, J.R. Bacteriocins: An Overview of Antimicrobial, Toxicity, and Biosafety Assessment by in vivo Models. Front. Microbiol. 2021, 12, 630695. [Google Scholar] [CrossRef]
- Patz, S.; Becker, Y.; Richert-Pöggeler, K.R.; Berger, B.; Ruppel, S.; Huson, D.H.; Becker, M. Phage tail-like particles are versatile bacterial nanomachines—A mini-review. J. Adv. Res. 2019, 19, 75–84. [Google Scholar] [CrossRef]
- Quinten, T.A.; Kuhn, A. Membrane Interaction of the Portal Protein gp20 of Bacteriophage T4. J. Virol. 2012, 86, 11107–11114. [Google Scholar] [CrossRef]
- Arisaka, F. Assembly and infection process of bacteriophage T4. Chaos Interdiscip. J. Nonlinear Sci. 2005, 15, 047502. [Google Scholar] [CrossRef]
- Leiman, P.G.; Kanamaru, S.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure and morphogenesis of bacteriophage T4. Cell. Mol. Life Sci. 2003, 60, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety; European Centre for Disease Prevention and Control. The European Union Summary Report on Antimicrobial Resistance in zoonotic and indicator bacteria from humans, animals and food in 2019–2020. EFSA J. 2022, 20, e07209. [Google Scholar]
- Volozhantsev, N.V.; Verevkin, V.V.; Krasilnikova, V.M.; Kislichkina, A.A.; Popova, A.V. Complete Genome Sequence of Klebsiella pneumoniae Bacteriophage KpS110, Encoding Five Tail-Associated Proteins with Putative Polysaccharide Depolymerase Domains. Microbiol. Resour. Announc. 2023, 12, e0015323. [Google Scholar] [CrossRef]
- Sørensen, A.N.; Woudstra, C.; Sørensen, M.C.H.; Brøndsted, L. Subtypes of tail spike proteins predicts the host range of Acker-mannviridae phages. Comput. Struct. Biotechnol. J. 2021, 19, 4854–4867. [Google Scholar] [CrossRef] [PubMed]
- Herridge, W.P.; Shibu, P.; O’shea, J.; Brook, T.C.; Hoyles, L. Bacteriophages of Klebsiella spp., their diversity and potential therapeutic uses. J. Med. Microbiol. 2020, 69, 176–194. [Google Scholar]
- Ho, T.D.; Slauch, J.M. Ompc is the receptor for gifsy-1 and gifsy-2 bacteriophages of Salmonella. J. Bacteriol. 2001, 183, 1495–1498. [Google Scholar] [CrossRef]
- Shin, H.; Lee, J.-H.; Kim, H.; Choi, Y.; Heu, S.; Ryu, S. Receptor diversity and host interaction of bacteriophages infecting Salmonella enterica serovar typhimurium. PLoS ONE 2012, 7, e43392. [Google Scholar] [CrossRef]
- Garcia-Russell, N.; Elrod, B.; Dominguez, K. Stress-induced prophage DNA replication in Salmonella enterica serovar Typhi-murium. Infect. Genet. Evol. 2009, 9, 889–895. [Google Scholar] [CrossRef]
- Jakočiūnė, D.; Moodley, A. A rapid bacteriophage DNA extraction method. Methods Protoc. 2018, 1, 27. [Google Scholar] [CrossRef]
- Li, Q.; Sun, B.; Chen, J.; Zhang, Y.; Jiang, Y.U.; Yang, S. A modified pCas/pTargetF system for CRISPR-Cas9-assisted genome editing in Escherichia coli. Acta Biochim. Biophys. Sin. 2021, 53, 620–627. [Google Scholar] [CrossRef]
- Fortier, L.C.; Moineau, S. Phage production and maintenance of stocks, including expected stock lifetimes. Methods Mol. Biol. 2009, 501, 203. [Google Scholar]
- Hoshiga, F.; Yoshizaki, K.; Takao, N.; Miyanaga, K.; Tanji, Y. Modification of T2 phage infectivity toward Escherichia coli O157:H7 via using CRISPR/Cas9. FEMS Microbiol. Lett. 2019, 366, fnz041. [Google Scholar] [CrossRef] [PubMed]
- Duong, M.M.; Carmody, C.M.; Ma, Q.; Peters, J.E.; Nugen, S.R. Optimization of T4 phage engineering via CRISPR/Cas9. Sci. Rep. 2020, 10, 18229. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E.M.; Skutt-Kakaria, K.; Blasdel, B.; El-Shibiny, A.; Castano, A.; Bryan, D.; Kropinski, A.M.; Villegas, A.; Ackermann, H.-W.; Toribio, A.L.; et al. Characterization of a Vil-like phage specific to Escherichia coli O157:H7. Virol. J. 2011, 8, 430. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Tie, D.; Sun, Y.; Jiang, J.; Huang, H.; Gong, Y.; Zhao, C. Characterization and Genomic Analysis of Escherichia coli O157:H7 Bacteriophage FEC14, a New Member of Genus Kuttervirus. Curr. Microbiol. 2021, 78, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.-D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 2015, 12, 982–988. [Google Scholar] [CrossRef]
- Kutter, E. Phage host range and efficiency of plating. Methods Mol. Biol. 2009, 501, 141–149. [Google Scholar] [CrossRef]
- Yamamoto, K.R.; Alberts, B.M.; Benzinger, R.; Lawhorne, L.; Treiber, G. Rapid bacteriophage sedimentation in the presence of poly-ethylene glycol and its application to large-scale virus purification. Virology 1970, 40, 734–744. [Google Scholar] [CrossRef]
- Morgan, G.; Lim, D.; Wong, P.; Tamboline, B. Electron microscopy to visualize T4 bacteriophage interactions with Escherichia coli strain DFB1655, an isogenic derivative of strain MG1655 engineered to express O16 antigen. Undergrad. J. Ex-Perimental Microbiol. Immunol. (UJEMI) 2019, 24, 1–13. [Google Scholar]
- Matilla, M.A.; Fang, X.; Salmond, G.P. Viunalikeviruses are environmentally common agents of horizontal gene transfer in pathogens and biocontrol bacteria. ISME J. 2014, 8, 2143–2147. [Google Scholar] [CrossRef]
- Islam, M.Z.; Fokine, A.; Mahalingam, M.; Zhang, Z.; Garcia-Doval, C.; van Raaij, M.J.; Rossmann, M.G.; Rao, V.B. Molecular Anatomy of the Receptor Binding Module of a Bacteriophage Long Tail Fiber. PLoS Pathog. 2019, 15, e1008193. [Google Scholar] [CrossRef] [PubMed]
- Verbeken, G.; Pirnay, J.-P. European regulatory aspects of phage therapy: Magistral phage preparations. Curr. Opin. Virol. 2022, 52, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Nolan, L.M.; Allsopp, L.P. Antimicrobial Weapons of Pseudomonas aeruginosa. In Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2022. [Google Scholar]
- Scholl, D. Phage Tail-Like Bacteriocins. Annu. Rev. Virol. 2017, 4, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Scholl, D.; Leiman, P.G.; Yu, X.; Miller, J.F.; Zhou, Z.H. Atomic structures of a bactericidal contractile nanotube in its pre- and postcontraction states. Nat. Struct. Mol. Biol. 2015, 22, 377–382. [Google Scholar] [CrossRef]
- Fernandez, M.; Godino, A.; Príncipe, A.; Morales, G.M.; Fischer, S. Effect of a Pseudomonas fluorescens tailocin against phyto-pathogenic Xanthomonas observed by atomic force microscopy. J. Biotechnol. 2017, 256, 13–20. [Google Scholar] [CrossRef]
- Woudstra, C.; Brøndsted, L. Producing Tailocins from Phages Using Osmotic Shock and Benzalkonium Chloride. PHAGE 2023, 4, 136–140. [Google Scholar] [CrossRef]
- Walker, S.A.; Klaenhammer, T.R. An Explosive Antisense RNA Strategy for Inhibition of a Lactococcal Bacteriophage. Appl. Environ. Microbiol. 2000, 66, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Netter, Z.; Boyd, C.M.; Silvas, T.V.; Seed, K.D. A phage satellite tunes inducing phage gene expression using a domesticated en-donuclease to balance inhibition and virion hijacking. Nucleic Acids Res. 2021, 49, 4386–4401. [Google Scholar] [CrossRef]
- Saifaldeen, M.; Al-Ansari, D.E.; Ramotar, D.; Aouida, M. CRISPR FokI Dead Cas9 System: Principles and Applications in Genome Engineering. Cells 2020, 9, 2518. [Google Scholar] [CrossRef]
- Scholl, D.; Martin, D.W., Jr. Antibacterial Efficacy of R-Type Pyocins towards Pseudomonas aeruginosa in a Murine Peritonitis Model. Antimicrob. Agents Chemother. 2008, 52, 1647–1652. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage therapy dosing: The problem(s) with multiplicity of infection (MOI). Bacteriophage 2016, 6, e1220348. [Google Scholar] [CrossRef] [PubMed]
- Dharmaraj, T.; Kratochvil, M.J.; Pourtois, J.D.; Chen, Q.; Hajfathalian, M.; Hargil, A.; Bollyky, P.L. Rapid assessment of changes in phage bioactivity using dynamic light scattering. bioRxiv 2023. Available online: http://biorxiv.org/content/early/2023/07/02/2023.07.02.547396.abstract (accessed on 8 November 2023).
- Emslander, Q.; Vogele, K.; Braun, P.; Stender, J.; Willy, C.; Joppich, M.; Hammerl, J.A.; Abele, M.; Meng, C.; Pichlmair, A.; et al. Cell-free production of personalized therapeutic phages targeting multidrug-resistant bacteria. Cell Chem. Biol. 2022, 29, 1434–1445.e7. [Google Scholar] [CrossRef]
- Garenne, D.; Bowden, S.; Noireaux, V. Cell-free expression and synthesis of viruses and bacteriophages: Applications to medicine and nanotechnology. Curr. Opin. Syst. Biol. 2021, 28, 100373. [Google Scholar] [CrossRef]
- Rustad, M.; Eastlund, A.; Jardine, P.; Noireaux, V. Cell-free TXTL synthesis of infectious bacteriophage T4 in a single test tube reaction. Synth. Biol. 2018, 3, ysy002. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | PAM | Strand | Position | On Target 1 | Off Target 2 | EOP LOG Reduction |
|---|---|---|---|---|---|---|---|
| gPortal1 | CTTCCTGTGTAATCTCCGCG | AGG | − | 1205 | 96 | 100 | 0 |
| gPortal2 | GGTTCTATAATCCTTCAGGG | CGG | + | 168 | 87.8 | 100 | 0 |
| gPortal3 | ATAGAATGCGCGTTTCTCAG | GGG | − | 865 | 70.7 | 100 | 0 |
| gPortal4 | CAATGATGTTGTCACCTGTG | AGG | + | 311 | 74 | 100 | 0 |
| gPortal5 | GCATTCTGACCTTCGCGGCG | CGG | − | 1036 | 62 | 100 | 0 |
| gPortal6 | TACCAGATTGGCCAGTGAAT | TGG | − | 654 | 34 | 100 | 0 |
| gPortal7 | ATGGGGTTCTTCGGTTTGTT | TGG | + | 36 | 0 | 100 | 0 |
| gPortal8 | CAACAATTCATCTTGTTTTT | GGG | − | 85 | 19 | 100 | 0 |
| gPortal9 | TCTCAGCGTTAAAACAGTTG | TGG | − | 230 | 32 | 100 | 0 |
| gPortal10 | CATAACTTCTTTGAAGCATT | CGG | − | 388 | 38 | 100 | 0 |
| gPortal11 | ATCCCACGAATAAAAAAGGC | GGG | + | 514 | 18 | 100 | 5 |
| gPortal12 | GAGAAGGCGATGCGTGAAGG | CGG | + | 591 | 80 | 100 | 0 |
| gPortal13 | GGTGAAGAGCCATTGGCCAA | TGG | + | 768 | 53 | 100 | 0 |
| gPortal14 | GAATACATGACCATGATGAT | GGG | + | 942 | 38 | 100 | 2 |
| gPortal15 | GTGAATTCAAACTTTATAAA | CGG | - | 1356 | 38 | 100 | 0 |
| gPortal16 | TTGGCCAATGGTATTGTCCC | AGG | + | 760 | 45 | 100 | 1 |
| gPortal17 | CAACGCATATGACCGCACCAC | TGG | + | 960 | 64 | 100 | 0 |
| gPortal18 | AAGAGCCGCCTCCAAGAGGA | AGG | + | 1156 | 62 | 100 | 0 |
| gPortal19 | GTTAATTTTGAAAGGCGTTA | CGG | + | 1302 | 44 | 100 | 2 |
| gPortal20 | AAGGTTCAAGCAGATGAAAC | TGG | + | 1576 | 51 | 100 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woudstra, C.; Sørensen, A.N.; Brøndsted, L. Engineering of Salmonella Phages into Novel Antimicrobial Tailocins. Cells 2023, 12, 2637. https://doi.org/10.3390/cells12222637
Woudstra C, Sørensen AN, Brøndsted L. Engineering of Salmonella Phages into Novel Antimicrobial Tailocins. Cells. 2023; 12(22):2637. https://doi.org/10.3390/cells12222637
Chicago/Turabian StyleWoudstra, Cedric, Anders Nørgaard Sørensen, and Lone Brøndsted. 2023. "Engineering of Salmonella Phages into Novel Antimicrobial Tailocins" Cells 12, no. 22: 2637. https://doi.org/10.3390/cells12222637