
11,12-EET Regulates PPAR-γ Expression to Modulate TGF-β-Mediated Macrophage Polarization

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Monocyte Isolation and Macrophage Polarization
2.3. RNA Isolation and Quantitative Real Time PCR (RT-qPCR)
2.4. RNA Sequencing
2.5. Phagocytosis Assays
2.6. PPAR-γ Activity
2.7. Immunoblotting
2.8. Statistical Analyses
2.9. Data and Material Availability
3. Results
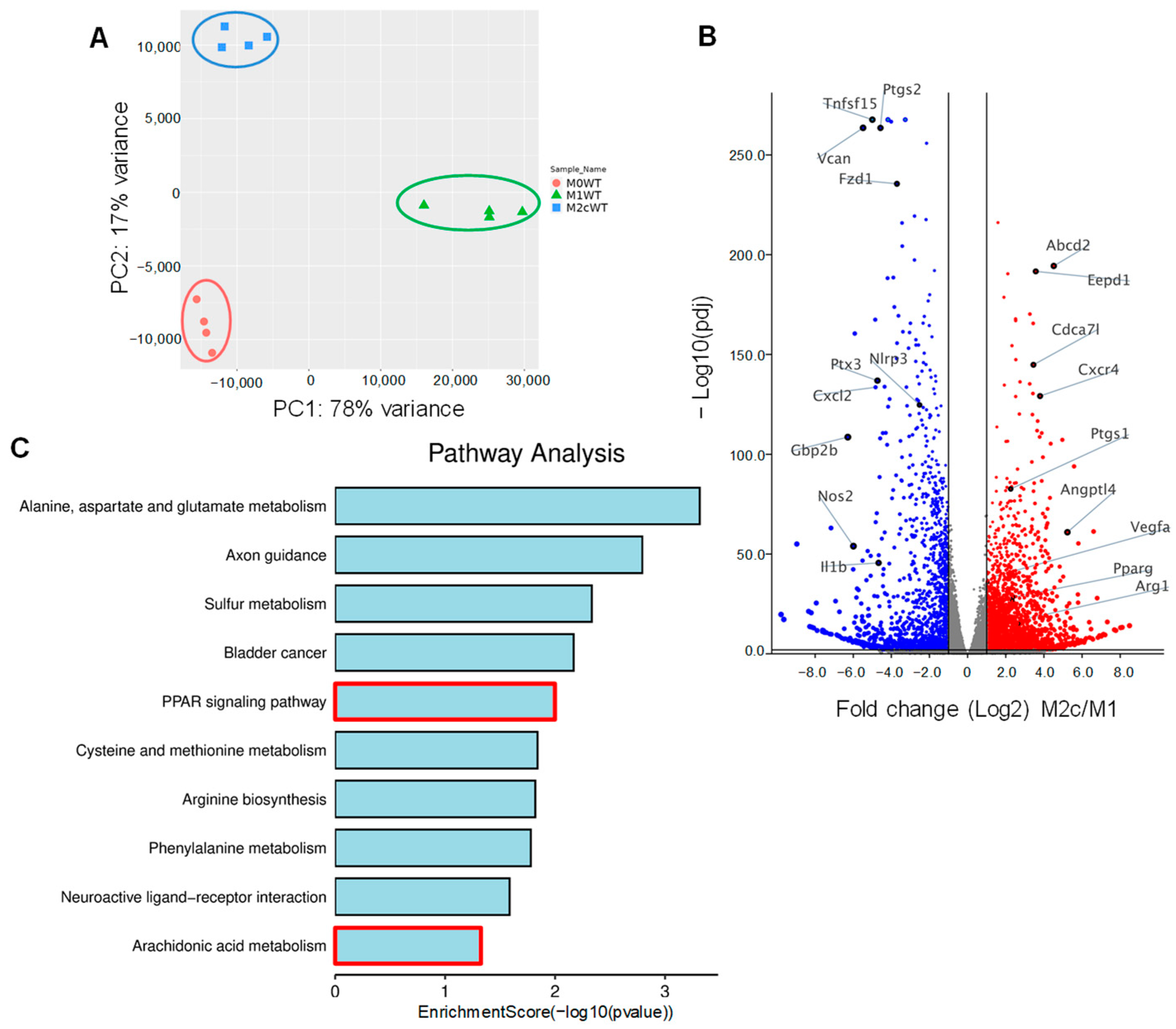
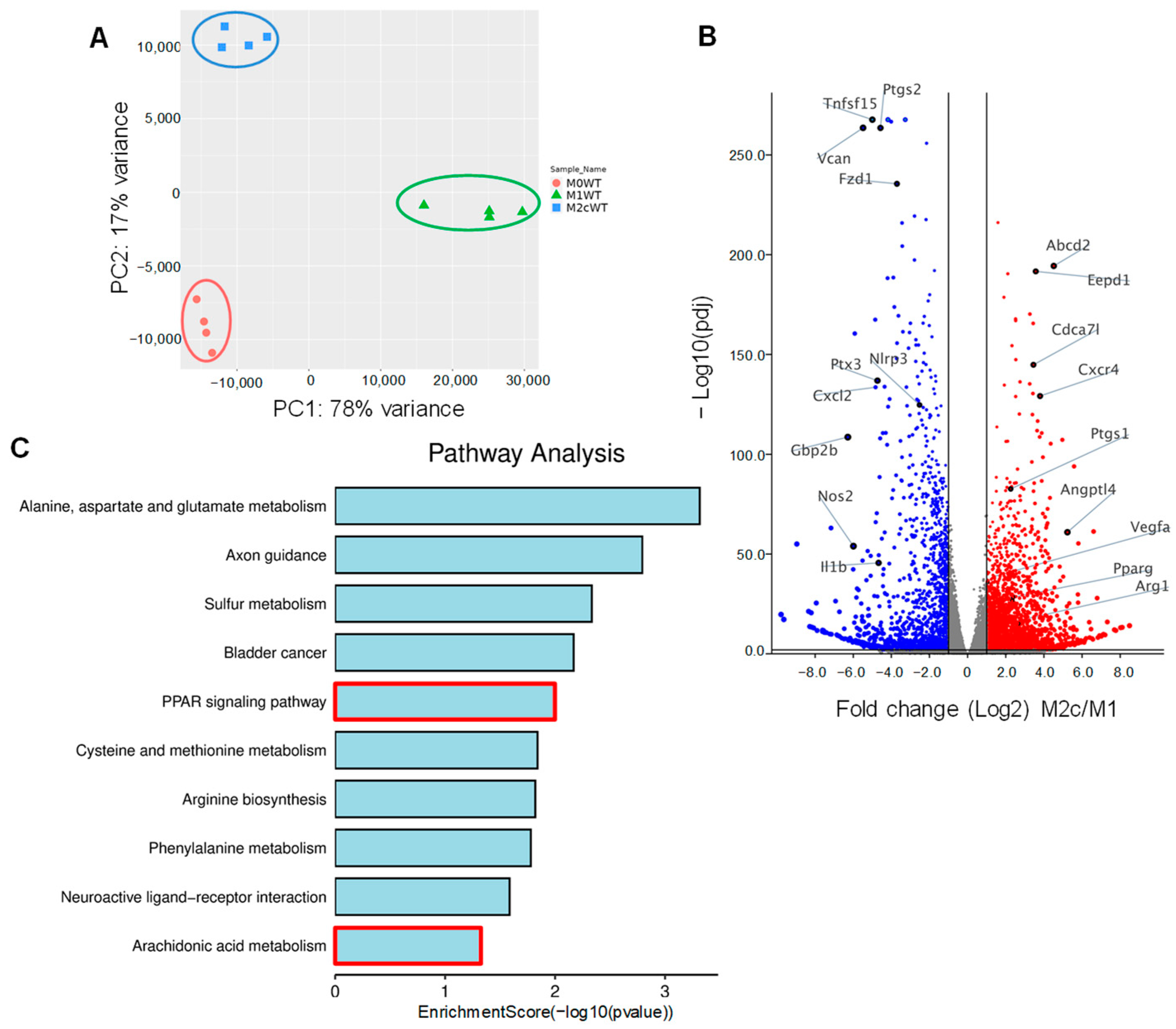
3.1. Impact of TGF-β-Induced Macrophage Repolarization on Gene Expression
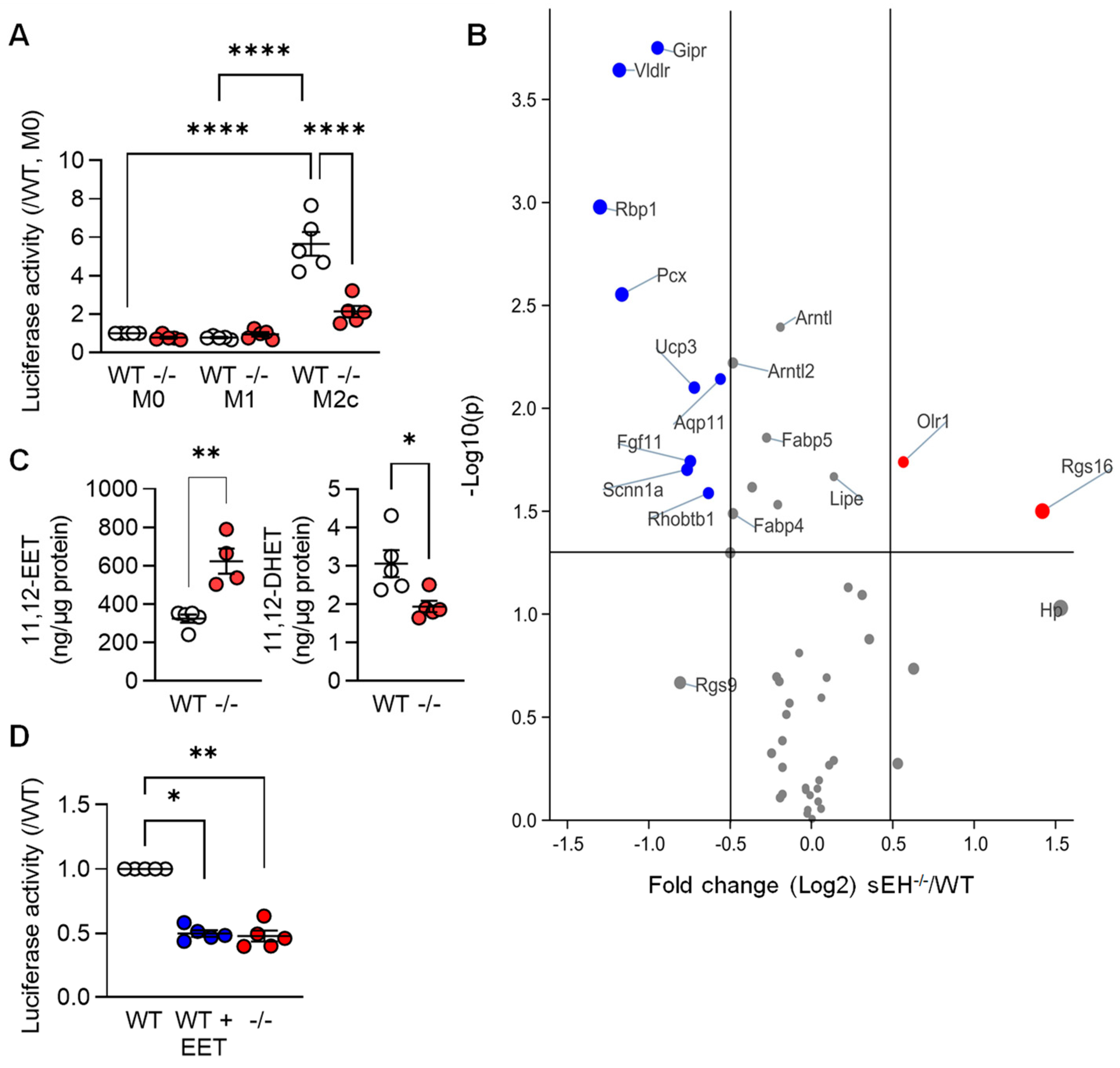
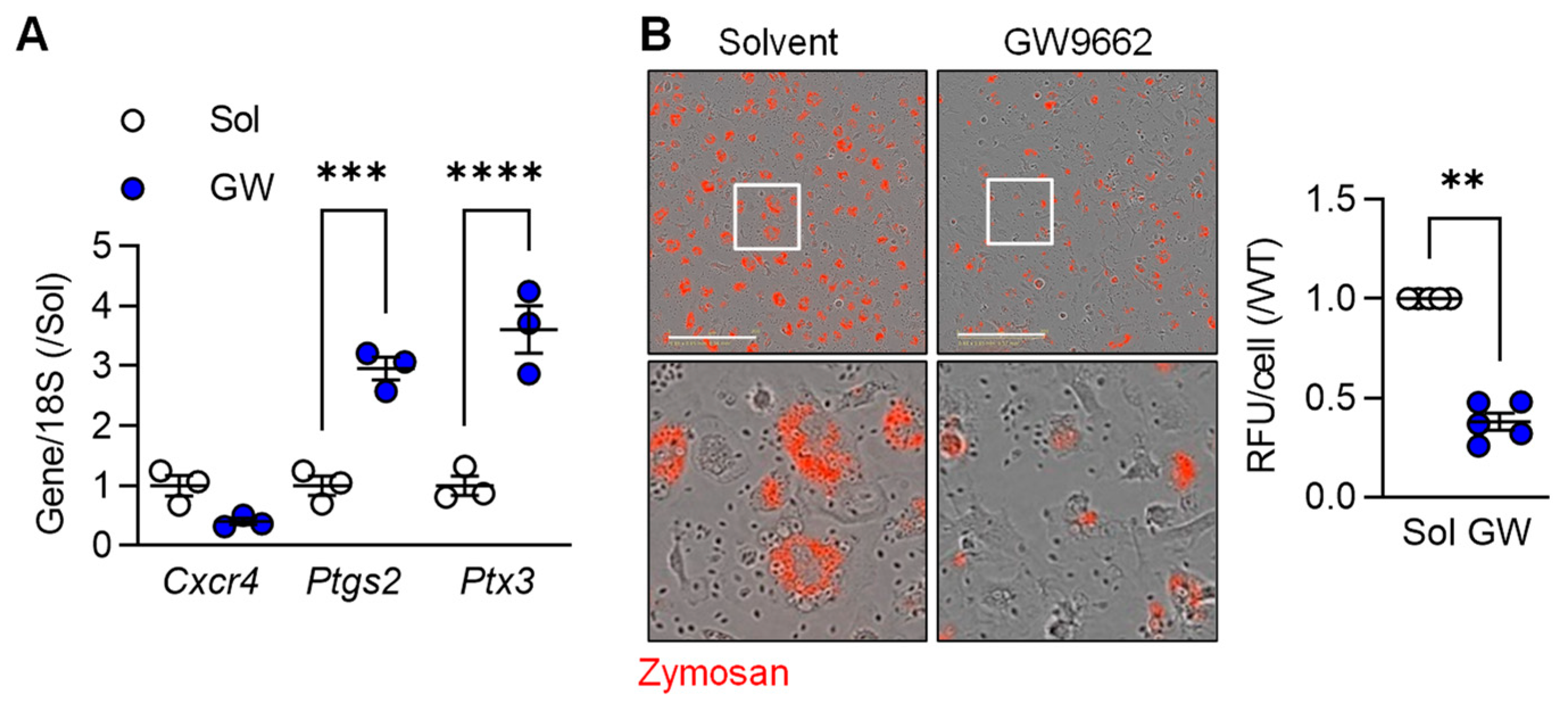
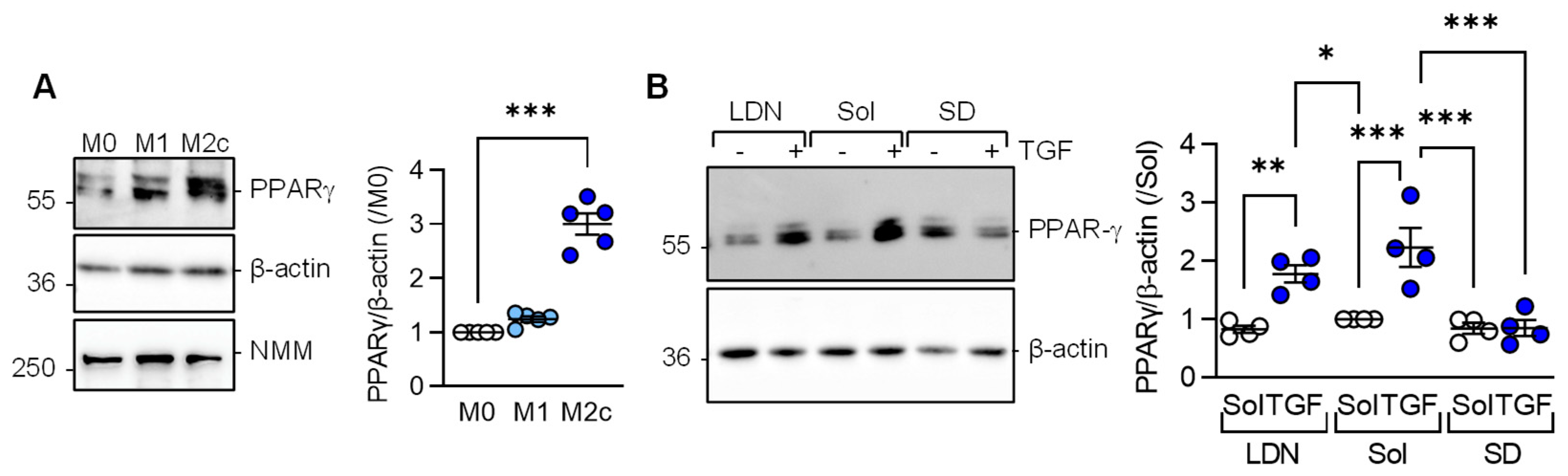
3.2. TGF-β-induced M2c Macrophage Polarization Relies on PPAR-γ and Alk5 Activation
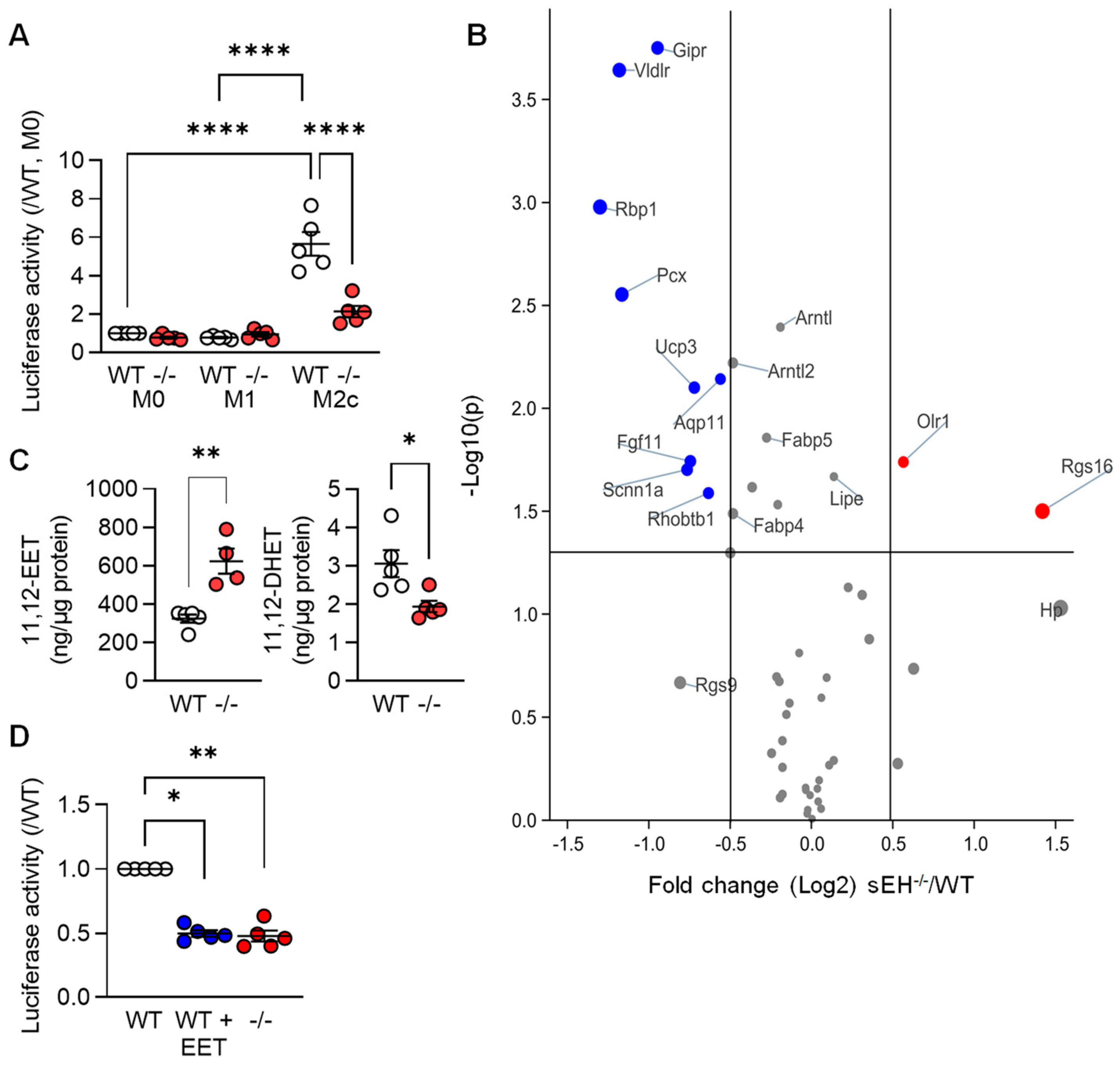
3.3. PPARγ Activity in Differentially Polarized Macrophages from Wild-Type and sEH−/− Mice
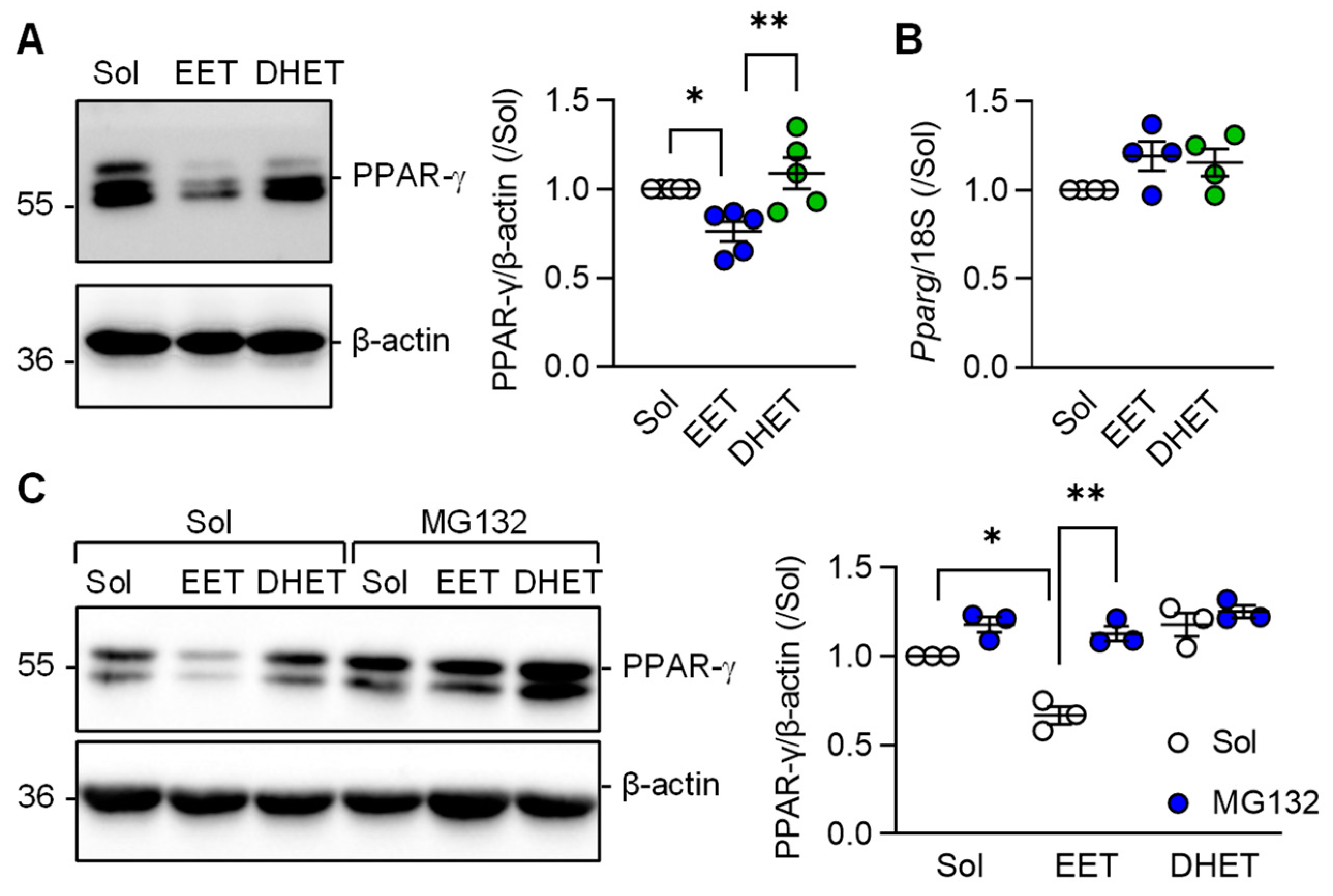
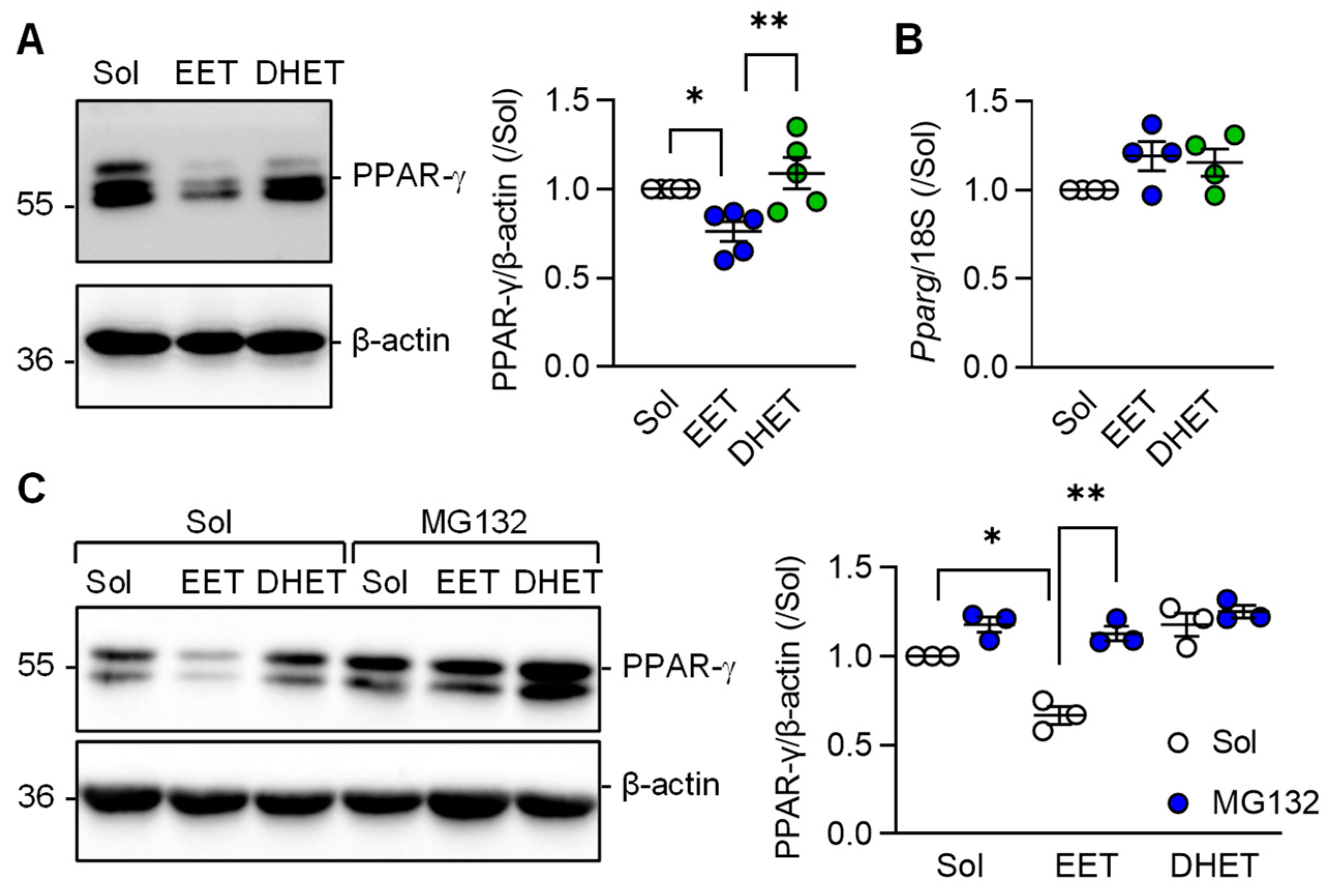
3.4. Regulation of PPAR-γ Levels by 11,12-EET
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage phenotypes in atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Wang, L.-X.; Zhang, S.-X.; Wu, H.-J.; Rong, X.-L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Ukan, Ü.; Delgado Lagos, F.; Kempf, S.; Günther, S.; Siragusa, M.; Fisslthaler, B.; Fleming, I. Effect of thrombin on the metabolism and function of murine macrophages. Cells 2022, 11, 1718. [Google Scholar] [CrossRef]
- Larson, C.; Oronsky, B.; Carter, C.A.; Oronsky, A.; Knox, S.J.; Sher, D.; Reid, T.R. TGF-beta: A master immune regulator. Expert Opin. Ther. Targets 2020, 24, 427–438. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming growth factor-β signaling in immunity and cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Cilfone, N.A.; Kauffman, K.J.; Sun, L.; et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 2019, 1, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Tedgui, A.; Mallat, Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goumans, M.-J.; Dijke, P. ten. TGF-β signaling in control of cardiovascular function. Cold Spring Harb. Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Popp, R.; Frömel, T.; Ehling, M.; Awwad, K.; Adams, R.H.; Hammes, H.-P.; Fleming, I. Müller glia cells regulate Notch signaling and retinal angiogenesis via the generation of 19,20-dihydroxydocosapentaenoic acid. J. Exp. Med. 2014, 211, 281–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- UniProt Consortium. Activities at the Universal Protein Resource (UniProt). Nucleic. Acids Res. 2014, 42, D191–D198. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I.; Fisslthaler, B.; Dixit, M.; Busse, R. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci 2005, 118, 4103–4111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Vanella, L.; Inoue, K.; Burgess, A.; Gotlinger, K.; Manthati, V.L.; Koduru, S.R.; Zeldin, D.C.; Falck, J.R.; Schwartzman, M.L.; et al. Epoxyeicosatrienoic acid agonist regulates human mesenchymal stem cell-derived adipocytes through activation of HO-1-pAKT signaling and a decrease in PPARγ. Stem Cells Dev. 2010, 19, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Samokhvalov, V.; Vriend, J.; Jamieson, K.L.; Akhnokh, M.K.; Manne, R.; Falck, J.R.; Seubert, J.M. PPARγ signaling is required for mediating EETs protective effects in neonatal cardiomyocytes exposed to LPS. Front. Pharmacol. 2014, 5, 242. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xu, X.; Chen, C.; Wang, Y.; Gruzdev, A.; Zeldin, D.C.; Wang, D.W. CYP2J2 attenuates metabolic dysfunction in diabetic mice by reducing hepatic inflammation via the PPARγ. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E270–E282. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhu, F.; Meng, W.; Zhang, F.; Hong, J.; Zhang, G.; Wang, F. CYP2J2/EET reduces vulnerability to atrial fibrillation in chronic pressure overload mice. J. Cell Mol. Med. 2020, 24, 862–874. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, C.; Qiu, H.; Yang, J.; Wu, K.; Ding, S.; Jiang, C.; Jiang, Q. 14,15-EET involved in the development of diabetic cardiac hypertrophy mediated by PPARs. Prostaglandins Other Lipid Mediat. 2022, 159, 106620. [Google Scholar] [CrossRef]
- Li, J.J.; Wang, R.; Lama, R.; Wang, X.; Floyd, Z.E.; Park, E.A.; Liao, F.-F. Ubiquitin ligase NEDD4 regulates PPARγ stability and adipocyte differentiation in 3T3-L1 cells. Sci. Rep. 2016, 6, 38550. [Google Scholar] [CrossRef] [Green Version]
- Nelson, V.L.; Nguyen, H.C.B.; Garcìa-Cañaveras, J.C.; Briggs, E.R.; Ho, W.Y.; DiSpirito, J.R.; Marinis, J.M.; Hill, D.A.; Lazar, M.A. PPARγ is a nexus controlling alternative activation of macrophages via glutamine metabolism. Genes Dev. 2018, 32, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Kökény, G.; Calvier, L.; Hansmann, G. PPARγ and TGFβ-major regulators of metabolism, inflammation, and fibrosis in the lungs and kidneys. Int. J. Mol. Sci. 2021, 22, 10431. [Google Scholar] [CrossRef]
- Yu, X.; Buttgereit, A.; Lelios, I.; Utz, S.G.; Cansever, D.; Becher, B.; Greter, M. The cytokine TGF-β promotes the development and homeostasis of alveolar macrophages. Immunity 2017, 47, 903–912.e4. [Google Scholar] [CrossRef] [Green Version]
- Calvier, L.; Chouvarine, P.; Legchenko, E.; Hoffmann, N.; Geldner, J.; Borchert, P.; Jonigk, D.; Mozes, M.M.; Hansmann, G. PPARγ Llinks BMP2 and TGFβ1 pathways in vascular smooth muscle cells, regulating cell proliferation and glucose metabolism. Cell Metab. 2017, 25, 1118–1134.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Marion-Letellier, R.; Savoye, G.; Ghosh, S. Fatty acids, eicosanoids and PPAR gamma. Eur. J. Pharmacol. 2016, 785, 44–49. [Google Scholar] [CrossRef]
- Wray, J.; Bishop-Bailey, D. Epoxygenases and peroxisome proliferator-activated receptors in mammalian vascular biology. Exp. Physiol. 2008, 93, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Ulu, A.; Davis, B.B.; Tsai, H.-J.; Kim, I.-H.; Morisseau, C.; Inceoglu, B.; Fiehn, O.; Hammock, B.D.; Weiss, R.H. Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J. Cardiovasc. Pharmacol. 2008, 52, 314–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.-N.; Vincelette, J.; Cheng, Y.; Mehra, U.; Chen, D.; Anandan, S.-K.; Gless, R.; Webb, H.K.; Wang, Y.-X.J. Inhibition of soluble epoxide hydrolase attenuated atherosclerosis, abdominal aortic aneurysm formation, and dyslipidemia. Arterioscler Thromb. Vasc. Biol. 2009, 29, 1265–1270. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Wang, X.; Edwards, S.; Dai, M.; Li, J.; Wu, L.; Xu, R.; Han, J.; Yuan, H. NLRX1 knockout aggravates lipopolysaccharide (LPS)-induced heart injury and attenuates the anti-LPS cardioprotective effect of CYP2J2/11,12-EET by enhancing activation of NF-κB and NLRP3 inflammasome. Eur. J. Pharmacol. 2020, 881, 173276. [Google Scholar] [CrossRef]
- Rahman, K.; Vengrenyuk, Y.; Ramsey, S.A.; Vila, N.R.; Girgis, N.M.; Liu, J.; Gusarova, V.; Gromada, J.; Weinstock, A.; Moore, K.J.; et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J. Clin. Investig. 2017, 127, 2904–2915. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| 18S | ctttggtcgctcgctcctc | ctgaccgggttggttttgat |
| Pparg | acaagagctgacccaatggt | tgaggcctgttgtagagctg |
| Cxcr4 | atggaaccgatcagtgtgagt | tagatggtgggcaggaagatc |
| Ptgs2 | gctgtacaagcagtggcaaa | ccccaaagatagcatctgga |
| Ptx3 | cctgctttgtgctctctggt | tctccagcatgatgaacagc |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Kempf, S.; Günther, S.; Hu, J.; Fleming, I. 11,12-EET Regulates PPAR-γ Expression to Modulate TGF-β-Mediated Macrophage Polarization. Cells 2023, 12, 700. https://doi.org/10.3390/cells12050700
Li X, Kempf S, Günther S, Hu J, Fleming I. 11,12-EET Regulates PPAR-γ Expression to Modulate TGF-β-Mediated Macrophage Polarization. Cells. 2023; 12(5):700. https://doi.org/10.3390/cells12050700
Chicago/Turabian StyleLi, Xiaoming, Sebastian Kempf, Stefan Günther, Jiong Hu, and Ingrid Fleming. 2023. "11,12-EET Regulates PPAR-γ Expression to Modulate TGF-β-Mediated Macrophage Polarization" Cells 12, no. 5: 700. https://doi.org/10.3390/cells12050700