
LIM Kinases, LIMK1 and LIMK2, Are Crucial Node Actors of the Cell Fate: Molecular to Pathological Features

 , ,
, ,  and
and 
Abstract
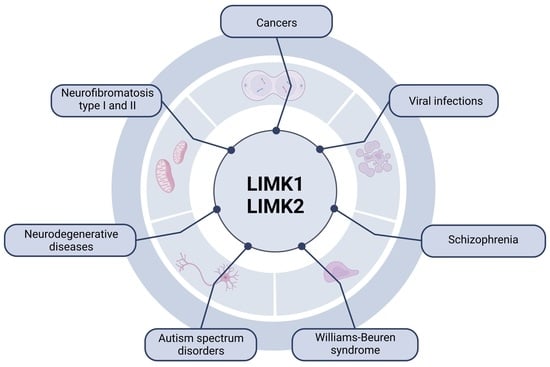
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Gene Description and Encoded Proteins
3. Regulation of LIM Kinases—Partner Network
3.1. LIMKs, Downstream Effectors of Small Rho GTPases
3.2. Other Activators of LIMKs
3.3. Negative Regulators of LIMKs
3.4. Other Partners of LIMKs
4. LIMK Substrates
5. LIMK Physiological Functions
5.1. Cell Migration
5.2. Cell Cycle
5.3. Apoptosis
5.4. Neurodevelopment and Synaptic Plasticity
5.5. Membrane Trafficking
6. Involvement of LIMKs in Different Pathologies
6.1. Cancer
6.1.1. Breast Cancer
6.1.2. Prostate Cancer
6.1.3. Leukaemia
6.1.4. Osteosarcoma
6.1.5. Glioblastoma
6.2. Neuronal Diseases
6.2.1. Alzheimer’s Disease
6.2.2. Parkinson’s Disease
6.2.3. Autism Spectrum Disorders
6.2.4. Schizophrenia
6.2.5. Williams–Beuren Syndrome
6.2.6. Amyotrophic Lateral Sclerosis
6.3. Neurofibromatosis
6.3.1. Neurofibromatosis Type 1
6.3.2. Neurofibromatosis Type 2
6.4. Viral Infections
6.5. Reproduction Troubles
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mizuno, K.; Okano, I.; Ohashi, K.; Nunoue, K.; Kuma, K.; Miyata, T.; Nakamura, T. Identification of a Human CDNA Encoding a Novel Protein Kinase with Two Repeats of the LIM/Double Zinc Finger Motif. Oncogene 1994, 9, 1605–1612. [Google Scholar] [PubMed]
- Bernard, O.; Ganiatsas, S.; Kannourakis, G.; Dringen, R. Kiz-1, a Protein with LIM Zinc Finger and Kinase Domains, Is Expressed Mainly in Neurons. Cell Growth Differ. 1994, 5, 1159–1171. [Google Scholar]
- Sumi, T.; Hashigasako, A.; Matsumoto, K.; Nakamura, T. Different Activity Regulation and Subcellular Localization of LIMK1 and LIMK2 during Cell Cycle Transition. Exp. Cell Res. 2006, 312, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, K.; Moussi, N.; Li, R.; Soo, P.; Bernard, O. LIM Kinase 2 Is Widely Expressed in All Tissues. J. Histochem. Cytochem. 2006, 54, 487–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foletta, V.C.; Moussi, N.; Sarmiere, P.D.; Bamburg, J.R.; Bernard, O. LIM Kinase 1, a Key Regulator of Actin Dynamics, Is Widely Expressed in Embryonic and Adult Tissues. Exp. Cell Res. 2004, 294, 392–405. [Google Scholar] [CrossRef]
- Pröschel, C.; Blouin, M.J.; Gutowski, N.J.; Ludwig, R.; Noble, M. Limk1 Is Predominantly Expressed in Neural Tissues and Phosphorylates Serine, Threonine and Tyrosine Residues in Vitro. Oncogene 1995, 11, 1271–1281. [Google Scholar] [PubMed]
- Sumi, T.; Matsumoto, K.; Takai, Y.; Nakamura, T. Cofilin Phosphorylation and Actin Cytoskeletal Dynamics Regulated by Rho- and Cdc42-Activated Lim-Kinase 2. J. Cell Biol. 1999, 147, 1519–1532. [Google Scholar] [CrossRef] [Green Version]
- Sumi, T.; Matsumoto, K.; Shibuya, A.; Nakamura, T. Activation of LIM Kinases by Myotonic Dystrophy Kinase-Related Cdc42-Binding Kinase α. J. Biol. Chem. 2001, 276, 23092–23096. [Google Scholar] [CrossRef] [Green Version]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of Actin Dynamics through Phosphorylation of Cofilin by LIM-Kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef]
- Yang, N.; Higuchi, O.; Ohashi, K.; Nagata, K.; Wada, A.; Kangawa, K.; Nishida, E.; Mizuno, K. Cofilin Phosphorylation by LIM-Kinase 1 and Its Role in Rac-Mediated Actin Reorganization. Nature 1998, 393, 809–812. [Google Scholar] [CrossRef]
- Gorovoy, M.; Niu, J.; Bernard, O.; Profirovic, J.; Minshall, R.; Neamu, R.; Voyno-Yasenetskaya, T. LIM Kinase 1 Coordinates Microtubule Stability and Actin Polymerization in Human Endothelial Cells. J. Biol. Chem. 2005, 280, 26533–26542. [Google Scholar] [CrossRef] [Green Version]
- Po’uha, S.T.; Shum, M.S.Y.; Goebel, A.; Bernard, O.; Kavallaris, M. LIM-Kinase 2, a Regulator of Actin Dynamics, Is Involved in Mitotic Spindle Integrity and Sensitivity to Microtubule-Destabilizing Drugs. Oncogene 2010, 29, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Okano, I.; Hiraoka, J.; Otera, H.; Nunoue, K.; Ohashi, K.; Iwashita, S.; Hirai, M.; Mizuno, K. Identification and Characterization of a Novel Family of Serine/Threonine Kinases Containing Two N-Terminal LIM Motifs. J. Biol. Chem. 1995, 270, 31321–31330. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.W.; Olson, M.F. LIM Kinases: Function, Regulation and Association with Human Disease. J. Mol. Med. 2007, 85, 555–568. [Google Scholar] [CrossRef]
- Osada, H.; Hasada, K.; Inazawa, J.; Uchida, K.; Ueda, R.; Takahashi, T.; Takahashi, T. Subcellular Localization and Protein Interaction of the Human LIMK2 Gene Expressing Alternative Transcripts with Tissue-Specific Regulation. Biochem. Biophys. Res. Commun. 1996, 229, 582–589. [Google Scholar] [CrossRef]
- Vallée, B.; Cuberos, H.; Doudeau, M.; Godin, F.; Gosset, D.; Vourc’h, P.; Andres, C.R.; Bénédetti, H. LIMK2-1, a New Isoform of Human LIMK2, Regulates Actin Cytoskeleton Remodeling via a Different Signaling Pathway than That of Its Two Homologs, LIMK2a and LIMK2b. Biochem. J. 2018, 475, 3745–3761. [Google Scholar] [CrossRef] [PubMed]
- Vallée, B.; Doudeau, M.; Godin, F.; Bénédetti, H. Characterization at the Molecular Level Using Robust Biochemical Approaches of a New Kinase Protein. JoVE 2019, 148, e59820. [Google Scholar] [CrossRef]
- Takahashi, H.; Koshimizu, U.; Miyazaki, J.; Nakamura, T. Impaired Spermatogenic Ability of Testicular Germ Cells in Mice Deficient in the LIM-Kinase 2 Gene. Dev. Biol. 2002, 241, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Koshimizu, U.; Nakamura, T. A Novel Transcript Encoding Truncated LIM Kinase 2 Is Specifically Expressed in Male Germ Cells Undergoing Meiosis. Biochem. Biophys. Res. Commun. 1998, 249, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Manetti, F. LIM Kinases Are Attractive Targets with Many Macromolecular Partners and Only a Few Small Molecule Regulators: LIM KINASES ARE ATTRACTIVE TARGETS. Med. Res. Rev. 2012, 32, 968–998. [Google Scholar] [CrossRef]
- Yang, N.; Mizuno, K. Nuclear Export of LIM-Kinase 1, Mediated by Two Leucine-Rich Nuclear-Export Signals within the PDZ Domain. Biochem. J. 1999, 338, 793–798. [Google Scholar] [CrossRef]
- Goyal, P.; Pandey, D.; Siess, W. Phosphorylation-Dependent Regulation of Unique Nuclear and Nucleolar Localization Signals of LIM Kinase 2 in Endothelial Cells. J. Biol. Chem. 2006, 281, 25223–25230. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, D.; Preuss, F.; Dederer, V.; Knapp, S.; Mathea, S. Structural Aspects of LIMK Regulation and Pharmacology. Cells 2022, 11, 142. [Google Scholar] [CrossRef]
- Nadella, K.S.; Saji, M.; Jacob, N.K.; Pavel, E.; Ringel, M.D.; Kirschner, L.S. Regulation of Actin Function by Protein Kinase A-mediated Phosphorylation of Limk1. EMBO Rep. 2009, 10, 599–605. [Google Scholar] [CrossRef]
- Kobayashi, M.; Nishita, M.; Mishima, T.; Ohashi, K.; Mizuno, K. MAPKAPK-2-Mediated LIM-Kinase Activation Is Critical for VEGF-Induced Actin Remodeling and Cell Migration. EMBO J. 2006, 25, 713–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, F.-F.; Lin, T.-Y.; Chen, J.-Y.; Shieh, S.-Y. P53-Mediated Transactivation of LIMK2b Links Actin Dynamics to Cell Cycle Checkpoint Control. Oncogene 2010, 29, 2864–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, D.R.; Crighton, D.; Samuel, M.S.; Lourenco, F.C.; Munro, J.; Wood, J.; Bensaad, K.; Vousden, K.H.; Sansom, O.J.; Ryan, K.M.; et al. P53-Mediated Transcriptional Regulation and Activation of the Actin Cytoskeleton Regulatory RhoC to LIMK2 Signaling Pathway Promotes Cell Survival. Cell Res. 2011, 21, 666–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkenfeld, J.; Betz, H.; Roth, D. Identification of Cofilin and LIM-Domain-Containing Protein Kinase 1 as Novel Interaction Partners of 14-3-3 Zeta. Biochem. J. 2003, 369, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Gohla, A.; Bokoch, G.M. 14-3-3 Regulates Actin Dynamics by Stabilizing Phosphorylated Cofilin. Curr. Biol. 2002, 12, 1704–1710. [Google Scholar] [CrossRef] [Green Version]
- Vlachos, P.; Joseph, B. The Cdk Inhibitor P57Kip2 Controls LIM-Kinase 1 Activity and Regulates Actin Cytoskeleton Dynamics. Oncogene 2009, 28, 4175–4188. [Google Scholar] [CrossRef] [Green Version]
- Yokoo, T.; Toyoshima, H.; Miura, M.; Wang, Y.; Iida, K.T.; Suzuki, H.; Sone, H.; Shimano, H.; Gotoda, T.; Nishimori, S.; et al. P57Kip2 Regulates Actin Dynamics by Binding and Translocating LIM-Kinase 1 to the Nucleus. J. Biol. Chem. 2003, 278, 52919–52923. [Google Scholar] [CrossRef] [Green Version]
- Dong, Q.; Ji, Y.-S.; Cai, C.; Chen, Z.-Y. LIM Kinase 1 (LIMK1) Interacts with Tropomyosin-Related Kinase B (TrkB) and Mediates Brain-Derived Neurotrophic Factor (BDNF)-Induced Axonal Elongation. J. Biol. Chem. 2012, 287, 41720–41731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.O.; Chang, K.-H.; Ghosh, S.; Venkatesh, C.; Giger, K.; Low, P.S.; Shah, K. LIMK2 Is a Crucial Regulator and Effector of Aurora-A-Kinase-Mediated Malignancy. J. Cell Sci. 2012, 125, 1204–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Milosavljevic, T.; Alahari, S.K. Nischarin Inhibits LIM Kinase To Regulate Cofilin Phosphorylation and Cell Invasion. Mol. Cell Biol. 2008, 28, 3742–3756. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Baranwal, S.; Dong, S.; Struckhoff, A.P.; Worthylake, R.A.; Alahari, S.K. Integrin-Binding Protein Nischarin Interacts with Tumor Suppressor Liver Kinase B1 (LKB1) to Regulate Cell Migration of Breast Epithelial Cells. J. Biol. Chem. 2013, 288, 15495–15509. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yu, K.; Hao, Y.; Li, D.; Stewart, R.; Insogna, K.L.; Xu, T. LATS1 Tumour Suppressor Affects Cytokinesis by Inhibiting LIMK1. Nat. Cell Biol. 2004, 6, 609–617. [Google Scholar] [CrossRef]
- Soosairajah, J.; Maiti, S.; Wiggan, O.; Sarmiere, P.; Moussi, N.; Sarcevic, B.; Sampath, R.; Bamburg, J.R.; Bernard, O. Interplay between Components of a Novel LIM Kinase–Slingshot Phosphatase Complex Regulates Cofilin. EMBO J. 2005, 24, 473–486. [Google Scholar] [CrossRef]
- Zoudilova, M.; Kumar, P.; Ge, L.; Wang, P.; Bokoch, G.M.; DeFea, K.A. β-Arrestin-Dependent Regulation of the Cofilin Pathway Downstream of Protease-Activated Receptor-2. J. Biol. Chem. 2007, 282, 20634–20646. [Google Scholar] [CrossRef] [Green Version]
- Tursun, B.; Schlüter, A.; Peters, M.A.; Viehweger, B.; Ostendorff, H.P.; Soosairajah, J.; Drung, A.; Bossenz, M.; Johnsen, S.A.; Schweizer, M.; et al. The Ubiquitin Ligase Rnf6 Regulates Local LIM Kinase 1 Levels in Axonal Growth Cones. Genes Dev. 2005, 19, 2307–2319. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Macara, I.G. Par-3 Mediates the Inhibition of LIM Kinase 2 to Regulate Cofilin Phosphorylation and Tight Junction Assembly. J. Cell Biol. 2006, 172, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, P.; Carpentier, R.; Ségard, P.; Olivé-Cren, C.; Lefebvre, P. Multiple Signaling Pathways Regulate the Transcriptional Activity of the Orphan Nuclear Receptor NURR1. Nucleic Acids Res. 2006, 34, 5515–5527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayo, A.; Parsons, M.; Adams, J.C. A Novel Rho-Dependent Pathway That Drives Interaction of Fascin-1 with p-Lin-11/Isl-1/Mec-3 Kinase (LIMK) 1/2 to Promote Fascin-1/Actin Binding and Filopodia Stability. BMC Biol. 2012, 10, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, A.V.; Steel, R.; Bernard, O. Rho-Associated Coiled-Coil Kinase (ROCK) Protein Controls Microtubule Dynamics in a Novel Signaling Pathway That Regulates Cell Migration. J. Biol. Chem. 2012, 287, 43620–43629. [Google Scholar] [CrossRef] [Green Version]
- Janke, C.; Montagnac, G. Causes and Consequences of Microtubule Acetylation. Curr. Biol. 2017, 27, R1287–R1292. [Google Scholar] [CrossRef] [Green Version]
- Schofield, A.V.; Gamell, C.; Bernard, O. LIMK1/TPPP1/HDAC6 Is a Dual Actin and Microtubule Regulatory Complex That Promotes Drug Resistance. Adv. Biosci. Biotechnol. 2014, 05, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.J.; Yoon, J.-H.; Min, D.S.; Chung, K.C. LIM Kinase 1 Activates CAMP-Responsive Element-Binding Protein during the Neuronal Differentiation of Immortalized Hippocampal Progenitor Cells. J. Biol. Chem. 2004, 279, 8903–8910. [Google Scholar] [CrossRef] [Green Version]
- Côté, M.C.; Lavoie, J.R.; Houle, F.; Poirier, A.; Rousseau, S.; Huot, J. Regulation of Vascular Endothelial Growth Factor-Induced Endothelial Cell Migration by LIM Kinase 1-Mediated Phosphorylation of Annexin 1. J. Biol. Chem. 2010, 285, 8013–8021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White-Grindley, E.; Li, L.; Mohammad Khan, R.; Ren, F.; Saraf, A.; Florens, L.; Si, K. Contribution of Orb2A Stability in Regulated Amyloid-Like Oligomerization of Drosophila Orb2. PLoS Biol. 2014, 12, e1001786. [Google Scholar] [CrossRef] [Green Version]
- Lagoutte, E.; Villeneuve, C.; Lafanechère, L.; Wells, C.M.; Jones, G.E.; Chavrier, P.; Rossé, C. LIMK Regulates Tumor-Cell Invasion and Matrix Degradation Through Tyrosine Phosphorylation of MT1-MMP. Sci. Rep. 2016, 6, 24925. [Google Scholar] [CrossRef] [Green Version]
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 10, a018267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridley, A.J. Rho GTPase Signalling in Cell Migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, C.D.; Ridley, A.J. Rho GTPase Signaling Complexes in Cell Migration and Invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seetharaman, S.; Etienne-Manneville, S. Cytoskeletal Crosstalk in Cell Migration. Trends. Cell Biol. 2020, 30, 720–735. [Google Scholar] [CrossRef] [PubMed]
- Zebda, N.; Bernard, O.; Bailly, M.; Welti, S.; Lawrence, D.S.; Condeelis, J.S. Phosphorylation of Adf/Cofilin Abolishes Egf-Induced Actin Nucleation at the Leading Edge and Subsequent Lamellipod Extension. J. Cell Biol. 2000, 151, 1119–1128. [Google Scholar] [CrossRef] [Green Version]
- Nishita, M.; Tomizawa, C.; Yamamoto, M.; Horita, Y.; Ohashi, K.; Mizuno, K. Spatial and Temporal Regulation of Cofilin Activity by LIM Kinase and Slingshot Is Critical for Directional Cell Migration. J. Cell Biol. 2005, 171, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Nishita, M.; Aizawa, H.; Mizuno, K. Stromal Cell-Derived Factor 1α Activates LIM Kinase 1 and Induces Cofilin Phosphorylation for T-Cell Chemotaxis. Mol. Cell. Biol. 2002, 22, 774–783. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.Y.; Raft, S.; Bailly, M.; Wyckoff, J.B.; Segall, J.E.; Condeelis, J.S. EGF Stimulates an Increase in Actin Nucleation and FIlament Number at the Leading Edge of the Lamellipod in Mammary Adenocarcinoma Cells. J. Cell Sci. 1998, 111, 199–211. [Google Scholar] [CrossRef]
- Yoshioka, K.; Foletta, V.; Bernard, O.; Itoh, K. A Role for LIM Kinase in Cancer Invasion. Proc. Natl. Acad. Sci. USA 2003, 100, 7247–7252. [Google Scholar] [CrossRef] [Green Version]
- You, T.; Gao, W.; Wei, J.; Jin, X.; Zhao, Z.; Wang, C.; Li, Y. Overexpression of LIMK1 Promotes Tumor Growth and Metastasis in Gastric Cancer. Biomed. Pharmacother. 2015, 69, 96–101. [Google Scholar] [CrossRef]
- Chen, J.; Ananthanarayanan, B.; Springer, K.S.; Wolf, K.J.; Sheyman, S.M.; Tran, V.D.; Kumar, S. Suppression of LIM Kinase 1 and LIM Kinase 2 Limits Glioblastoma Invasion. Cancer Res. 2020, 80, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, Y.; Xing, F.; Wang, J.; Wang, Y.; Wang, H.; Yang, Y.; Gao, Z. Overexpression of LIMK1 Promotes Migration Ability of Multidrug-Resistant Osteosarcoma Cells. Oncol. Res. 2011, 19, 501–509. [Google Scholar] [CrossRef]
- Berabez, R.; Routier, S.; Bénédetti, H.; Plé, K.; Vallée, B. LIM Kinases, Promising but Reluctant Therapeutic Targets: Chemistry and Preclinical Validation In Vivo. Cells 2022, 11, 2090. [Google Scholar] [CrossRef] [PubMed]
- Rizzelli, F.; Malabarba, M.G.; Sigismund, S.; Mapelli, M. The Crosstalk between Microtubules, Actin and Membranes Shapes Cell Division. Open Biol. 2020, 10, 190314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, R.; Jones, J.L.; Oelschlager, D.K.; Tapia, T.; Tousson, A.; Grizzle, W.E. Phosphorylated LIM Kinases Colocalize with Gamma-Tubulin in Centrosomes During Early Stages of Mitosis. Cell Cycle 2007, 6, 2944–2952. [Google Scholar] [CrossRef] [Green Version]
- Kaji, N.; Ohashi, K.; Shuin, M.; Niwa, R.; Uemura, T.; Mizuno, K. Cell Cycle-Associated Changes in Slingshot Phosphatase Activity and Roles in Cytokinesis in Animal Cells. J. Biol. Chem. 2003, 278, 33450–33455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumi, T.; Matsumoto, K.; Nakamura, T. Mitosis-Dependent Phosphorylation and Activation of LIM-Kinase 1. Biochem. Biophys. Res. Commun. 2002, 290, 1315–1320. [Google Scholar] [CrossRef]
- Amano, T.; Kaji, N.; Ohashi, K.; Mizuno, K. Mitosis-Specific Activation of LIM Motif-Containing Protein Kinase and Roles of Cofilin Phosphorylation and Dephosphorylation in Mitosis. J. Biol. Chem. 2002, 277, 22093–22102. [Google Scholar] [CrossRef] [Green Version]
- Kaji, N.; Muramoto, A.; Mizuno, K. LIM Kinase-Mediated Cofilin Phosphorylation during Mitosis Is Required for Precise Spindle Positioning. J. Biol. Chem. 2008, 283, 4983–4992. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.; Tan, M.-H.; Weng, T.; Li, H.; Koh, C.-G. LIM Kinase1 Regulates Mitotic Centrosome Integrity via Its Activity on Dynein Light Intermediate Chains. Open Biol. 2018, 8, 170202. [Google Scholar] [CrossRef] [Green Version]
- Prudent, R.; Vassal-Stermann, E.; Nguyen, C.-H.; Pillet, C.; Martinez, A.; Prunier, C.; Barette, C.; Soleilhac, E.; Filhol, O.; Beghin, A.; et al. Pharmacological Inhibition of LIM Kinase Stabilizes Microtubules and Inhibits Neoplastic Growth. Cancer Res. 2012, 72, 4429–4439. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Ren, W.; Zhao, W.; Cao, L.; Huang, J. Involvement of the Actin Machinery in Programmed Cell Death. Front. Cell Dev. Biol. 2021, 8, 634849. [Google Scholar] [CrossRef] [PubMed]
- Tomiyoshi, G.; Horita, Y.; Nishita, M.; Ohashi, K.; Mizuno, K. Caspase-Mediated Cleavage and Activation of LIM-Kinase 1 and Its Role in Apoptotic Membrane Blebbing. Genes Cells 2004, 9, 591–600. [Google Scholar] [CrossRef]
- Papadopoulou, N.; Charalampopoulos, I.; Alevizopoulos, K.; Gravanis, A.; Stournaras, C. Rho/ROCK/Actin Signaling Regulates Membrane Androgen Receptor Induced Apoptosis in Prostate Cancer Cells. Exp. Cell Res. 2008, 314, 3162–3174. [Google Scholar] [CrossRef]
- Ivanovska, J.; Tregubova, A.; Mahadevan, V.; Chakilam, S.; Gandesiri, M.; Benderska, N.; Ettle, B.; Hartmann, A.; Söder, S.; Ziesché, E.; et al. Identification of DAPK as a Scaffold Protein for the LIMK/Cofilin Complex in TNF-Induced Apoptosis. Int. J. Biochem. Cell Biol. 2013, 45, 1720–1729. [Google Scholar] [CrossRef]
- Kavanagh, E.; Vlachos, P.; Emourgeon, V.; Rodhe, J.; Joseph, B. P57KIP2 Control of Actin Cytoskeleton Dynamics Is Responsible for Its Mitochondrial Pro-Apoptotic Effect. Cell Death Dis. 2012, 3, e311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dan, S.; Tsunoda, T.; Kitahara, O.; Yanagawa, R.; Zembutsu, H.; Katagiri, T.; Yamazaki, K.; Nakamura, Y.; Yamori, T. An Integrated Database of Chemosensitivity to 55 Anticancer Drugs and Gene Expression Profiles of 39 Human Cancer Cell Lines. Cancer Res. 2002, 62, 1139–1147. [Google Scholar] [PubMed]
- Kim, J.-E.; Ryu, H.J.; Kim, M.J.; Kang, T.-C. LIM Kinase-2 Induces Programmed Necrotic Neuronal Death via Dysfunction of DRP1-Mediated Mitochondrial Fission. Cell Death Differ 2014, 21, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.-R.; Hyun, H.-W.; Min, S.-J.; Kim, J.-E.; Kang, T.-C. Endothelin-1 Induces LIMK2-Mediated Programmed Necrotic Neuronal Death Independent of NOS Activity. Mol. Brain 2015, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Ribba, A.-S.; Fraboulet, S.; Sadoul, K.; Lafanechère, L. The Role of LIM Kinases during Development: A Lens to Get a Glimpse of Their Implication in Pathologies. Cells 2022, 11, 403. [Google Scholar] [CrossRef]
- Cuberos, H.; Vallée, B.; Vourc’h, P.; Tastet, J.; Andres, C.R.; Bénédetti, H. Roles of LIM Kinases in Central Nervous System Function and Dysfunction. FEBS Lett. 2015, 589, 3795–3806. [Google Scholar] [CrossRef]
- Ben Zablah, Y.; Zhang, H.; Gugustea, R.; Jia, Z. LIM-Kinases in Synaptic Plasticity, Memory, and Brain Diseases. Cells 2021, 10, 2079. [Google Scholar] [CrossRef] [PubMed]
- Halasz, E.; Townes-Anderson, E.; Wang, W. LIM Kinases in Synaptic Plasticity and Their Potential as Therapeutic Targets. Neural Regen. Res. 2020, 15, 1471. [Google Scholar] [CrossRef]
- Meng, Y.; Zhang, Y.; Tregoubov, V.; Janus, C.; Cruz, L.; Jackson, M.; Lu, W.Y.; MacDonald, J.F.; Wang, J.Y.; Falls, D.L.; et al. Abnormal Spine Morphology and Enhanced LTP in LIMK-1 Knockout Mice. Neuron 2002, 35, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Takahashi, H.; Meng, J.; Zhang, Y.; Lu, G.; Asrar, S.; Nakamura, T.; Jia, Z. Regulation of ADF/Cofilin Phosphorylation and Synaptic Function by LIM-Kinase. Neuropharmacology 2004, 47, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, C.; Li, J.; Zhou, Z.; Ju, X.; Xia, S.; Li, Y.; Liu, A.; Teng, H.; Zhang, K.; et al. PAK2 Haploinsufficiency Results in Synaptic Cytoskeleton Impairment and Autism-Related Behavior. Cell Rep. 2018, 24, 2029–2041. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.L.; Choi, S.-Y.; Rao, B.S.S.; Jung, H.-Y.; Lee, H.-K.; Zhang, D.; Chattarji, S.; Kirkwood, A.; Tonegawa, S. Altered Cortical Synaptic Morphology and Impaired Memory Consolidation in Forebrain- Specific Dominant-Negative PAK Transgenic Mice. Neuron 2004, 42, 773–787. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Meng, Y.; Asrar, S.; Todorovski, Z.; Jia, Z. A Critical Role of Rho-Kinase ROCK2 in the Regulation of Spine and Synaptic Function. Neuropharmacology 2009, 56, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Todorovski, Z.; Asrar, S.; Liu, J.; Saw, N.M.N.; Joshi, K.; Cortez, M.A.; Snead, O.C.; Xie, W.; Jia, Z. LIMK1 Regulates Long-Term Memory and Synaptic Plasticity via the Transcriptional Factor CREB. Mol. Cell Biol 2015, 35, 1316–1328. [Google Scholar] [CrossRef] [Green Version]
- Rosso, S.; Bollati, F.; Bisbal, M.; Peretti, D.; Sumi, T.; Nakamura, T.; Quiroga, S.; Ferreira, A.; Cáceres, A. LIMK1 Regulates Golgi Dynamics, Traffic of Golgi-Derived Vesicles, and Process Extension in Primary Cultured Neurons. Mol. Biol. Cell 2004, 15, 3433–3449. [Google Scholar] [CrossRef] [Green Version]
- Salvarezza, S.B.; Deborde, S.; Schreiner, R.; Campagne, F.; Kessels, M.M.; Qualmann, B.; Caceres, A.; Kreitzer, G.; Rodriguez-Boulan, E. LIM Kinase 1 and Cofilin Regulate Actin Filament Population Required for Dynamin-Dependent Apical Carrier Fission from the Trans -Golgi Network. Mol. Biol. Cell 2009, 20, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davila, M.; Frost, A.R.; Grizzle, W.E.; Chakrabarti, R. LIM Kinase 1 Is Essential for the Invasive Growth of Prostate Epithelial Cells: Implications in Prostate Cancer. J. Biol. Chem. 2003, 278, 36868–36875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikhil, K.; Chang, L.; Viccaro, K.; Jacobsen, M.; McGuire, C.; Satapathy, S.R.; Tandiary, M.; Broman, M.M.; Cresswell, G.; He, Y.J.; et al. Identification of LIMK2 as a Therapeutic Target in Castration Resistant Prostate Cancer. Cancer Lett. 2019, 448, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, I.; Pirker, C.; Bilban, M.; Berger, W.; Losert, D.; Marosi, C.; Haas, O.A.; Wolff, K.; Pehamberger, H. Seven Novel and Stable Translocations Associated with Oncogenic Gene Expression in Malignant Melanoma. Neoplasia 2005, 7, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Narayan, A.K.; Lee, C.I.; Lehman, C.D. Screening for Breast Cancer. Med. Clin. N. Am. 2020, 104, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Malvi, P.; Janostiak, R.; Chava, S.; Manrai, P.; Yoon, E.; Singh, K.; Harigopal, M.; Gupta, R.; Wajapeyee, N. LIMK2 Promotes the Metastatic Progression of Triple-Negative Breast Cancer by Activating SRPK1. Oncogenesis 2020, 9, 77. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Tse, A.; Schwartz, G.K. Aurora Kinases: New Targets for Cancer Therapy. Clin. Cancer Res. 2006, 12, 6869–6875. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-C.; Lin, C.-Y.; Tarn, W.-Y.; Li, F.-Y. Elevated SRPK1 Lessens Apoptosis in Breast Cancer Cells through RBM4-Regulated Splicing Events. RNA 2014, 20, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhou, Z.; Subhramanyam, C.S.; Cao, Q.; Heng, Z.S.L.; Liu, W.; Fu, X.; Hu, Q. SRPK1 Acetylation Modulates Alternative Splicing to Regulate Cisplatin Resistance in Breast Cancer Cells. Commun. Biol. 2020, 3, 268. [Google Scholar] [CrossRef]
- van Roosmalen, W.; Le Dévédec, S.E.; Golani, O.; Smid, M.; Pulyakhina, I.; Timmermans, A.M.; Look, M.P.; Zi, D.; Pont, C.; de Graauw, M.; et al. Tumor Cell Migration Screen Identifies SRPK1 as Breast Cancer Metastasis Determinant. J. Clin. Investig. 2015, 125, 1648–1664. [Google Scholar] [CrossRef] [Green Version]
- Prunier, C.; Josserand, V.; Vollaire, J.; Beerling, E.; Petropoulos, C.; Destaing, O.; Montemagno, C.; Hurbin, A.; Prudent, R.; de Koning, L.; et al. LIM Kinase Inhibitor Pyr1 Reduces the Growth and Metastatic Load of Breast Cancers. Cancer Res. 2016, 76, 3541–3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Nikhil, K.; Haymour, H.S.; Kamra, M.; Shah, K. Phosphorylation-Dependent Regulation of SPOP by LIMK2 Promotes Castration-Resistant Prostate Cancer. Br. J. Cancer 2021, 124, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Nikhil, K.; Kamra, M.; Raza, A.; Haymour, H.S.; Shah, K. Molecular Interplay between AURKA and SPOP Dictates CRPC Pathogenesis via Androgen Receptor. Cancers 2021, 12, 3247. [Google Scholar] [CrossRef]
- Sooreshjani, M.A.; Nikhil, K.; Kamra, M.; Nguyen, D.N.; Kumar, D.; Shah, K. LIMK2-NKX3.1 Engagement Promotes Castration-Resistant Prostate Cancer. Cancers 2021, 13, 2324. [Google Scholar] [CrossRef]
- Zhao, Z.; Rahman, M.A.; Chen, Z.G.; Shin, D.M. Multiple Biological Functions of Twist1 in Various Cancers. Oncotarget 2017, 8, 20380–20393. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, N.C.; Cianchetta, G.; Burgoon, H.A.; Healy, J.; Mabon, R.; Strobel, E.D.; Allen, J.; Wang, S.; Hamman, B.D.; Rawlins, D.B. Discovery of a Type III Inhibitor of LIM Kinase 2 That Binds in a DFG-Out Conformation. ACS Med. Chem. Lett. 2015, 6, 53–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Xu, Y.; Pan, C.; Yan, L.; Wang, Z.; Zhu, X. The Emerging Role of SPOP Protein in Tumorigenesis and Cancer Therapy. Mol. Cancer 2020, 19, 2. [Google Scholar] [CrossRef]
- Nikhil, K.; Kamra, M.; Raza, A.; Shah, K. Negative Cross Talk between LIMK2 and PTEN Promotes Castration Resistant Prostate Cancer Pathogenesis in Cells and in Vivo. Cancer Lett. 2021, 498, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bowen, C.; Ostrowski, M.C.; Leone, G.; Gelmann, E.P. Loss of PTEN Accelerates NKX3.1 Degradation to Promote Prostate Cancer Progression. Cancer Res. 2019, 79, 4124–4134. [Google Scholar] [CrossRef] [Green Version]
- Mardilovich, K.; Baugh, M.; Crighton, D.; Kowalczyk, D.; Gabrielsen, M.; Munro, J.; Croft, D.R.; Lourenco, F.; James, D.; Kalna, G.; et al. LIM Kinase Inhibitors Disrupt Mitotic Microtubule Organization and Impair Tumor Cell Proliferation. Oncotarget 2015, 6, 38469–38486. [Google Scholar] [CrossRef] [Green Version]
- Fujiuchi, Y.; Nagakawa, O.; Murakami, K.; Fuse, H.; Saiki, I. Effect of Hepatocyte Growth Factor on Invasion of Prostate Cancer Cell Lines. Oncol. Rep. 2003, 10, 1001–1006. [Google Scholar] [CrossRef]
- Wells, C.M.; Ahmed, T.; Masters, J.R.W.; Jones, G.E. Rho Family GTPases Are Activated during HGF-Stimulated Prostate Cancer-Cell Scattering. Cell Motil. Cytoskelet. 2005, 62, 180–194. [Google Scholar] [CrossRef]
- Ahmed, T.; Shea, K.; Masters, J.R.W.; Jones, G.E.; Wells, C.M. A PAK4-LIMK1 Pathway Drives Prostate Cancer Cell Migration Downstream of HGF. Cell Signal 2008, 20, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Juliusson, G.; Hough, R. Leukemia. In Progress in Tumor Research; Stark, D.P., Vassal, G., Eds.; S. Karger AG: Basel, Switzerland, 2016; Volume 43, pp. 87–100. ISBN 978-3-318-05911-3. [Google Scholar]
- Guo, H.; Gu, F.; Li, W.; Zhang, B.; Niu, R.; Fu, L.; Zhang, N.; Ma, Y. Reduction of Protein Kinase C Zeta Inhibits Migration and Invasion of Human Glioblastoma Cells. J. Neurochem. 2009, 109, 203–213. [Google Scholar] [CrossRef]
- Jensen, P.; Carlet, M.; Schlenk, R.F.; Weber, A.; Kress, J.; Brunner, I.; Słabicki, M.; Grill, G.; Weisemann, S.; Cheng, Y.-Y.; et al. Requirement for LIM Kinases in Acute Myeloid Leukemia. Leukemia 2020, 34, 3173–3185. [Google Scholar] [CrossRef]
- Ross-Macdonald, P.; de Silva, H.; Guo, Q.; Xiao, H.; Hung, C.-Y.; Penhallow, B.; Markwalder, J.; He, L.; Attar, R.M.; Lin, T.; et al. Identification of a Nonkinase Target Mediating Cytotoxicity of Novel Kinase Inhibitors. Mol. Cancer Ther. 2008, 7, 3490–3498. [Google Scholar] [CrossRef] [Green Version]
- Oku, Y.; Tareyanagi, C.; Takaya, S.; Osaka, S.; Ujiie, H.; Yoshida, K.; Nishiya, N.; Uehara, Y. Multimodal Effects of Small Molecule ROCK and LIMK Inhibitors on Mitosis, and Their Implication as Anti-Leukemia Agents. PLoS ONE 2014, 9, e92402. [Google Scholar] [CrossRef] [PubMed]
- Djamai, H.; Berrou, J.; Dupont, M.; Kaci, A.; Ehlert, J.E.; Weber, H.; Baruchel, A.; Paublant, F.; Prudent, R.; Gardin, C.; et al. Synergy of FLT3 Inhibitors and the Small Molecule Inhibitor of LIM Kinase1/2 CEL_Amide in FLT3-ITD Mutated Acute Myeloblastic Leukemia (AML) Cells. Leuk. Res. 2021, 100, 106490. [Google Scholar] [CrossRef] [PubMed]
- Berrou, J.; Dupont, M.; Djamai, H.; Adicéam, E.; Parietti, V.; Kaci, A.; Clappier, E.; Cayuela, J.-M.; Baruchel, A.; Paublant, F.; et al. Preclinical Evaluation of a Novel Small Molecule Inhibitor of LIM Kinases (LIMK) CEL_Amide in Philadelphia-Chromosome Positive (BCR::ABL+) Acute Lymphoblastic Leukemia (ALL). J. Clin. Med. 2022, 11, 6761. [Google Scholar] [CrossRef]
- Brion, R.; Regnier, L.; Mullard, M.; Amiaud, J.; Rédini, F.; Verrecchia, F. LIM Kinases in Osteosarcoma Development. Cells 2021, 10, 3542. [Google Scholar] [CrossRef]
- Ritter, J.; Bielack, S.S. Osteosarcoma. Ann. Oncol. 2010, 21, vii320–vii325. [Google Scholar] [CrossRef]
- Zhang, H.-S.; Zhao, J.-W.; Wang, H.; Zhang, H.-Y.; Ji, Q.-Y.; Meng, L.-J.; Xing, F.-J.; Yang, S.-T.; Wang, Y. LIM Kinase 1 Is Required for Insulin-Dependent Cell Growth of Osteosarcoma Cell Lines. Mol. Med. Rep. 2014, 9, 103–108. [Google Scholar] [CrossRef]
- Li, Z.; Yao, Y.; Zhao, Y.; Liu, Y.; Liu, Z.; Hu, P.; Zhu, Z. Effects of PAK4/LIMK1/Cofilin-1 Signaling Pathway on Proliferation, Invasion, and Migration of Human Osteosarcoma Cells. J. Clin. Lab. Anal. 2020, 34, e23362. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, L.; Chen, R.; Meng, L.; Gao, Y.; Ji, Q.; Wang, Y. LIM Kinase 1 Serves an Important Role in the Multidrug Resistance of Osteosarcoma Cells. Oncol. Lett. 2017, 15, 250–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, L.; Qu, R.; Zhang, L.; Huang, W. Rho A Regulates Epidermal Growth Factor-Induced Human Osteosarcoma MG63 Cell Migration. Int. J. Mol. Sci. 2018, 19, 1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, T.; Zheng, B.; Huang, Y.; Wang, S.; Bao, X.; Liu, K.; Guo, W. Osteosarcoma Cell Intrinsic PD-L2 Signals Promote Invasion and Metastasis via the RhoA-ROCK-LIMK2 and Autophagy Pathways. Cell Death Dis. 2019, 10, 261. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Zhou, C.; Qu, G.; Ren, C.; Yan, P.; Guo, W.; Yue, B. VEGFR2 Promotes Metastasis and PD-L2 Expression of Human Osteosarcoma Cells by Activating the STAT3 and RhoA-ROCK-LIMK2 Pathways. Front. Oncol. 2020, 10, 543562. [Google Scholar] [CrossRef]
- Wang, G.; Sun, M.; Jiang, Y.; Zhang, T.; Sun, W.; Wang, H.; Yin, F.; Wang, Z.; Sang, W.; Xu, J.; et al. Anlotinib, a Novel Small Molecular Tyrosine Kinase Inhibitor, Suppresses Growth and Metastasis via Dual Blockade of VEGFR2 and MET in Osteosarcoma. Int. J. Cancer 2019, 145, 979–993. [Google Scholar] [CrossRef]
- Wang, S.; Ren, T.; Jiao, G.; Huang, Y.; Bao, X.; Zhang, F.; Liu, K.; Zheng, B.; Sun, K.; Guo, W. BMPR2 Promotes Invasion and Metastasis via the RhoA-ROCK-LIMK2 Pathway in Human Osteosarcoma Cells. Oncotarget 2017, 8, 58625–58641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foletta, V.C.; Lim, M.A.; Soosairaiah, J.; Kelly, A.P.; Stanley, E.G.; Shannon, M.; He, W.; Das, S.; Massagué, J.; Bernard, O. Direct Signaling by the BMP Type II Receptor via the Cytoskeletal Regulator LIMK. J. Cell Biol. 2003, 162, 1089–1098. [Google Scholar] [CrossRef]
- Yang, J.-Z.; Ma, S.-R.; Rong, X.-L.; Zhu, M.-J.; Ji, Q.-Y.; Meng, L.-J.; Gao, Y.-Y.; Yang, Y.-D.; Wang, Y. Characterization of Multidrug-Resistant Osteosarcoma Sublines and the Molecular Mechanisms of Resistance. Mol. Med. Rep. 2016, 14, 3269–3276. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Chen, L.; Liu, Y.; Chen, M.; Zhang, S.; Kong, D. Sea Cucumber Cucumaria Frondosa Fucoidan Inhibits Osteosarcoma Adhesion and Migration by Regulating Cytoskeleton Remodeling. Oncol. Rep. 2020, 44, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, M.; Nakamura, S.; Sugiyama, Y.; Tamai, S.; Ishida, Y.; Sueyoshi, M.; Toda, Y.; Hosogi, S.; Yano, Y.; Ashihara, E. 6-Hydroxythiobinupharidine Inhibits Migration of LM8 Osteosarcoma Cells by Decreasing Expression of LIM Domain Kinase 1. Anticancer Res. 2019, 39, 6507–6513. [Google Scholar] [CrossRef]
- Davis, M. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-B.; Agnihotri, S.; Golbourn, B.; Bertrand, K.C.; Luck, A.; Sabha, N.; Smith, C.A.; Byron, S.; Zadeh, G.; Croul, S.; et al. Transcriptional Profiling of GBM Invasion Genes Identifies Effective Inhibitors of the LIM Kinase-Cofilin Pathway. Oncotarget 2014, 5, 9382–9395. [Google Scholar] [CrossRef] [Green Version]
- Schulze, M.; Fedorchenko, O.; Zink, T.G.; Knobbe-Thomsen, C.B.; Kraus, S.; Schwinn, S.; Beilhack, A.; Reifenberger, G.; Monoranu, C.M.; Sirén, A.-L.; et al. Chronophin Is a Glial Tumor Modifier Involved in the Regulation of Glioblastoma Growth and Invasiveness. Oncogene 2016, 35, 3163–3177. [Google Scholar] [CrossRef] [PubMed]
- Erkutlu, I.; Cigiloglu, A.; Kalender, M.E.; Alptekin, M.; Demiryurek, A.T.; Suner, A.; Ozkaya, E.; Ulasli, M.; Camci, C.; Ulasli, M.; et al. Correlation between Rho-Kinase Pathway Gene Expressions and Development and Progression of Glioblastoma Multiforme. Tumour Biol. 2013, 34, 1139–1144. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, B.; Li, W.; Liu, X.; Wang, J.; Ding, T.; Zhang, J.; Ying, G.; Fu, L.; Gu, F. Intersectin1-s Is Involved in Migration and Invasion of Human Glioma Cells. J. Neurosci. Res. 2011, 89, 1079–1090. [Google Scholar] [CrossRef]
- Koul, D. PTEN Signaling Pathways in Glioblastoma. Cancer Biol. Ther. 2008, 7, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Vallée, B.; Doudeau, M.; Godin, F.; Gombault, A.; Tchalikian, A.; de Tauzia, M.-L.; Bénédetti, H. Nf1 RasGAP Inhibition of LIMK2 Mediates a New Cross-Talk between Ras and Rho Pathways. PLoS ONE 2012, 7, e47283. [Google Scholar] [CrossRef] [Green Version]
- Starinsky-Elbaz, S.; Faigenbloom, L.; Friedman, E.; Stein, R.; Kloog, Y. The Pre-GAP-Related Domain of Neurofibromin Regulates Cell Migration through the LIM Kinase/Cofilin Pathway. Mol. Cell. Neurosci. 2009, 42, 278–287. [Google Scholar] [CrossRef]
- Schulze, M.; Hutterer, M.; Sabo, A.; Hoja, S.; Lorenz, J.; Rothhammer-Hampl, T.; Herold-Mende, C.; Floßbach, L.; Monoranu, C.; Riemenschneider, M.J. Chronophin Regulates Active Vitamin B6 Levels and Transcriptomic Features of Glioblastoma Cell Lines Cultured under Non-Adherent, Serum-Free Conditions. BMC Cancer 2018, 18, 524. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zou, S.; Ren, T.; Zhao, L.-J.; Yu, L.-F.; Li, X.-Y.; Yan, X.; Zhang, L.-J. Alantolactone Suppresses the Metastatic Phenotype and Induces the Apoptosis of Glioblastoma Cells by Targeting LIMK Kinase Activity and Activating the Cofilin/G-actin Signaling Cascade. Int. J. Mol. Med. 2021, 47, 68. [Google Scholar] [CrossRef]
- Hou, Y.; Zhou, L.; Yang, Q.D.; Du, X.P.; Li, M.; Yuan, M.; Zhou, Z.W. Changes in Hippocampal Synapses and Learning-Memory Abilities in a Streptozotocin-Treated Rat Model and Intervention by Using Fasudil Hydrochloride. Neuroscience 2012, 200, 120–129. [Google Scholar] [CrossRef]
- Mao, R.; Deng, R.; Wei, Y.; Han, L.; Meng, Y.; Xie, W.; Jia, Z. LIMK1 and LIMK2 Regulate Cortical Development through Affecting Neural Progenitor Cell Proliferation and Migration. Mol. Brain 2019, 12, 67. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association 2016 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2016, 12, 459–509. [CrossRef]
- Neugroschl, J.; Wang, S. Alzheimer’s Disease: Diagnosis and Treatment Across the Spectrum of Disease Severity: A LZHEIMER’S D ISEASE. Mt. Sinai. J. Med. 2011, 78, 596–612. [Google Scholar] [CrossRef] [Green Version]
- Lyketsos, C.G.; Carrillo, M.C.; Ryan, J.M.; Khachaturian, A.S.; Trzepacz, P.; Amatniek, J.; Cedarbaum, J.; Brashear, R.; Miller, D.S. Neuropsychiatric Symptoms in Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-L.; Hu, N.; Tan, M.-S.; Yu, J.-T.; Tan, L. Behavioral and Psychological Symptoms in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.-J. A Systemic View of Alzheimer Disease—Insights from Amyloid-β Metabolism beyond the Brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Heredia, L.; Helguera, P.; de Olmos, S.; Kedikian, G.; Sola Vigo, F.; LaFerla, F.; Staufenbiel, M.; de Olmos, J.; Busciglio, J.; Caceres, A.; et al. Phosphorylation of Actin-Depolymerizing Factor/Cofilin by LIM-Kinase Mediates Amyloid -Induced Degeneration: A Potential Mechanism of Neuronal Dystrophy in Alzheimer’s Disease. J. Neurosci. 2006, 26, 6533–6542. [Google Scholar] [CrossRef]
- Mendoza-Naranjo, A.; Contreras-Vallejos, E.; Henriquez, D.R.; Otth, C.; Bamburg, J.R.; Maccioni, R.B.; Gonzalez-Billault, C. Fibrillar Amyloid-β 1-42 Modifies Actin Organization Affecting the Cofilin Phosphorylation State: A Role for Rac1/Cdc42 Effector Proteins and the Slingshot Phosphatase. J. Alzheimer’s Dis. 2012, 29, 63–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamburg, J.R.; Bernstein, B.W. Actin Dynamics and Cofilin-Actin Rods in Alzheimer Disease: Cofilin-Actin Rods in Neuronal Stress. Cytoskeleton 2016, 73, 477–497. [Google Scholar] [CrossRef] [Green Version]
- Maciver, S.K.; Harrington, C.R. Two Actin Binding Proteins, Actin Depolymerizing Factor and Cofilin, Are Associated with Hirano Bodies: 1995, 6, 1985–1988. NeuroReport 1995, 6, 1985–1988. [Google Scholar] [CrossRef]
- Galloway, P.G.; Perry, G.; Gambetti, P. Hirano Body Filaments Contain Actin and Actin-Associated Proteins. J. Neuropathol. Exp. Neurol. 1987, 46, 185–199. [Google Scholar] [CrossRef]
- Heredia, L.; Lin, R.; Vigo, F.S.; Kedikian, G.; Busciglio, J.; Lorenzo, A. Deposition of Amyloid Fibrils Promotes Cell-Surface Accumulation of Amyloid Beta Precursor Protein. Neurobiol. Dis. 2004, 16, 617–629. [Google Scholar] [CrossRef]
- Rush, T.; Martinez-Hernandez, J.; Dollmeyer, M.; Frandemiche, M.L.; Borel, E.; Boisseau, S.; Jacquier-Sarlin, M.; Buisson, A. Synaptotoxicity in Alzheimer’s Disease Involved a Dysregulation of Actin Cytoskeleton Dynamics through Cofilin 1 Phosphorylation. J. Neurosci. 2018, 38, 10349–10361. [Google Scholar] [CrossRef] [Green Version]
- Henderson, B.W.; Gentry, E.G.; Rush, T.; Troncoso, J.C.; Thambisetty, M.; Montine, T.J.; Herskowitz, J.H. Rho-Associated Protein Kinase 1 (ROCK1) Is Increased in Alzheimer’s Disease and ROCK1 Depletion Reduces Amyloid-β Levels in Brain. J. Neurochem. 2016, 138, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Henderson, B.W.; Greathouse, K.M.; Ramdas, R.; Walker, C.K.; Rao, T.C.; Bach, S.V.; Curtis, K.A.; Day, J.J.; Mattheyses, A.L.; Herskowitz, J.H. Pharmacologic Inhibition of LIMK1 Provides Dendritic Spine Resilience against β-Amyloid. Sci. Signal. 2019, 12, eaaw9318. [Google Scholar] [CrossRef]
- Zhang, H.; Ben Zablah, Y.; Liu, A.; Lee, D.; Zhang, H.; Meng, Y.; Zhou, C.; Liu, X.; Wang, Y.; Jia, Z. Overexpression of LIMK1 in Hippocampal Excitatory Neurons Improves Synaptic Plasticity and Social Recognition Memory in APP/PS1 Mice. Mol. Brain 2021, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Chen, H.; Li, C.; Xu, C.; Xu, Y.-J. C-Terminal Fragments of Amyloid Precursor Proteins Increase Cofilin Phosphorylation by LIM Kinase in Cultured Rat Primary Neurons. NeuroReport 2019, 30, 38–45. [Google Scholar] [CrossRef]
- Yin, Y.; Zheng, K.; Eid, N.; Howard, S.; Jeong, J.-H.; Yi, F.; Guo, J.; Park, C.M.; Bibian, M.; Wu, W.; et al. Bis-Aryl Urea Derivatives as Potent and Selective LIM Kinase (Limk) Inhibitors. J. Med. Chem. 2015, 58, 1846–1861. [Google Scholar] [CrossRef] [Green Version]
- Moon, M.; Jung, E.S.; Jeon, S.G.; Cha, M.-Y.; Jang, Y.; Kim, W.; Lopes, C.; Mook-Jung, I.; Kim, K.-S. Nurr1 (NR4A2) Regulates Alzheimer’s Disease-Related Pathogenesis and Cognitive Function in the 5XFAD Mouse Model: XXXX. Aging Cell 2019, 18, e12866. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Lee, V.M.-Y. Parkin and the Molecular Pathways of Parkinson’s Disease. Neuron 2001, 31, 885–888. [Google Scholar] [CrossRef] [Green Version]
- Erro, R.; Stamelou, M. The Motor Syndrome of Parkinson’s Disease. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 132, pp. 25–32. ISBN 978-0-12-809714-4. [Google Scholar]
- Forno, L.S. Neuropathology of Parkinson’s Disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Engelender, S. Ubiquitination of α-Synuclein and Autophagy in Parkinson’s Disease. Autophagy 2008, 4, 372–374. [Google Scholar] [CrossRef] [Green Version]
- Oliveira da Silva, M.I.; Liz, M.A. Linking Alpha-Synuclein to the Actin Cytoskeleton: Consequences to Neuronal Function. Front. Cell Dev. Biol. 2020, 8, 787. [Google Scholar] [CrossRef]
- Sousa, V.L.; Bellani, S.; Giannandrea, M.; Yousuf, M.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. α-Synuclein and Its A30P Mutant Affect Actin Cytoskeletal Structure and Dynamics. Mol. Biol. Cell 2009, 20, 3725–3739. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Meng, L.; Dai, L.; Zhang, X.; Chen, G.; Zheng, Y.; Zha, Y.; Zeng, Y.; Zhang, Z. Cofilin 1 Promotes the Aggregation and Cell-to-Cell Transmission of α-Synuclein in Parkinson’s Disease. Biochem. Biophys. Res. Commun. 2020, 529, 1053–1060. [Google Scholar] [CrossRef]
- Bellani, S.; Mescola, A.; Ronzitti, G.; Tsushima, H.; Tilve, S.; Canale, C.; Valtorta, F.; Chieregatti, E. GRP78 Clustering at the Cell Surface of Neurons Transduces the Action of Exogenous Alpha-Synuclein. Cell Death Differ. 2014, 21, 1971–1983. [Google Scholar] [CrossRef]
- Lim, M.K.; Kawamura, T.; Ohsawa, Y.; Ohtsubo, M.; Asakawa, S.; Takayanagi, A.; Shimizu, N. Parkin Interacts with LIM Kinase 1 and Reduces Its Cofilin-Phosphorylation Activity via Ubiquitination. Exp. Cell Res. 2007, 313, 2858–2874. [Google Scholar] [CrossRef]
- Chu, Y.; Le, W.; Kompoliti, K.; Jankovic, J.; Mufson, E.J.; Kordower, J.H. Nurr1 in Parkinson’s Disease and Related Disorders. J. Comp. Neurol. 2006, 494, 495–514. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Li, S.; Mo, J.-L.; Cai, H.-B.; Le, W.-D. Nurr1-Based Therapies for Parkinson’s Disease. CNS Neurosci. Ther. 2016, 22, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Jankovic, J.; Chen, S.; Le, W.D. The Role of Nurr1 in the Development of Dopaminergic Neurons and Parkinson’s Disease. Prog. Neurobiol. 2005, 77, 128–138. [Google Scholar] [CrossRef]
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism Spectrum Disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X Syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Maurin, T.; Zongaro, S.; Bardoni, B. Fragile X Syndrome: From Molecular Pathology to Therapy. Neurosci. Biobehav. Rev. 2014, 46, 242–255. [Google Scholar] [CrossRef]
- Chidambaram, S.B.; Rathipriya, A.G.; Bolla, S.R.; Bhat, A.; Ray, B.; Mahalakshmi, A.M.; Manivasagam, T.; Thenmozhi, A.J.; Essa, M.M.; Guillemin, G.J.; et al. Dendritic Spines: Revisiting the Physiological Role. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 92, 161–193. [Google Scholar] [CrossRef]
- Nishiyama, J. Plasticity of Dendritic Spines: Molecular Function and Dysfunction in Neurodevelopmental Disorders. Psychiatry Clin. Neurosci. 2019, 73, 541–550. [Google Scholar] [CrossRef]
- Michaelsen-Preusse, K.; Feuge, J.; Korte, M. Imbalance of Synaptic Actin Dynamics as a Key to Fragile X Syndrome?: Synaptic Actin and Fragile X Syndrome. J. Physiol. 2018, 596, 2773–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Kay, Y.; Sadybekov, A.; Rao, S.; Katritch, V.; Herring, B.E. An Intellectual Disability-Related Missense Mutation in Rac1 Prevents LTP Induction. Front. Mol. Neurosci. 2018, 11, 223. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, S.; Folsom, T.D.; Kneeland, R.E.; Yousefi, M.K.; Liesch, S.B.; Thuras, P.D. Impairment of Fragile X Mental Retardation Protein-Metabotropic Glutamate Receptor 5 Signaling and Its Downstream Cognates Ras-Related C3 Botulinum Toxin Substrate 1, Amyloid Beta A4 Precursor Protein, Striatal-Enriched Protein Tyrosine Phosphatase, and Homer 1, in Autism: A Postmortem Study in Cerebellar Vermis and Superior Frontal Cortex. Mol. Autism. 2013, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Pyronneau, A.; He, Q.; Hwang, J.-Y.; Porch, M.; Contractor, A.; Zukin, R.S. Aberrant Rac1-Cofilin Signaling Mediates Defects in Dendritic Spines, Synaptic Function, and Sensory Perception in Fragile X Syndrome. Sci. Signal. 2017, 10, eaan0852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolan, B.M.; Duron, S.G.; Campbell, D.A.; Vollrath, B.; Rao, B.S.S.; Ko, H.-Y.; Lin, G.G.; Govindarajan, A.; Choi, S.-Y.; Tonegawa, S. Rescue of Fragile X Syndrome Phenotypes in Fmr1 KO Mice by the Small-Molecule PAK Inhibitor FRAX486. Proc. Natl. Acad. Sci. USA 2013, 110, 5671–5676. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.L.; Rao, B.S.S.; Seo, J.-S.; Choi, H.-S.; Dolan, B.M.; Choi, S.-Y.; Chattarji, S.; Tonegawa, S. Inhibition of P21-Activated Kinase Rescues Symptoms of Fragile X Syndrome in Mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11489–11494. [Google Scholar] [CrossRef] [Green Version]
- Kashima, R.; Roy, S.; Ascano, M.; Martinez-Cerdeno, V.; Ariza-Torres, J.; Kim, S.; Louie, J.; Lu, Y.; Leyton, P.; Bloch, K.D.; et al. Augmented Noncanonical BMP Type II Receptor Signaling Mediates the Synaptic Abnormality of Fragile X Syndrome. Sci. Signal. 2016, 9, ra58. [Google Scholar] [CrossRef] [Green Version]
- Lee-Hoeflich, S.T.; Causing, C.G.; Podkowa, M.; Zhao, X.; Wrana, J.L.; Attisano, L. Activation of LIMK1 by Binding to the BMP Receptor, BMPRII, Regulates BMP-Dependent Dendritogenesis. EMBO J. 2004, 23, 4792–4801. [Google Scholar] [CrossRef] [Green Version]
- Kashima, R.; Redmond, P.L.; Ghatpande, P.; Roy, S.; Kornberg, T.B.; Hanke, T.; Knapp, S.; Lagna, G.; Hata, A. Hyperactive Locomotion in a Drosophila Model Is a Functional Readout for the Synaptic Abnormalities Underlying Fragile X Syndrome. Sci. Signal. 2017, 10, eaai8133. [Google Scholar] [CrossRef] [Green Version]
- Yao, F.; Zhang, K.; Feng, C.; Gao, Y.; Shen, L.; Liu, X.; Ni, J. Protein Biomarkers of Autism Spectrum Disorder Identified by Computational and Experimental Methods. Front. Psychiatry 2021, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M.; et al. De Novo Mutations in Schizophrenia Implicate Synaptic Networks. Nature 2014, 506, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.; Trent, S.; Thomas, K.L.; O’Donovan, M.C.; Owen, M.J. Genetic Risk for Schizophrenia: Convergence on Synaptic Pathways Involved in Plasticity. Biol. Psychiatry 2015, 77, 52–58. [Google Scholar] [CrossRef]
- Hill, J.J.; Hashimoto, T.; Lewis, D.A. Molecular Mechanisms Contributing to Dendritic Spine Alterations in the Prefrontal Cortex of Subjects with Schizophrenia. Mol. Psychiatry 2006, 11, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Ide, M.; Lewis, D.A. Altered Cortical CDC42 Signaling Pathways in Schizophrenia: Implications for Dendritic Spine Deficits. Biol. Psychiatry 2010, 68, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, D.; Arion, D.; Corradi, J.P.; Lewis, D.A. Altered Expression of CDC42 Signaling Pathway Components in Cortical Layer 3 Pyramidal Cells in Schizophrenia. Biol. Psychiatry 2015, 78, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Genis-Mendoza, A.D.; Beltrán-Villalobos, I.; Nicolini-Sánchez, H. Behavioral Assessment of the “Schizophrenia-like” Phenotype in an Animal Model of Neonatal Lesion in the Ventral Hippocampus (NLVH) of Young and Adult Rats. Gac. Med. Mex. 2014, 150, 420–431. [Google Scholar]
- Genis-Mendoza, A.D.; Gallegos-Silva, R.I.; López-Casamichana, M.; López-Rubalcava, C.; Nicolini, H. Gene Expression Profiles of Nucleus Accumbens, Prefrontal Cortex and Hippocampus in an Animal Model of Schizophrenia: Proposed Candidate Genes. Actas. Esp. Psiquiatr. 2013, 41, 154–163. [Google Scholar]
- Foote, M.; Qiao, H.; Graham, K.; Wu, Y.; Zhou, Y. Inhibition of 14-3-3 Proteins Leads to Schizophrenia-Related Behavioral Phenotypes and Synaptic Defects in Mice. Biol. Psychiatry 2015, 78, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Xu, J.; Chen, J.; Kim, S.; Reimers, M.; Bacanu, S.-A.; Yu, H.; Liu, C.; Sun, J.; Wang, Q.; et al. Transcriptome Sequencing and Genome-Wide Association Analyses Reveal Lysosomal Function and Actin Cytoskeleton Remodeling in Schizophrenia and Bipolar Disorder. Mol. Psychiatry 2015, 20, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Zhou, Z.; Dang, R.; Zhu, Y.; Qi, J.; He, G.; Leung, C.; Pak, D.; Jia, Z.; Xie, W. Neuroligin 1 Regulates Spines and Synaptic Plasticity via LIMK1/Cofilin-Mediated Actin Reorganization. J. Cell Biol. 2016, 212, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Stefansson, H.; Petursson, H.; Sigurdsson, E.; Steinthorsdottir, V.; Bjornsdottir, S.; Sigmundsson, T.; Ghosh, S.; Brynjolfsson, J.; Gunnarsdottir, S.; Ivarsson, O.; et al. Neuregulin 1 and Susceptibility to Schizophrenia. Am. J. Hum. Genet. 2002, 71, 877–892. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Jing, H.; Xiong, M.; Zhang, Q.; Lin, D.; Ren, D.; Wang, S.; Yin, D.; Chen, Y.; Zhou, T.; et al. Spine Impairment in Mice High-Expressing Neuregulin 1 Due to LIMK1 Activation. Cell Death Dis. 2021, 12, 403. [Google Scholar] [CrossRef]
- Yin, D.-M.; Chen, Y.-J.; Lu, Y.-S.; Bean, J.C.; Sathyamurthy, A.; Shen, C.; Liu, X.; Lin, T.W.; Smith, C.A.; Xiong, W.-C.; et al. Reversal of Behavioral Deficits and Synaptic Dysfunction in Mice Overexpressing Neuregulin 1. Neuron 2013, 78, 644–657. [Google Scholar] [CrossRef] [Green Version]
- Gory-Fauré, S.; Powell, R.; Jonckheere, J.; Lanté, F.; Denarier, E.; Peris, L.; Nguyen, C.H.; Buisson, A.; Lafanechère, L.; Andrieux, A. Pyr1-Mediated Pharmacological Inhibition of LIM Kinase Restores Synaptic Plasticity and Normal Behavior in a Mouse Model of Schizophrenia. Front. Pharmacol. 2021, 12, 627995. [Google Scholar] [CrossRef]
- Kozel, B.A.; Barak, B.; Kim, C.A.; Mervis, C.B.; Osborne, L.R.; Porter, M.; Pober, B.R. Williams Syndrome. Nat. Rev. Dis. Prim. 2021, 7, 42. [Google Scholar] [CrossRef]
- Gregory, M.D.; Mervis, C.B.; Elliott, M.L.; Kippenhan, J.S.; Nash, T.; Czarapata, J.B.; Prabhakaran, R.; Roe, K.; Eisenberg, D.P.; Kohn, P.D.; et al. Williams Syndrome Hemideletion and LIMK1 Variation Both Affect Dorsal Stream Functional Connectivity. Brain 2019, 142, 3963–3974. [Google Scholar] [CrossRef]
- Frangiskakis, J.M.; Ewart, A.K.; Morris, C.A.; Mervis, C.B.; Bertrand, J.; Robinson, B.F.; Klein, B.P.; Ensing, G.J.; Everett, L.A.; Green, E.D.; et al. LIM-Kinase1 Hemizygosity Implicated in Impaired Visuospatial Constructive Cognition. Cell 1996, 86, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Sivadasan, R.; Hornburg, D.; Drepper, C.; Frank, N.; Jablonka, S.; Hansel, A.; Lojewski, X.; Sterneckert, J.; Hermann, A.; Shaw, P.J.; et al. C9ORF72 Interaction with Cofilin Modulates Actin Dynamics in Motor Neurons. Nat. Neurosci. 2016, 19, 1610–1618. [Google Scholar] [CrossRef]
- Hensel, N.; Claus, P. The Actin Cytoskeleton in SMA and ALS: How Does It Contribute to Motoneuron Degeneration? Neuroscientist 2018, 24, 54–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, K.; Wharton, K.A. BMP/TGF-β Signaling as a Modulator of Neurodegeneration in ALS. Dev. Dyn. 2021, 251, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Rossie, S. Tale of the Good and the Bad Cdk5: Remodeling of the Actin Cytoskeleton in the Brain. Mol. Neurobiol. 2018, 55, 3426–3438. [Google Scholar] [CrossRef] [PubMed]
- Gillardon, F.; Steinlein, P.; Bürger, E.; Hildebrandt, T.; Gerner, C. Phosphoproteome and Transcriptome Analysis of the Neuronal Response to a CDK5 Inhibitor. Proteomics 2005, 5, 1299–1307. [Google Scholar] [CrossRef]
- Conti, A.; Riva, N.; Pesca, M.; Iannaccone, S.; Cannistraci, C.V.; Corbo, M.; Previtali, S.C.; Quattrini, A.; Alessio, M. Increased Expression of Myosin Binding Protein H in the Skeletal Muscle of Amyotrophic Lateral Sclerosis Patients. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimino, P.J.; Gutmann, D.H. Neurofibromatosis Type 1. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 148, pp. 799–811. ISBN 978-0-444-64076-5. [Google Scholar]
- Hyman, S.L.; Shores, A.; North, K.N. The Nature and Frequency of Cognitive Deficits in Children with Neurofibromatosis Type 1. Neurology 2005, 65, 1037–1044. [Google Scholar] [CrossRef]
- Viskochil, D.; Buchberg, A.M.; Xu, G.; Cawthon, R.M.; Stevens, J.; Wolff, R.K.; Culver, M.; Carey, J.C.; Copeland, N.G.; Jenkins, N.A.; et al. Deletions and a Translocation Interrupt a Cloned Gene at the Neurofibromatosis Type 1 Locus. Cell 1990, 62, 187–192. [Google Scholar] [CrossRef]
- Daston, M.M.; Scrable, H.; Nordlund, M.; Sturbaum, A.K.; Nissen, L.M.; Ratner, N. The Protein Product of the Neurofibromatosis Type 1 Gene Is Expressed at Highest Abundance in Neurons, Schwann Cells, and Oligodendrocytes. Neuron 1992, 8, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef]
- Ozawa, T.; Araki, N.; Yunoue, S.; Tokuo, H.; Feng, L.; Patrakitkomjorn, S.; Hara, T.; Ichikawa, Y.; Matsumoto, K.; Fujii, K.; et al. The Neurofibromatosis Type 1 Gene Product Neurofibromin Enhances Cell Motility by Regulating Actin Filament Dynamics via the Rho-ROCK-LIMK2-Cofilin Pathway. J. Biol. Chem. 2005, 280, 39524–39533. [Google Scholar] [CrossRef] [Green Version]
- Asthagiri, A.R.; Parry, D.M.; Butman, J.A.; Kim, H.J.; Tsilou, E.T.; Zhuang, Z.; Lonser, R.R. Neurofibromatosis Type 2. Lancet 2009, 373, 1974–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.; Giancotti, F.G. Molecular Insights into NF2 /Merlin Tumor Suppressor Function. FEBS Lett. 2014, 588, 2743–2752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, M.; Shevde, L.A. Merlin Regulates Signaling Events at the Nexus of Development and Cancer. Cell Commun. Signal 2020, 18, 63. [Google Scholar] [CrossRef] [Green Version]
- Kissil, J.L.; Wilker, E.W.; Johnson, K.C.; Eckman, M.S.; Yaffe, M.B.; Jacks, T. Merlin, the Product of the Nf2 Tumor Suppressor Gene, Is an Inhibitor of the P21-Activated Kinase, Pak1. Mol. Cell 2003, 12, 841–849. [Google Scholar] [CrossRef]
- Petrilli, A.; Copik, A.; Posadas, M.; Chang, L.-S.; Welling, D.B.; Giovannini, M.; Fernández-Valle, C. LIM Domain Kinases as Potential Therapeutic Targets for Neurofibromatosis Type 2. Oncogene 2014, 33, 3571–3582. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.P.; Koyuncu, O.O.; Enquist, L.W. Subversion of the Actin Cytoskeleton during Viral Infection. Nat. Rev. Microbiol. 2011, 9, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.; Naghavi, M.H. Exploitation of Cytoskeletal Networks during Early Viral Infection. Trends Microbiol. 2019, 27, 39–50. [Google Scholar] [CrossRef]
- Manetti, F. HIV-1 Proteins Join the Family of LIM Kinase Partners. New Roads Open up for HIV-1 Treatment. Drug Discov. Today 2012, 17, 81–88. [Google Scholar] [CrossRef]
- Ospina Stella, A.; Turville, S. All-Round Manipulation of the Actin Cytoskeleton by HIV. Viruses 2018, 10, 63. [Google Scholar] [CrossRef] [Green Version]
- Stolp, B.; Fackler, O.T. How HIV Takes Advantage of the Cytoskeleton in Entry and Replication. Viruses 2011, 3, 293–311. [Google Scholar] [CrossRef] [Green Version]
- Vorster, P.J.; Guo, J.; Yoder, A.; Wang, W.; Zheng, Y.; Xu, X.; Yu, D.; Spear, M.; Wu, Y. LIM Kinase 1 Modulates Cortical Actin and CXCR4 Cycling and Is Activated by HIV-1 to Initiate Viral Infection. J. Biol. Chem. 2011, 286, 12554–12564. [Google Scholar] [CrossRef] [Green Version]
- Yoder, A.; Yu, D.; Dong, L.; Iyer, S.R.; Xu, X.; Kelly, J.; Liu, J.; Wang, W.; Vorster, P.J.; Agulto, L.; et al. HIV Envelope-CXCR4 Signaling Activates Cofilin to Overcome Cortical Actin Restriction in Resting CD4 T Cells. Cell 2008, 134, 782–792. [Google Scholar] [CrossRef] [Green Version]
- Cameron, P.U.; Saleh, S.; Sallmann, G.; Solomon, A.; Wightman, F.; Evans, V.A.; Boucher, G.; Haddad, E.K.; Sekaly, R.-P.; Harman, A.N.; et al. Establishment of HIV-1 Latency in Resting CD4+ T Cells Depends on Chemokine-Induced Changes in the Actin Cytoskeleton. Proc. Natl. Acad. Sci. USA 2010, 107, 16934–16939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Yoder, A.; Yu, D.; Wang, W.; Liu, J.; Barrett, T.; Wheeler, D.; Schlauch, K. Cofilin Activation in Peripheral CD4 T Cells of HIV-1 Infected Patients: A Pilot Study. Retrovirology 2008, 5, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishaq, M.; Lin, B.-R.; Bosche, M.; Zheng, X.; Yang, J.; Huang, D.; Lempicki, R.A.; Natarajan, V. LIM Kinase 1-Dependent Cofilin 1 Pathway and Actin Dynamics Mediate Nuclear Retinoid Receptor Function in T Lymphocytes. BMC Mol. Biol. 2011, 12, 41. [Google Scholar] [CrossRef] [Green Version]
- Gladnikoff, M.; Shimoni, E.; Gov, N.S.; Rousso, I. Retroviral Assembly and Budding Occur through an Actin-Driven Mechanism. Biophys. J. 2009, 97, 2419–2428. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.; Ding, L.; Wang, J.-J.; Qi, M.; Hammonds, J.; Chu, H.; Chen, X.; Hunter, E.; Spearman, P. ROCK1 and LIM Kinase Modulate Retrovirus Particle Release and Cell-Cell Transmission Events. J. Virol. 2014, 88, 6906–6921. [Google Scholar] [CrossRef] [Green Version]
- Van den Broeke, C.; Favoreel, H.W. Actin’ up: Herpesvirus Interactions with Rho GTPase Signaling. Viruses 2011, 3, 278–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Zheng, K.; Ju, H.; Wang, S.; Pei, Y.; Ding, W.; Chen, Z.; Wang, Q.; Qiu, X.; Zhong, M.; et al. Cofilin 1-Mediated Biphasic F-Actin Dynamics of Neuronal Cells Affect Herpes Simplex Virus 1 Infection and Replication. J. Virol. 2012, 86, 8440–8451. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Xiang, Y.; Wang, X.; Wang, Q.; Zhong, M.; Wang, S.; Wang, X.; Fan, J.; Kitazato, K.; Wang, Y. Epidermal Growth Factor Receptor-PI3K Signaling Controls Cofilin Activity To Facilitate Herpes Simplex Virus 1 Entry into Neuronal Cells. mBio 2014, 5, e00958-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Song, X.; Su, G.; Wang, Y.; Wang, Z.; Jia, J.; Qing, S.; Huang, L.; Wang, Y.; Zheng, K.; et al. Amentoflavone Inhibits HSV-1 and ACV-Resistant Strain Infection by Suppressing Viral Early Infection. Viruses 2019, 11, 466. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.; Guo, J.; Dabbagh, D.; Spear, M.; He, S.; Kehn-Hall, K.; Fontenot, J.; Yin, Y.; Bibian, M.; Park, C.M.; et al. Discovery of Novel Small-Molecule Inhibitors of LIM Domain Kinase for Inhibiting HIV-1. J. Virol. 2017, 91, 21. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Song, X.; Wang, Y.; Huang, L.; Luo, W.; Li, F.; Qin, S.; Wang, Y.; Xiao, J.; Wu, Y.; et al. Dysregulation of Cofilin-1 Activity—the Missing Link between Herpes Simplex Virus Type-1 Infection and Alzheimer’s Disease. Crit. Rev. Microbiol. 2020, 46, 381–396. [Google Scholar] [CrossRef]
- Park, J.; Kim, S.W.; Cho, M.C. The Role of LIM Kinase in the Male Urogenital System. Cells 2021, 11, 78. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villalonga, E.; Mosrin, C.; Normand, T.; Girardin, C.; Serrano, A.; Žunar, B.; Doudeau, M.; Godin, F.; Bénédetti, H.; Vallée, B. LIM Kinases, LIMK1 and LIMK2, Are Crucial Node Actors of the Cell Fate: Molecular to Pathological Features. Cells 2023, 12, 805. https://doi.org/10.3390/cells12050805
Villalonga E, Mosrin C, Normand T, Girardin C, Serrano A, Žunar B, Doudeau M, Godin F, Bénédetti H, Vallée B. LIM Kinases, LIMK1 and LIMK2, Are Crucial Node Actors of the Cell Fate: Molecular to Pathological Features. Cells. 2023; 12(5):805. https://doi.org/10.3390/cells12050805
Chicago/Turabian StyleVillalonga, Elodie, Christine Mosrin, Thierry Normand, Caroline Girardin, Amandine Serrano, Bojan Žunar, Michel Doudeau, Fabienne Godin, Hélène Bénédetti, and Béatrice Vallée. 2023. "LIM Kinases, LIMK1 and LIMK2, Are Crucial Node Actors of the Cell Fate: Molecular to Pathological Features" Cells 12, no. 5: 805. https://doi.org/10.3390/cells12050805
APA StyleVillalonga, E., Mosrin, C., Normand, T., Girardin, C., Serrano, A., Žunar, B., Doudeau, M., Godin, F., Bénédetti, H., & Vallée, B. (2023). LIM Kinases, LIMK1 and LIMK2, Are Crucial Node Actors of the Cell Fate: Molecular to Pathological Features. Cells, 12(5), 805. https://doi.org/10.3390/cells12050805