
S1P1 Threonine 236 Phosphorylation Mediates the Invasiveness of Triple-Negative Breast Cancer and Sensitivity to FTY720

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Zebrafish Maintenance and Husbandry
2.2. Subcloning
2.3. Cell Line Maintenance and Lentivirus Transduction
2.4. Cell Proliferation and Wound-Scratch Assays
2.5. Zebrafish Transplantation and Imaging
2.6. Small Molecule Treatment
2.7. Protein Extraction and Western Blotting
2.8. Gene Expression Analysis
2.9. Statistical Analysis
3. Results
3.1. T236 Phosphorylation but Not Total S1P1 Is Elevated in Human TNBC Cells
3.2. TNBC Progression Positively Correlates with S1P1 T236 Phosphorylation
3.3. Phosphorylation of S1P1 T236 Is Essential for TNBC Migration and Invasion
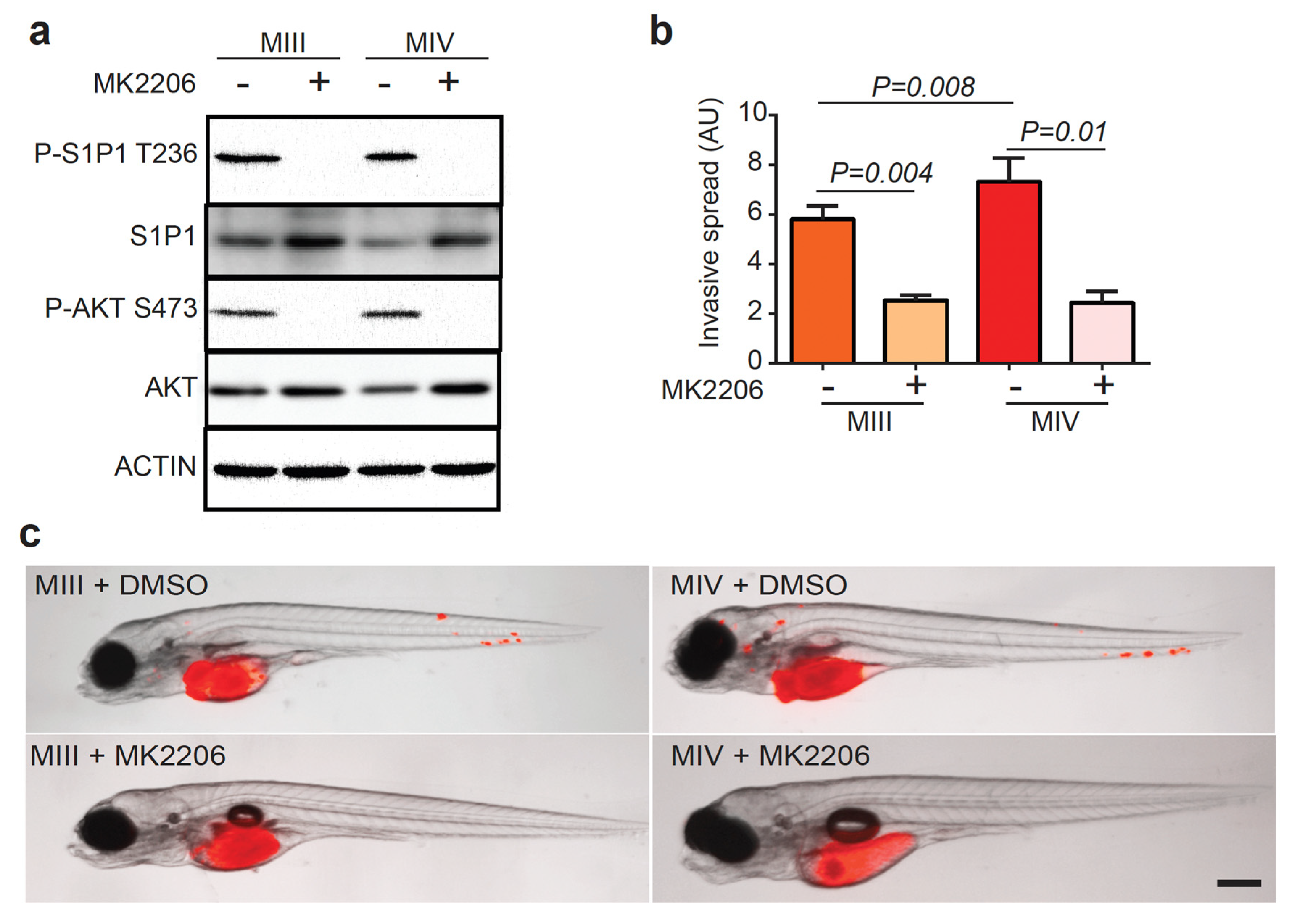
3.4. AKT Phosphorylates S1P1 T236 in TNBC Cells to Promote Disease Invasion
3.5. FTY720 Sensitizes TNBC Cells with Elevated P-S1P1 T236 to Apoptosis and Reduces Their Invasion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 2017, 8, 1913–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer 2016, 8, 93–107. [Google Scholar]
- Qiu, D.; Zhang, G.; Yan, X.; Xiao, X.; Ma, X.; Lin, S.; Wu, J.; Li, X.; Wang, W.; Liu, J.; et al. Prospects of Immunotherapy for Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 797092. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.F.; Jameel, F.; Irfan, M. Immunotherapy for Triple-Negative Breast Cancer: Latest Research and Clinical Prospects. Crit. Rev. Immunol. 2019, 39, 211–221. [Google Scholar] [CrossRef]
- Yuan, N.; Meng, M.; Liu, C.; Feng, L.; Hou, L.; Ning, Q.; Xin, G.; Pei, L.; Gu, S.; Li, X.; et al. Clinical characteristics and prognostic analysis of triple-negative breast cancer patients. Mol. Clin. Oncol. 2014, 2, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Anders, C.K.; Abramson, V.; Tan, T.; Dent, R. The evolution of triple-negative breast cancer: Form biology to novel therapeutics. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 34–42. [Google Scholar] [CrossRef]
- Pyne, N.J.; Tonelli, F.; Lim, K.G.; Long, J.S.; Edwards, J.; Pyne, S. Sphingosine 1-phosphate signalling in cancer. Biochem. Soc. Trans. 2012, 40, 94–100. [Google Scholar] [CrossRef]
- Patmanathan, S.N.; Wang, W.; Yap, L.F.; Herr, D.R.; Paterson, I.C. Mechanisms of sphingosine 1-phosphate receptor signalling in cancer. Cell. Signal. 2017, 34, 66–75. [Google Scholar] [CrossRef]
- Datta, A.; Loo, S.Y.; Huang, B.; Wong, L.; Tan, S.S.L.; Tan, T.Z.; Lee, S.; Thiery, J.P.; Lim, Y.C.; Yong, W.P.; et al. SPHK1 regulates proliferation and survival responses in triple-negative breast cancer. Oncotarget 2014, 5, 5920–5932. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.L.; Julovi, S.M.; Lin, M.Z.; de Silva, H.C.; Boyle, F.M.; Baxter, R.C. Inhibition of basal-like breast cancer growth by FTY720 in combination with epidermal growth factor receptor kinase blockade. Breast Cancer Res. 2017, 19, 90. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Liu, W.; Tian, M.; Fan, J.; Shi, Y. The SphKs/S1P/S1PR1 axis in immunity and cancer: More ore to be mined. World J. Surg. Oncol. 2016, 14, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hla, T.; Maciag, T. An abundant transcript induced in differentiating human endothelial cells encodes a polypeptide with structural similarities to G-protein-coupled receptors. J. Biol. Chem. 1990, 265, 9308–9313. [Google Scholar] [CrossRef]
- Lee, M.; Thangada, S.; Paik, J.; Sapkota, G.P.; Ancellin, N.; Chae, S.; Wu, M.; Morales-Ruiz, M.; Sessa, W.C.; Alessi, D.R.; et al. Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Mol. Cell 2001, 8, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Mizugishi, K.; Li, C.; Olivera, A.; Bielawski, J.; Bielawska, A.; Deng, C.; Proia, R.L. Maternal disturbance in activated sphingolipid metabolism causes pregnancy loss in mice. J. Clin. Invest. 2007, 117, 2993–3006. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Sanchez, T.; Yamase, H.; Hla, T.; Oo, M.L.; Pappalardo, A.; Lynch, K.R.; Lin, C.; Ferrer, F. S1P/S1P1 signaling stimulates cell migration and invasion in Wilms tumor. Cancer Lett. 2009, 276, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Fisher, K.E.; Pop, A.; Koh, W.; Anthis, N.J.; Saunders, W.B.; Davis, G.E. Tumor cell invasion of collagen matrices requires coordinate lipid agonist-induced G-protein and membrane-type matrix metalloproteinase-I-dependent signaling. Mol. Cancer 2006, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Young, N.; Pearl, D.K.; Brocklyn, J.R.V. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cry61. Mol. Cancer Res. 2009, 7, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Chae, S.; Paik, J.; Furneaux, H.; Hla, T. Requirement for sphingosine 1-phosphate receptor-1 in tumor angiogenesis demonstrated by in vivo RNA interference. J. Clin. Invest. 2004, 114, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Watson, C.; Long, J.S.; Orange, C.; Tannahill, C.L.; Mallon, E.; McGlynn, L.M.; Pyne, S.; Pyne, N.J.; Edwards, J. High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am. J. Pathol. 2010, 177, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Jiao, J.; Xu, C.; Dong, Y.; Li, D.; He, D.; Zhao, D.; Yu, J.; Sun, Y.; Zhang, W.; et al. The targetable nanoparticle BAF312@cRGD-CaP-NP represses tumor growth and angiogenesis by downregulating the S1PR1/P-STAT3/VEGFA axis in triple-negative breast cancer. J. Nanobiotechnology 2021, 19, 165. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Jiang, Y.X.; Chen, L.; Huang, L.; Tian, X.H.; Zhou, Y.D.; Nagle, D.G.; Zhang, D.D. Coix Seed Oil Exerts an Anti-Triple-Negative Breast Cancer Effect by Disrupting miR-205/S1PR1 Axis. Front. Pharmacol. 2020, 11, 529962. [Google Scholar] [CrossRef]
- Wang, Y.; Klijn, J.G.M.; Sieuwerts, A.M.; Look, M.P.; Yang, F.; Talantov, D.; Timmermans, M.; Meijer-van Gelder, M.E.; Yu, J.; Jatkoe, T.; et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005, 365, 671–679. [Google Scholar] [CrossRef]
- Imbalzano, K.M.; Tatarkova, I.; Imbalzano, A.N.; Nickerson, J.A. Increasingly transformed MCF-10A cells have a progressively tumor-like phenotype in three-dimensional basement membrane culture. Cancer Cell Int. 2009, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Xie, X.; Walker, S.; White, D.T.; Mumm, J.S.; Cowell, J.K. Evaluating human cancer cell metastasis in zebrafish. BMC Cancer 2013, 13, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertman, J.; Veinotte, C.J.; Dellaire, G.; Berman, J.N. The zebrafish xenograft platform: Evolution of a novel cancer model and preclinical screening tool. Adv. Exp. Med. Biol. 2016, 916, 289–314. [Google Scholar]
- Veinotte, C.J.; Dellaire, G.; Berman, J.N. Hooking the big one: The potential of zebrafish xenotransplantation to reform cancer drug screening in the genomic era. Dis. Model. Mech. 2014, 7, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Sossey-Alaoui, K.; Safina, A.; Li, X.; Vaughan, M.M.; Hicks, D.G.; Bakin, A.V.; Cowell, J.K. Down-regulation of WAVE3, a metastasis promoter gene, inhibits invasion and metastasis of breast cancer cells. Am. J. Pathol. 2007, 170, 2112–2121. [Google Scholar] [CrossRef] [Green Version]
- Bos, P.D.; Zhang, X.H.F.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; Van de Vijver, M.; Gerald, W.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef] [Green Version]
- Sangai, T.; Akcakanat, A.; Chen, H.; Tarco, E.; Wu, Y.; Do, K.; Miller, T.W.; Arteaga, C.L.; Mills, G.B.; Gonzalez-Angulo, A.M.; et al. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin. Cancer Res. 2012, 18, 5816–5828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.; Alshaker, H.; Cooper, C.; Winkler, M.; Pchejetski, D. The emerging role of FTY720 (Fingolimod) in cancer treatment. Oncotarget 2016, 7, 23106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, J.; Hartung, H. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [Green Version]
- O’Hurley, G.; Daly, E.; O’Grady, A.; Cummins, R.; Quinn, C.; Flanagan, L.; Pierce, A.; Fan, Y.; Lynn, M.A.; Rafferty, M.; et al. Investigation of molecular alterations of AKT-3 in triple-negative breast cancer. Histopathology 2014, 64, 660–670. [Google Scholar] [CrossRef]
- Urbano, M.; Guerrero, M.; Rosen, H.; Roberts, E. Modulators of the Sphingosine 1-phosphate receptor 1. Bioorg. Med. Chem. Lett. 2013, 23, 6377–6389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluk, M.J.; Ryan, K.; Wang, B.; Zhang, G.; Rodig, S.J.; Sanchez, T. Sphingosine-1-phosphate receptor 1 in classical hodgkin lymphoma: Assessment of expression and role in cell migration. Lab. Investig. 2013, 93, 462–471. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Ryu, J.; Yoon, G.; Jeon, H.; Cho, Y.; Choi, J.; Song, S.Y.; Do, I.; Lee, Y.; Kim, T.; et al. Sphingosine kinase 1 as a potential therapeutic target in epithelial ovarian cancer. Int. J. Cancer 2015, 137, 221–229. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laroche, F.J.F.; Li, S.; Shen, N.; Hwang, S.K.; Nguyen, G.; Yu, W.; Wong, C.K.; Quinton, R.J.; Berman, J.N.; Liu, C.-T.; et al. S1P1 Threonine 236 Phosphorylation Mediates the Invasiveness of Triple-Negative Breast Cancer and Sensitivity to FTY720. Cells 2023, 12, 980. https://doi.org/10.3390/cells12070980
Laroche FJF, Li S, Shen N, Hwang SK, Nguyen G, Yu W, Wong CK, Quinton RJ, Berman JN, Liu C-T, et al. S1P1 Threonine 236 Phosphorylation Mediates the Invasiveness of Triple-Negative Breast Cancer and Sensitivity to FTY720. Cells. 2023; 12(7):980. https://doi.org/10.3390/cells12070980
Chicago/Turabian StyleLaroche, Fabrice J. F., Sheng Li, Ning Shen, Soo Kyung Hwang, Gina Nguyen, Wenling Yu, Chen Khuan Wong, Ryan J. Quinton, Jason N. Berman, Ching-Ti Liu, and et al. 2023. "S1P1 Threonine 236 Phosphorylation Mediates the Invasiveness of Triple-Negative Breast Cancer and Sensitivity to FTY720" Cells 12, no. 7: 980. https://doi.org/10.3390/cells12070980