
Partial Inhibition of Complex I Restores Mitochondrial Morphology and Mitochondria-ER Communication in Hippocampus of APP/PS1 Mice

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and CP2 Synthesis
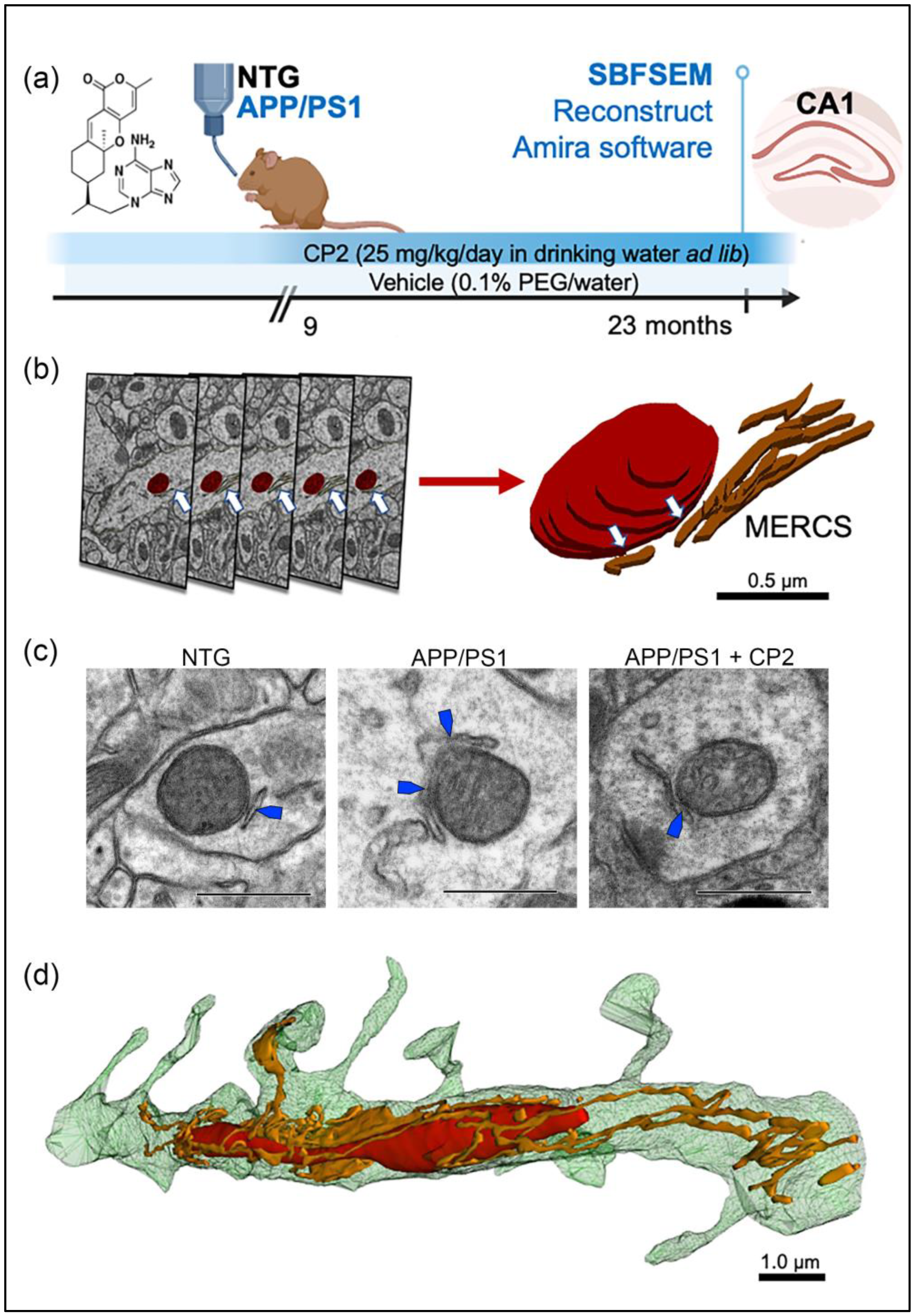
2.2. Mice and Chronic In Vivo CP2 Treatment
2.3. Tissue Dissection for Electron Microscopy Examination
2.4. Serial Block-Face Scanning Electron Microscopy (SBFSEM) and 3D EM Reconstruction
2.5. Image Segmentation and Quantitative Morphometric Analysis Using 3D EM
2.6. Mitochondrial Fractionation
2.7. Western Blot Analysis
2.8. Next-Generation RNA Sequencing
2.9. Statistics
3. Results
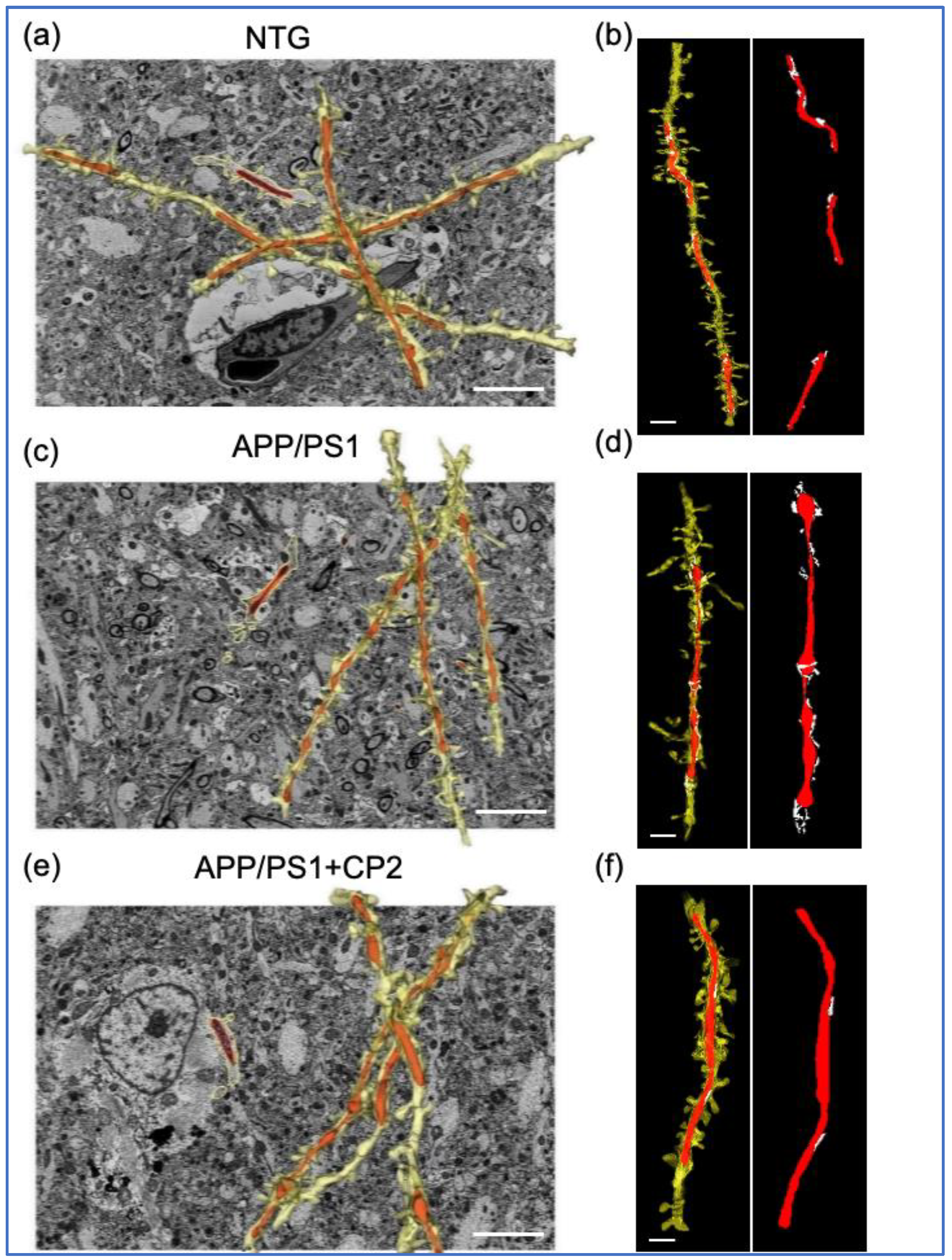
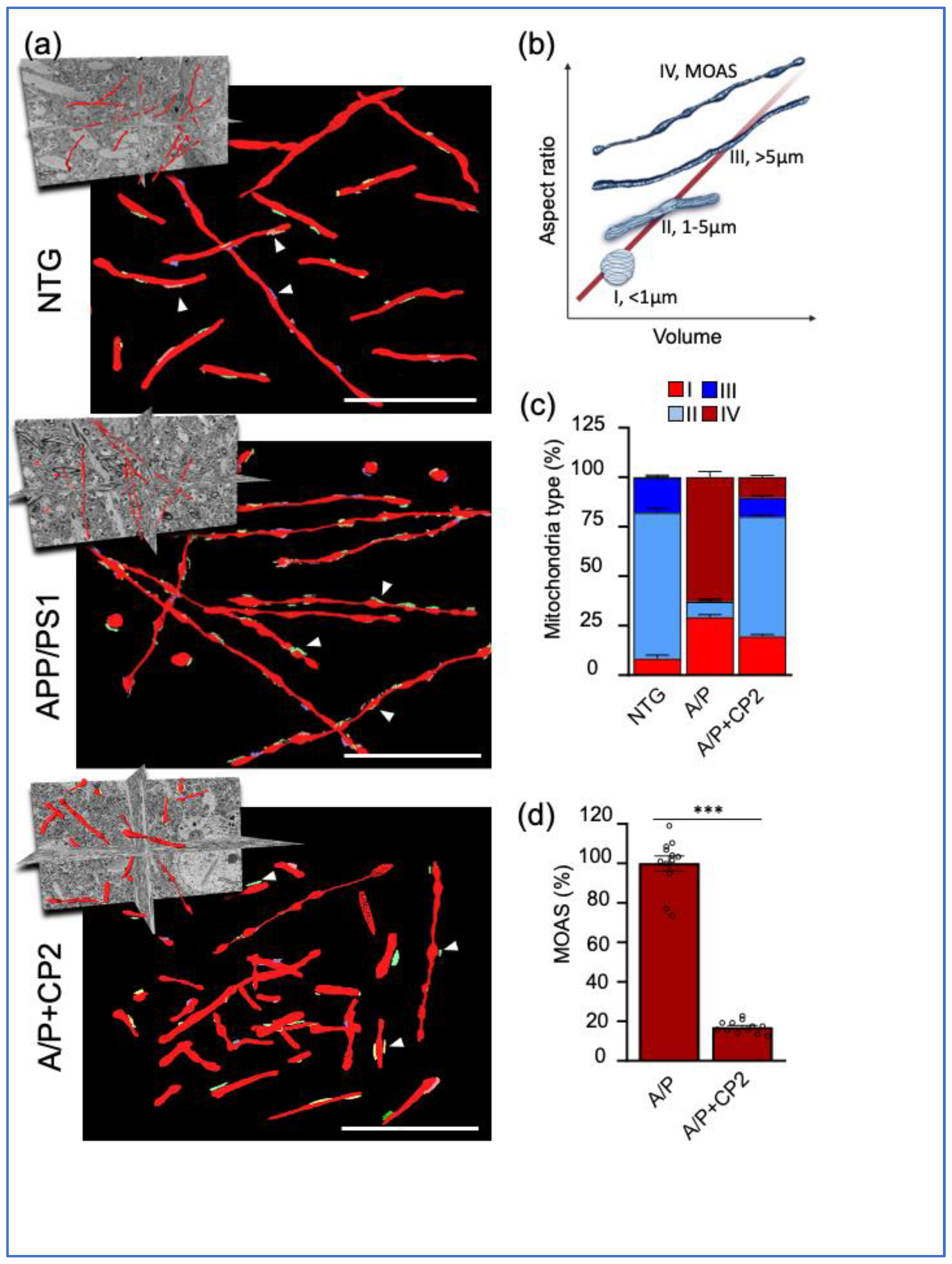
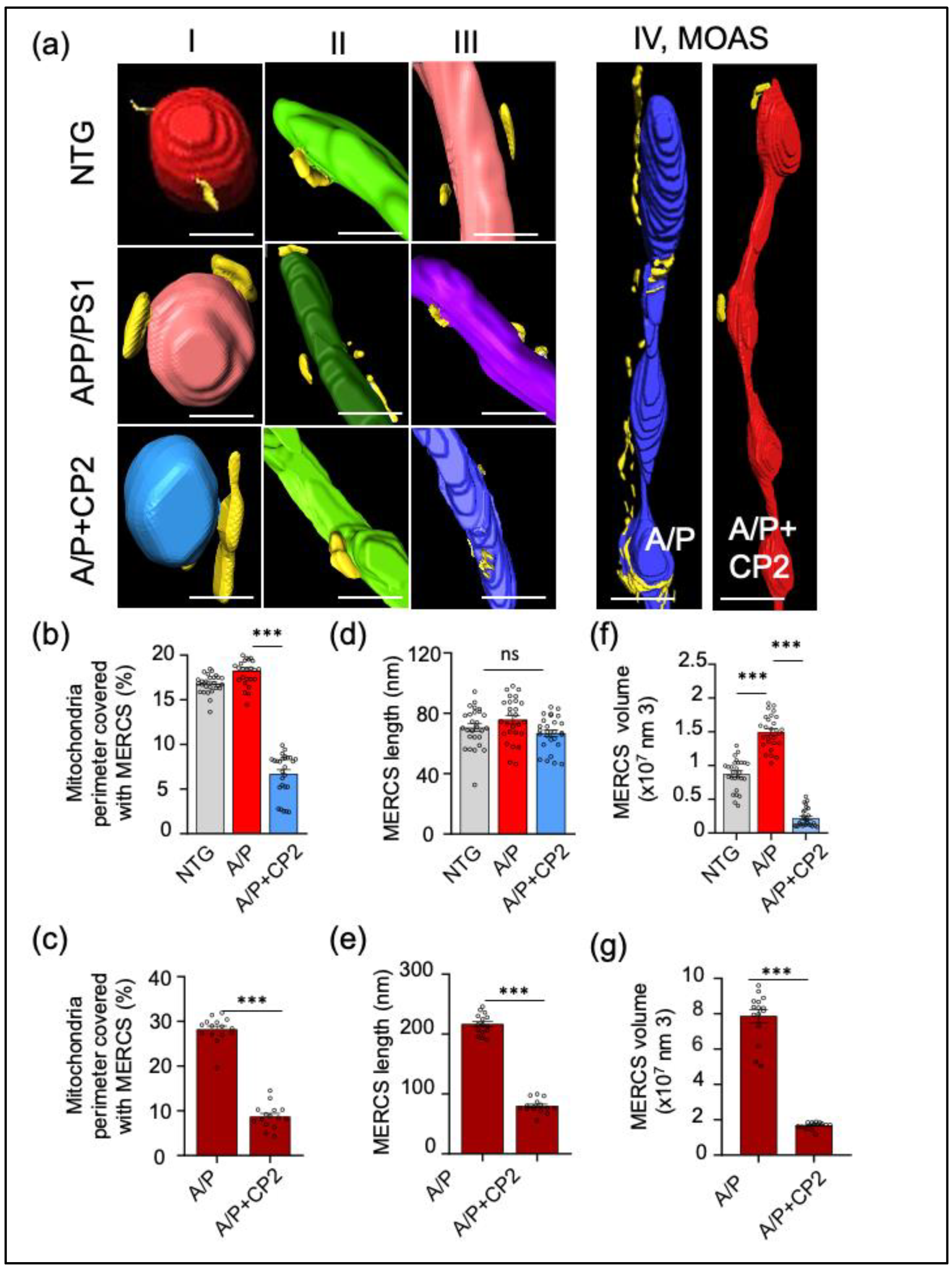
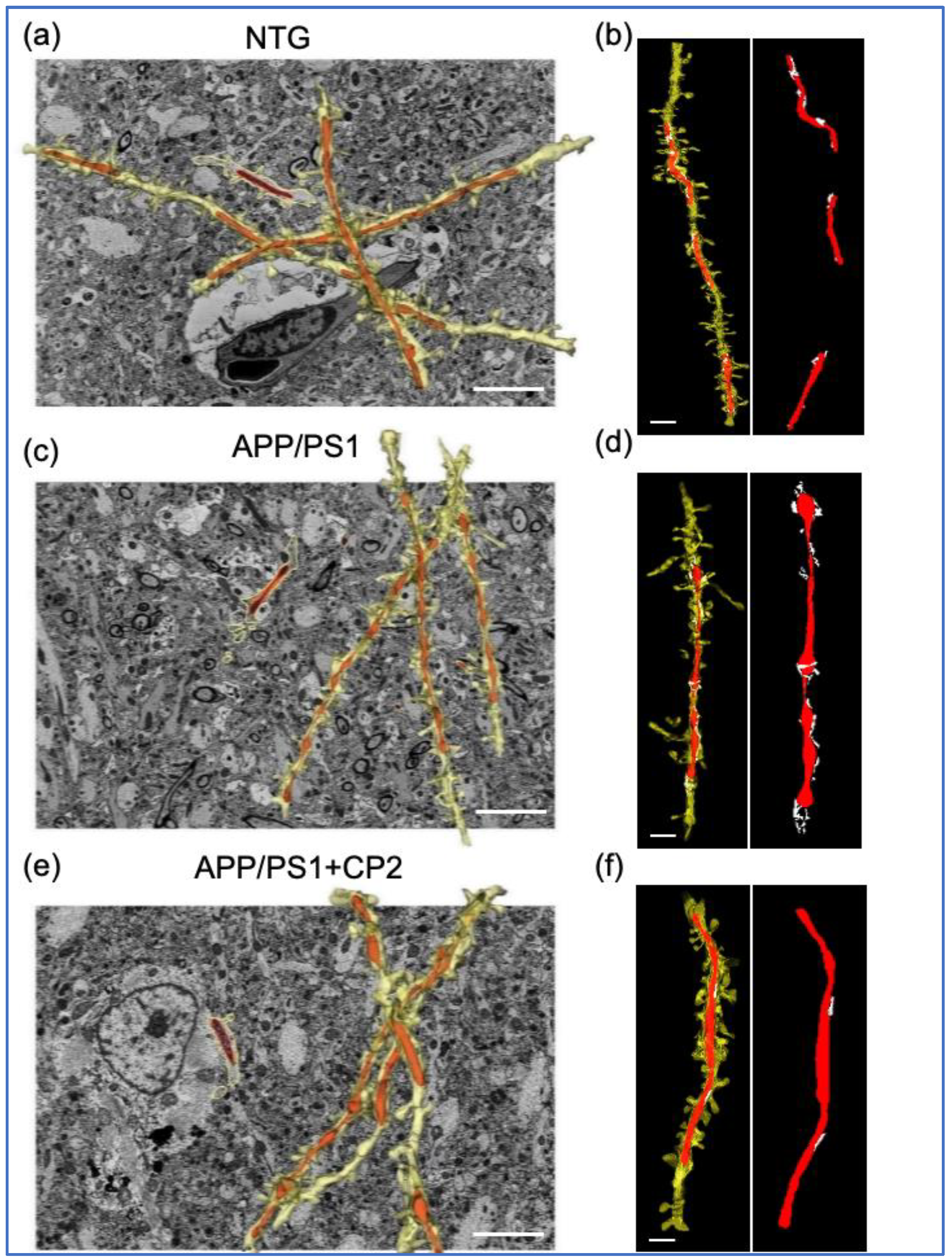
3.1. Evaluation of Mitochondrial Morphology and MERCS in Brain Tissue of APP/PS1 and NTG Mice Using SBFSEM and 3D EM Reconstruction
3.2. CP2 Treatment Restores Mitochondrial Morphology in Symptomatic APP/PS1 Mice
3.3. CP2 Treatment Reduces MERCS in APP/PS1 Mice
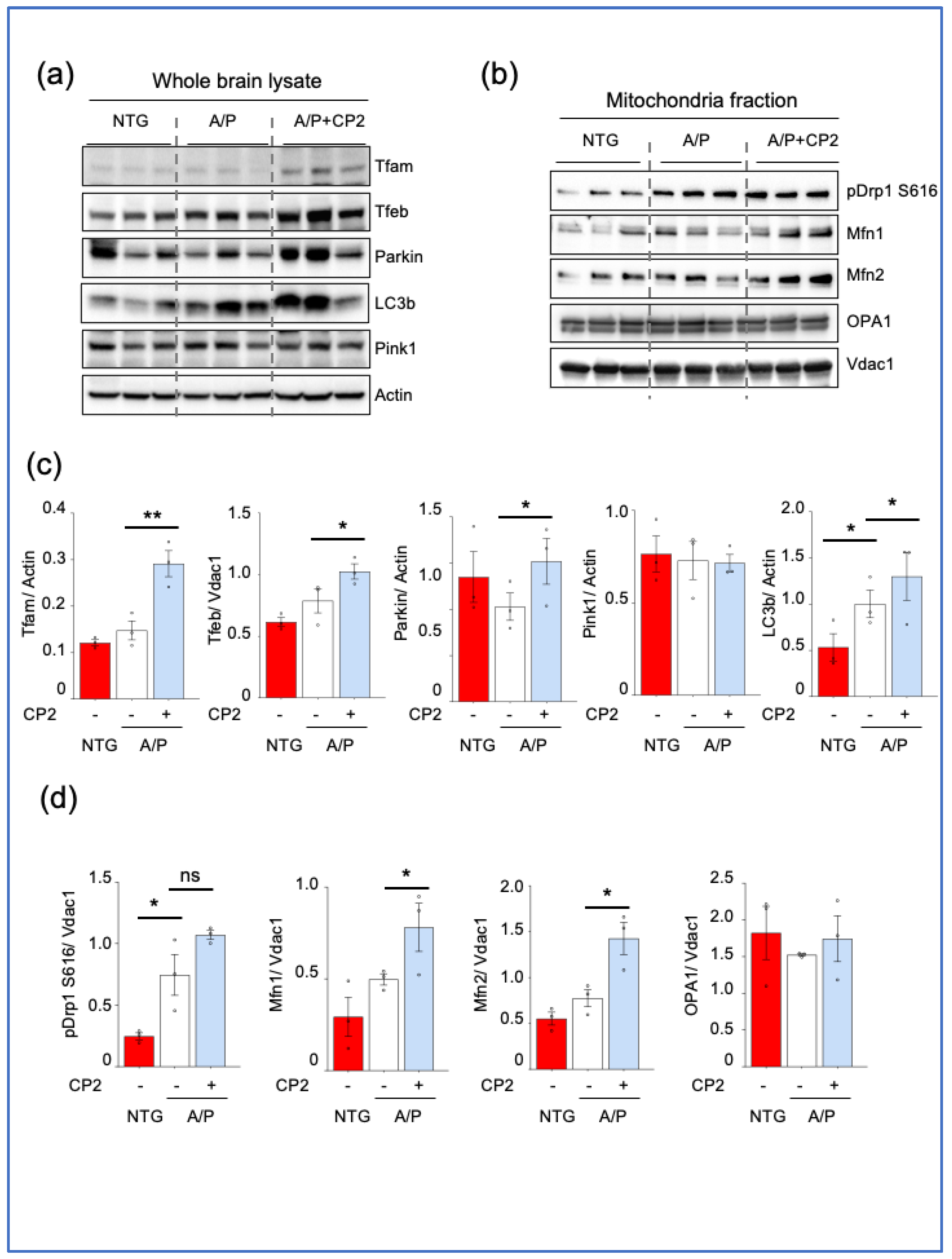
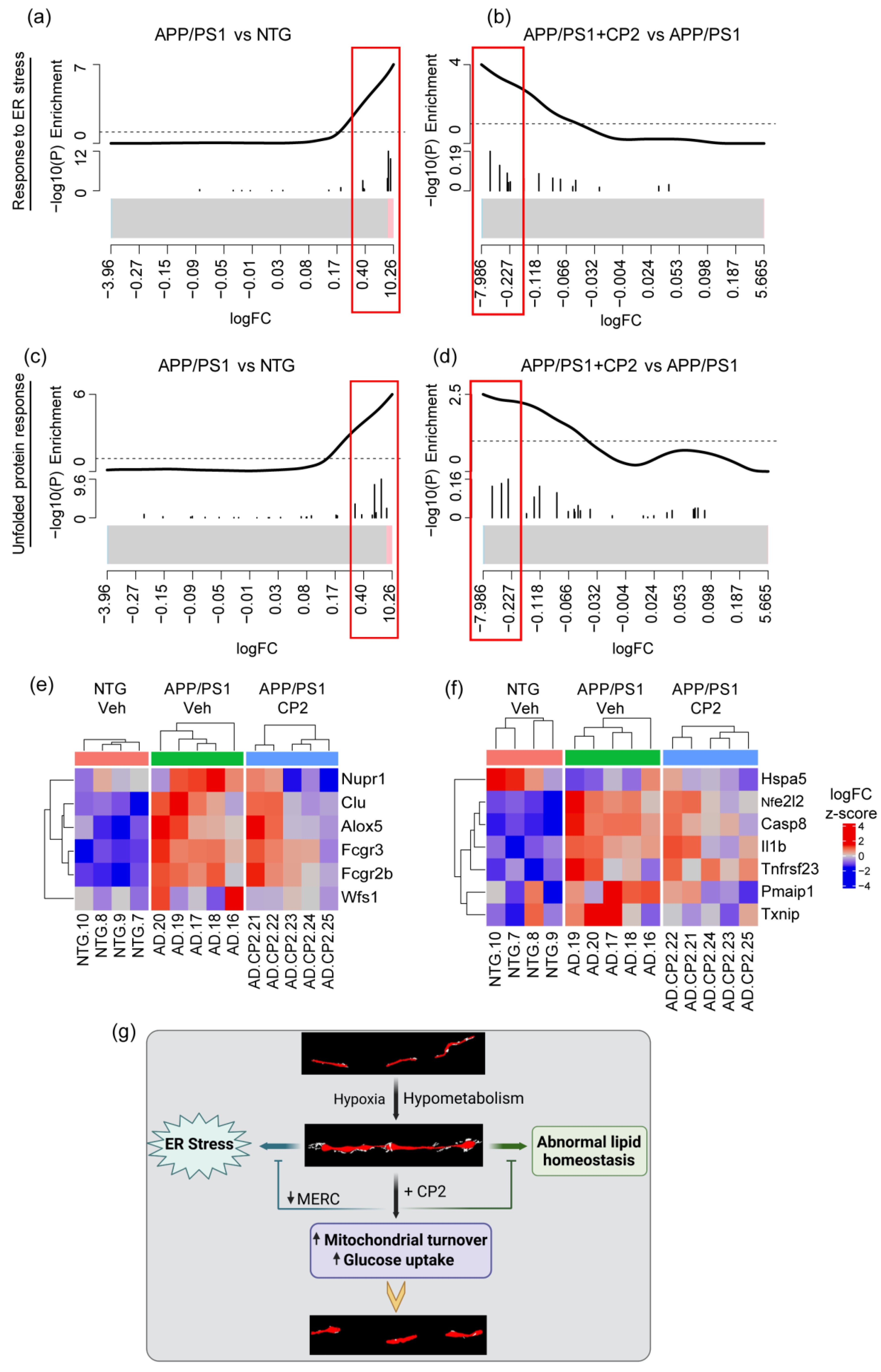
3.4. CP2 Treatment Augmented Mitochondrial Biogenesis and Turnover and Reduced ER Stress in APP/PS1 Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef]
- Mukherjee, S.; Heath, L.; Preuss, C.; Jayadev, S.; Garden, G.A.; Greenwood, A.K.; Sieberts, S.K.; De Jager, P.L.; Ertekin-Taner, N.; Carter, G.W.; et al. Molecular estimation of neurodegeneration pseudotime in older brains. Nat. Commun. 2020, 11, 5781. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Tian, Q.; Xu, Q.; Fu, Z.; Fu, Q. Mitochondrial dysfunction: A potential target for Alzheimer’s disease intervention and treatment. Drug Discov. Today 2021, 26, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 811–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial Dysfunction in Alzheimer’s Disease and Progress in Mitochondria-Targeted Therapeutics. Curr. Behav. Neurosci. Rep. 2019, 6, 88–102. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Carter, E.K.; Dammer, E.B.; Duong, D.M.; Gerasimov, E.S.; Liu, Y.; Liu, J.; Betarbet, R.; Ping, L.; Yin, L.; et al. Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 2022, 25, 213–225. [Google Scholar] [CrossRef]
- Torres, A.K.; Jara, C.; Park-Kang, H.S.; Polanco, C.M.; Tapia, D.; Alarcon, F.; de la Pena, A.; Llanquinao, J.; Vargas-Mardones, G.; Indo, J.A.; et al. Synaptic Mitochondria: An Early Target of Amyloid-beta and Tau in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 84, 1391–1414. [Google Scholar] [CrossRef]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020, 330, 113321. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and cell bioenergetics: Increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid. Redox Signal. 2012, 16, 1434–1455. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, A.P.; Weidling, I.; Ranjan, A.; Wang, X.; Novikova, L.; Chowdhury, S.R.; Menta, B.; Berkowicz, A.; Wilkins, H.M.; Peterson, K.R.; et al. Mitochondria Profoundly Influence Apolipoprotein E Biology. J. Alzheimers Dis. 2023, 92, 591–604. [Google Scholar] [CrossRef]
- Weidling, I.W.; Wilkins, H.M.; Koppel, S.J.; Hutfles, L.; Wang, X.; Kalani, A.; Menta, B.W.; Ryan, B.; Perez-Ortiz, J.; Gamblin, T.C.; et al. Mitochondrial DNA Manipulations Affect Tau Oligomerization. J. Alzheimers Dis. 2020, 77, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Strope, T.A.; Wilkins, H.M. Amyloid precursor protein and mitochondria. Curr. Opin. Neurobiol. 2023, 78, 102651. [Google Scholar] [CrossRef]
- Chandel, N.S. Evolution of Mitochondria as Signaling Organelles. Cell Metab. 2015, 22, 204–206. [Google Scholar] [CrossRef] [Green Version]
- Luan, Y.; Luan, Y.; Yuan, R.X.; Feng, Q.; Chen, X.; Yang, Y. Structure and Function of Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs) and Their Role in Cardiovascular Diseases. Oxid. Med. Cell Longev. 2021, 2021, 4578809. [Google Scholar] [CrossRef]
- Bernhard, W.; Rouiller, C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J. Biophys. Biochem. Cytol. 1956, 2, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Barazzuol, L.; Giamogante, F.; Cali, T. Mitochondria Associated Membranes (MAMs): Architecture and physiopathological role. Cell Calcium 2021, 94, 102343. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [Green Version]
- Zelnik, I.D.; Ventura, A.E.; Kim, J.L.; Silva, L.C.; Futerman, A.H. The role of ceramide in regulating endoplasmic reticulum function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158489. [Google Scholar] [CrossRef]
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J. Alzheimers Dis. 2012, 29, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Custodia, A.; Romaus-Sanjurjo, D.; Aramburu-Nunez, M.; Alvarez-Rafael, D.; Vazquez-Vazquez, L.; Camino-Castineiras, J.; Leira, Y.; Pias-Peleteiro, J.M.; Aldrey, J.M.; Sobrino, T.; et al. Ceramide/Sphingosine 1-Phosphate Axis as a Key Target for Diagnosis and Treatment in Alzheimer’s Disease and Other Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 8082. [Google Scholar] [CrossRef]
- Reddy, P.H.; Reddy, T.P.; Manczak, M.; Calkins, M.J.; Shirendeb, U.; Mao, P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 2011, 67, 103–118. [Google Scholar] [CrossRef] [Green Version]
- Trushina, E. A shape shifting organelle: Unusual mitochondrial phenotype determined with three-dimensional electron microscopy reconstruction. Neural Regen. Res. 2016, 11, 900–901. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in Mitochondrial Dynamics and Metabolomic Signatures of Evolving Energetic Stress in Mouse Models of Familial Alzheimer’s Disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 18725. [Google Scholar] [CrossRef] [Green Version]
- Morozov, Y.M.; Datta, D.; Paspalas, C.D.; Arnsten, A.F. Ultrastructural evidence for impaired mitochondrial fission in the aged rhesus monkey dorsolateral prefrontal cortex. Neurobiol. Aging 2017, 51, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavorato, M.; Loro, E.; Debattisti, V.; Khurana, T.S.; Franzini-Armstrong, C. Elongated mitochondrial constrictions and fission in muscle fatigue. J. Cell Sci. 2018, 131, jcs221028. [Google Scholar] [CrossRef] [Green Version]
- Kirov, S.A.; Fomitcheva, I.V.; Sword, J. Rapid Neuronal Ultrastructure Disruption and Recovery during Spreading Depolarization-Induced Cytotoxic Edema. Cereb. Cortex 2020, 30, 5517–5531. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, S.; Maezawa, I.; Trushin, S.; Minhas, P.; Pinto, M.; Jin, L.W.; Prasain, K.; Nguyen, T.D.; Yamazaki, Y.; et al. Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s Disease. EBioMedicine 2015, 2, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Stojakovic, A.; Trushin, S.; Sheu, A.; Khalili, L.; Chang, S.Y.; Li, X.; Christensen, T.; Salisbury, J.L.; Geroux, R.E.; Gateno, B.; et al. Partial inhibition of mitochondrial complex I ameliorates Alzheimer’s disease pathology and cognition in APP/PS1 female mice. Commun. Biol. 2021, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Maezawa, I.; Trushin, S.; Minhas, P.; Pinto, M.; Jin, L.W.; Prasain, K.; Nguyen, T.D.T.; Yamazaki, Y.; et al. Corrigendum to “Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s disease” [EBioMedicine 2 (2015) 294-305]. EBioMedicine 2019, 42, 532. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Tripathi, U.; Trushin, S.; Okromelidze, L.; Pichurin, N.P.; Wei, L.; Zhuang, Y.; Wang, L.; Trushina, E. A Genome-Wide Association Study in Human Lymphoblastoid Cells Supports Safety of Mitochondrial Complex I Inhibitor. Mitochondrion, 2021; in press. [Google Scholar] [CrossRef]
- Stojakovic, A.; Chang, S.Y.; Nesbitt, J.; Pichurin, N.P.; Ostroot, M.A.; Aikawa, T.; Kanekiyo, T.; Trushina, E. Partial Inhibition of Mitochondrial Complex I Reduces Tau Pathology and Improves Energy Homeostasis and Synaptic Function in 3xTg-AD Mice. J. Alzheimers Dis. 2021, 79, 335–353. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Drouin-Ouellet, J. Mitochondrial complex I deficiency and Parkinson disease. Nat. Rev. Neurosci. 2023, 24, 193. [Google Scholar] [CrossRef]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grunewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Park. Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef]
- Kim, H.W.; Choi, W.S.; Sorscher, N.; Park, H.J.; Tronche, F.; Palmiter, R.D.; Xia, Z. Genetic reduction of mitochondrial complex I function does not lead to loss of dopamine neurons in vivo. Neurobiol. Aging 2015, 36, 2617–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhubl, S.R. Why have antioxidants failed in clinical trials? Am. J. Cardiol. 2008, 101, 14D–19D. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Karamanlidis, G.; Tian, R. Novel targets for mitochondrial medicine. Sci. Transl. Med. 2016, 8, 326rv323. [Google Scholar] [CrossRef] [Green Version]
- Trushina, E.; Trushin, S.; Hasan, M.F. Mitochondrial complex I as a therapeutic target for Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 483–495. [Google Scholar] [CrossRef] [PubMed]
- de Cabo, R.; Mattson, M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, L. The effects of caloric restriction and its mimetics in Alzheimer’s disease through autophagy pathways. Food Funct. 2020, 11, 1211–1224. [Google Scholar] [CrossRef]
- Mottis, A.; Herzig, S.; Auwerx, J. Mitocellular communication: Shaping health and disease. Science 2019, 366, 827–832. [Google Scholar] [CrossRef]
- Meng, Q.; Lin, M.S.; Tzeng, I.S. Relationship Between Exercise and Alzheimer’s Disease: A Narrative Literature Review. Front. Neurosci. 2020, 14, 131. [Google Scholar] [CrossRef] [Green Version]
- Holcomb, L.; Gordon, M.N.; McGowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D.; et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef]
- Hinton, A., Jr.; Katti, P.; Christensen, T.A.; Mungai, M.; Shao, J.; Zhang, L.; Trushin, S.; Alghanem, A.; Jaspersen, A.; Geroux, R.E.; et al. A comprehensive approach to artifact-free sample preparation and the assessment of mitochondrial morphology in tissue and cultured cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Deerinck, T.J.; Bushong, E.A.; Ellisman, M.; Thor, A. Preparation of Biological Tissues for Serial Block Face Scanning Electron Microscopy (SBEM) v2. protocols.io; NCMIR; University of California: San Diego, CA, USA, 2022. [Google Scholar] [CrossRef]
- Fiala, J.C. Reconstruct: A free editor for serial section microscopy. J. Microsc. 2005, 218, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Garza-Lopez, E.; Vue, Z.; Katti, P.; Neikirk, K.; Biete, M.; Lam, J.; Beasley, H.K.; Marshall, A.G.; Rodman, T.A.; Christensen, T.A.; et al. Protocols for Generating Surfaces and Measuring 3D Organelle Morphology Using Amira. Cells 2021, 11, 65. [Google Scholar] [CrossRef]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef]
- Rao, Y.L.; Ganaraja, B.; Murlimanju, B.V.; Joy, T.; Krishnamurthy, A.; Agrawal, A. Hippocampus and its involvement in Alzheimer’s disease: A review. 3 Biotech 2022, 12, 55. [Google Scholar] [CrossRef]
- Faitg, J.; Lacefield, C.; Davey, T.; White, K.; Laws, R.; Kosmidis, S.; Reeve, A.K.; Kandel, E.R.; Vincent, A.E.; Picard, M. 3D neuronal mitochondrial morphology in axons, dendrites, and somata of the aging mouse hippocampus. Cell Rep. 2021, 36, 109509. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, B.; Hedskog, L.; Wiehager, B.; Ankarcrona, M. Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimers Dis. 2015, 43, 369–374. [Google Scholar] [CrossRef]
- Area-Gomez, E.; de Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 335. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Jin, H.; Huang, Y. Mitochondria-associated membranes (MAMs): A potential therapeutic target for treating Alzheimer’s disease. Clin. Sci. 2021, 135, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.K.; Georgescu, S.P.; Cano, C.; Aronovitz, M.J.; Iovanna, J.L.; Patten, R.D.; Kyriakis, J.M.; Goruppi, S. Deficiency of the transcriptional regulator p8 results in increased autophagy and apoptosis, and causes impaired heart function. Mol. Biol. Cell 2010, 21, 1335–1349. [Google Scholar] [CrossRef] [Green Version]
- Borrello, M.T.; Santofimia-Castano, P.; Bocchio, M.; Listi, A.; Fraunhoffer, N.; Soubeyran, P.; Chevet, E.; Pin, C.; Iovanna, J. NUPR1 interacts with eIF2alpha and is required for resolution of the ER stress response in pancreatic tissue. FEBS J. 2021, 288, 4081–4097. [Google Scholar] [CrossRef]
- Liu, J.; Song, X.; Kuang, F.; Zhang, Q.; Xie, Y.; Kang, R.; Kroemer, G.; Tang, D. NUPR1 is a critical repressor of ferroptosis. Nat. Commun. 2021, 12, 647. [Google Scholar] [CrossRef]
- Yang, L.; Nao, J. Ferroptosis: A potential therapeutic target for Alzheimer’s disease. Rev. Neurosci. 2022, online. [Google Scholar] [CrossRef]
- Poon, S.; Easterbrook-Smith, S.B.; Rybchyn, M.S.; Carver, J.A.; Wilson, M.R. Clusterin is an ATP-independent chaperone with very broad substrate specificity that stabilizes stressed proteins in a folding-competent state. Biochemistry 2000, 39, 15953–15960. [Google Scholar] [CrossRef] [PubMed]
- Hatters, D.M.; Wilson, M.R.; Easterbrook-Smith, S.B.; Howlett, G.J. Suppression of apolipoprotein C-II amyloid formation by the extracellular chaperone, clusterin. Eur. J. Biochem. 2002, 269, 2789–2794. [Google Scholar] [CrossRef] [PubMed]
- Zoubeidi, A.; Ettinger, S.; Beraldi, E.; Hadaschik, B.; Zardan, A.; Klomp, L.W.; Nelson, C.C.; Rennie, P.S.; Gleave, M.E. Clusterin facilitates COMMD1 and I-kappaB degradation to enhance NF-kappaB activity in prostate cancer cells. Mol. Cancer Res. 2010, 8, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Kim, J.K.; Edwards, C.A.; Xu, Z.; Taichman, R.; Wang, C.Y. Clusterin inhibits apoptosis by interacting with activated Bax. Nat. Cell Biol. 2005, 7, 909–915. [Google Scholar] [CrossRef]
- Li, N.; Zoubeidi, A.; Beraldi, E.; Gleave, M.E. GRP78 regulates clusterin stability, retrotranslocation and mitochondrial localization under ER stress in prostate cancer. Oncogene 2013, 32, 1933–1942. [Google Scholar] [CrossRef]
- Smyrniotis, C.J.; Barbour, S.R.; Xia, Z.; Hixon, M.S.; Holman, T.R. ATP allosterically activates the human 5-lipoxygenase molecular mechanism of arachidonic acid and 5(S)-hydroperoxy-6(E),8(Z),11(Z),14(Z)-eicosatetraenoic acid. Biochemistry 2014, 53, 4407–4419. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.F.; Pillai, P.S.; Recchiuti, A.; Yang, R.; Serhan, C.N. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Investig. 2011, 121, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Zhang, T.; Hwang, I.; Kim, A.; Nitschke, L.; Kim, M.; Scott, J.M.; Kamimura, Y.; Lanier, L.L.; Kim, S. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 2015, 42, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Baskin, J.G.; Mangan, E.K.; Su, K.; Gibson, A.W.; Ji, C.; Edberg, J.C.; Kimberly, R.P. The unique cytoplasmic domain of human FcgammaRIIIA regulates receptor-mediated function. J. Immunol. 2012, 189, 4284–4294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, D.; Ishihara, H.; Yamaguchi, S.; Yamada, T.; Tamura, A.; Katagiri, H.; Maruyama, Y.; Oka, Y. WFS1 protein modulates the free Ca(2+) concentration in the endoplasmic reticulum. FEBS Lett. 2006, 580, 5635–5640. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Liu, J.; Liu, D.; Zhou, Y.; Guo, Y.; Wang, Z.; Yang, S.; He, W.; Chen, P.; Wang, X.; et al. Glucose-regulated protein 78 modulates cell growth, epithelial-mesenchymal transition, and oxidative stress in the hyperplastic prostate. Cell Death Dis. 2022, 13, 78. [Google Scholar] [CrossRef]
- Chutkow, W.A.; Patwari, P.; Yoshioka, J.; Lee, R.T. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J. Biol. Chem. 2008, 283, 2397–2406. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.H.; Han, Q.X.; Ding, X.N.; Yan, J.Y.; Li, Q.; Zhang, D.; Zhu, H.Y. Critical hubs of renal ischemia-reperfusion injury: Endoplasmic reticulum-mitochondria tethering complexes. Chin. Med. J. 2020, 133, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Bogar, F.; Fulop, L.; Penke, B. Novel Therapeutic Target for Prevention of Neurodegenerative Diseases: Modulation of Neuroinflammation with Sig-1R Ligands. Biomolecules 2022, 12, 363. [Google Scholar] [CrossRef]
- Ismael, S.; Wajidunnisa; Sakata, K.; McDonald, M.P.; Liao, F.F.; Ishrat, T. ER stress associated TXNIP-NLRP3 inflammasome activation in hippocampus of human Alzheimer’s disease. Neurochem. Int. 2021, 148, 105104. [Google Scholar] [CrossRef] [PubMed]
- Kamboh, M.I.; Demirci, F.Y.; Wang, X.; Minster, R.L.; Carrasquillo, M.M.; Pankratz, V.S.; Younkin, S.G.; Saykin, A.J.; for the Alzheimer’s Disease Neuroimaging Initiative; Jun, G.; et al. Genome-wide association study of Alzheimer’s disease. Transl. Psychiatry 2012, 2, e117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523.e1515. [Google Scholar] [CrossRef]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Gleave, J.A.; Arathoon, L.R.; Trinh, D.; Lizal, K.E.; Giguere, N.; Barber, J.H.M.; Najarali, Z.; Khan, M.H.; Thiele, S.L.; Semmen, M.S.; et al. Sirtuin 3 rescues neurons through the stabilisation of mitochondrial biogenetics in the virally-expressing mutant alpha-synuclein rat model of parkinsonism. Neurobiol. Dis. 2017, 106, 133–146. [Google Scholar] [CrossRef]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M.; et al. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Kislin, M.; Sword, J.; Fomitcheva, I.V.; Croom, D.; Pryazhnikov, E.; Lihavainen, E.; Toptunov, D.; Rauvala, H.; Ribeiro, A.S.; Khiroug, L.; et al. Reversible Disruption of Neuronal Mitochondria by Ischemic and Traumatic Injury Revealed by Quantitative Two-Photon Imaging in the Neocortex of Anesthetized Mice. J. Neurosci. 2017, 37, 333–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, D.V.; Martin, N.; Bernard, D. Cellular senescence links mitochondria-ER contacts and aging. Commun. Biol. 2021, 4, 1323. [Google Scholar] [CrossRef]
- Baumgart, M.; Priebe, S.; Groth, M.; Hartmann, N.; Menzel, U.; Pandolfini, L.; Koch, P.; Felder, M.; Ristow, M.; Englert, C.; et al. Longitudinal RNA-Seq Analysis of Vertebrate Aging Identifies Mitochondrial Complex I as a Small-Molecule-Sensitive Modifier of Lifespan. Cell Syst. 2016, 2, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Madrigal-Perez, L.A.; Ramos-Gomez, M. Resveratrol Inhibition of Cellular Respiration: New Paradigm for an Old Mechanism. Int. J. Mol. Sci. 2016, 17, 368. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Xie, F.; Xiao, Y.; Lu, C.; Zhong, J.; Huang, D.; Chen, J.; Wei, J.; Jiang, Y.; Zhong, T. Metformin: A Potential Candidate for Targeting Aging Mechanisms. Aging Dis. 2021, 12, 480–493. [Google Scholar] [CrossRef]
- Porquet, D.; Casadesus, G.; Bayod, S.; Vicente, A.; Canudas, A.M.; Vilaplana, J.; Pelegri, C.; Sanfeliu, C.; Camins, A.; Pallas, M.; et al. Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age 2013, 35, 1851–1865. [Google Scholar] [CrossRef] [Green Version]
- Morgado-Caceres, P.; Liabeuf, G.; Calle, X.; Briones, L.; Riquelme, J.A.; Bravo-Sagua, R.; Parra, V. The aging of ER-mitochondria communication: A journey from undifferentiated to aged cells. Front. Cell Dev. Biol. 2022, 10, 946678. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Lindholm, D.; Ren, J.; Pratico, D. ER stress and UPR in Alzheimer’s disease: Mechanisms, pathogenesis, treatments. Cell Death Dis. 2022, 13, 706. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinangeli, C.; Didier, S.; Ahmed, T.; Caillerez, R.; Domise, M.; Laloux, C.; Begard, S.; Carrier, S.; Colin, M.; Marchetti, P.; et al. AMP-Activated Protein Kinase Is Essential for the Maintenance of Energy Levels during Synaptic Activation. iScience 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vingtdeux, V.; Chandakkar, P.; Zhao, H.; d’Abramo, C.; Davies, P.; Marambaud, P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J. 2011, 25, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Saito, M.; Das, B.C. Involvement of AMP-activated protein kinase in neuroinflammation and neurodegeneration in the adult and developing brain. Int. J. Dev. Neurosci. 2019, 77, 48–59. [Google Scholar] [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12, 585. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, H.; Zhang, L.; Lin, X.; Li, X.; Zhuang, H.; Fan, H.; Meng, T.; He, Z.; Huang, H.; et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 2021, 17, 1142–1156. [Google Scholar] [CrossRef]
- Assefa, B.T.; Tafere, G.G.; Wondafrash, D.Z.; Gidey, M.T. The Bewildering Effect of AMPK Activators in Alzheimer’s Disease: Review of the Current Evidence. Biomed. Res. Int. 2020, 2020, 9895121. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panes, J.; Nguyen, T.K.O.; Gao, H.; Christensen, T.A.; Stojakovic, A.; Trushin, S.; Salisbury, J.L.; Fuentealba, J.; Trushina, E. Partial Inhibition of Complex I Restores Mitochondrial Morphology and Mitochondria-ER Communication in Hippocampus of APP/PS1 Mice. Cells 2023, 12, 1111. https://doi.org/10.3390/cells12081111
Panes J, Nguyen TKO, Gao H, Christensen TA, Stojakovic A, Trushin S, Salisbury JL, Fuentealba J, Trushina E. Partial Inhibition of Complex I Restores Mitochondrial Morphology and Mitochondria-ER Communication in Hippocampus of APP/PS1 Mice. Cells. 2023; 12(8):1111. https://doi.org/10.3390/cells12081111
Chicago/Turabian StylePanes, Jessica, Thi Kim Oanh Nguyen, Huanyao Gao, Trace A. Christensen, Andrea Stojakovic, Sergey Trushin, Jeffrey L. Salisbury, Jorge Fuentealba, and Eugenia Trushina. 2023. "Partial Inhibition of Complex I Restores Mitochondrial Morphology and Mitochondria-ER Communication in Hippocampus of APP/PS1 Mice" Cells 12, no. 8: 1111. https://doi.org/10.3390/cells12081111
APA StylePanes, J., Nguyen, T. K. O., Gao, H., Christensen, T. A., Stojakovic, A., Trushin, S., Salisbury, J. L., Fuentealba, J., & Trushina, E. (2023). Partial Inhibition of Complex I Restores Mitochondrial Morphology and Mitochondria-ER Communication in Hippocampus of APP/PS1 Mice. Cells, 12(8), 1111. https://doi.org/10.3390/cells12081111