
BACH2: The Future of Induced T-Regulatory Cell Therapies

Abstract
:1. Introduction
2. BACH2 Structure
3. BACH2 Cell Type-Specific Roles
3.1. BACH2 in T-Cell Differentiation
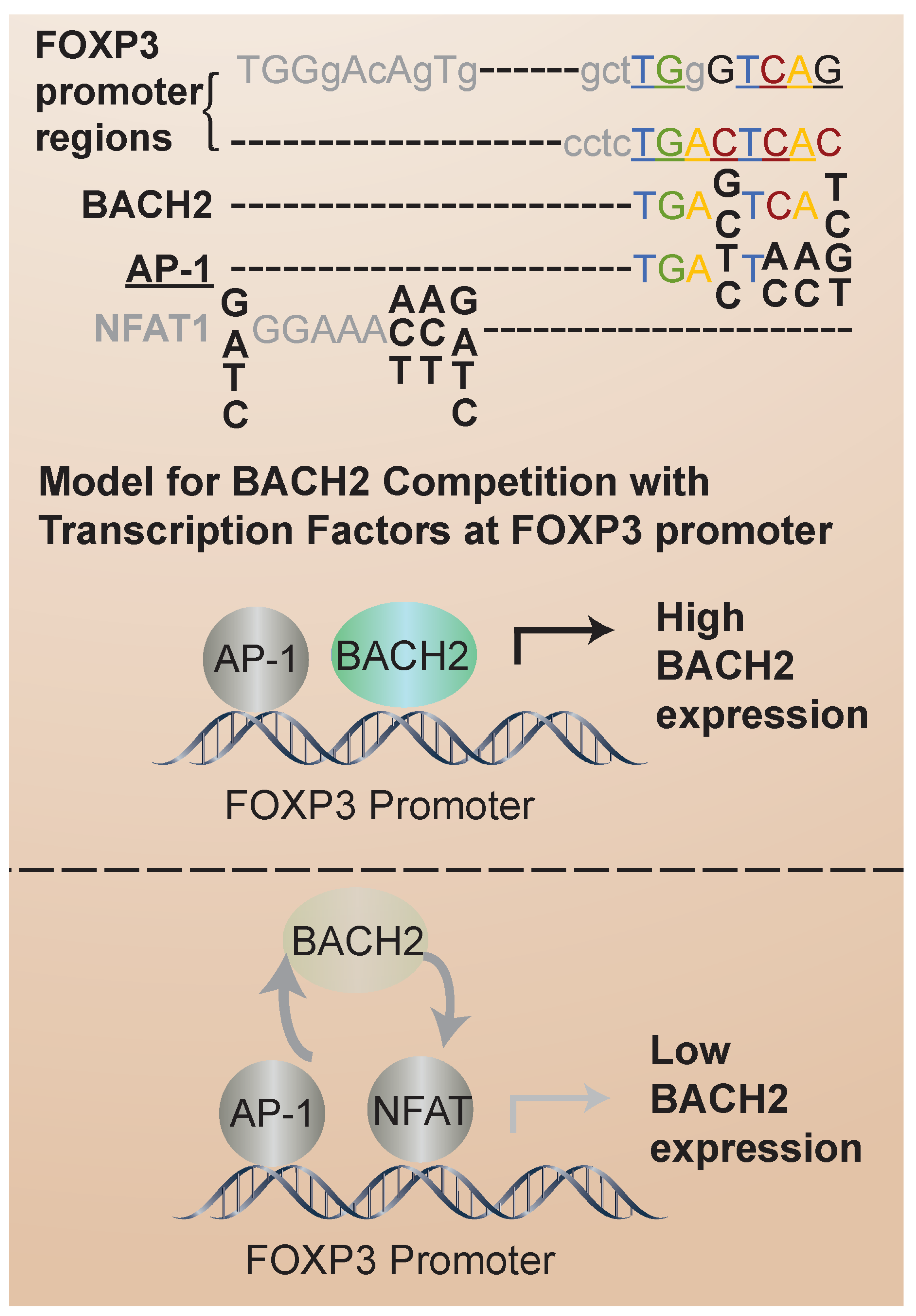
3.2. BACH2 in iTregs: Regulation of FOXP3 Expression and iTreg Stability in Humans
3.3. BACH2 in T-Cell Exhaustion
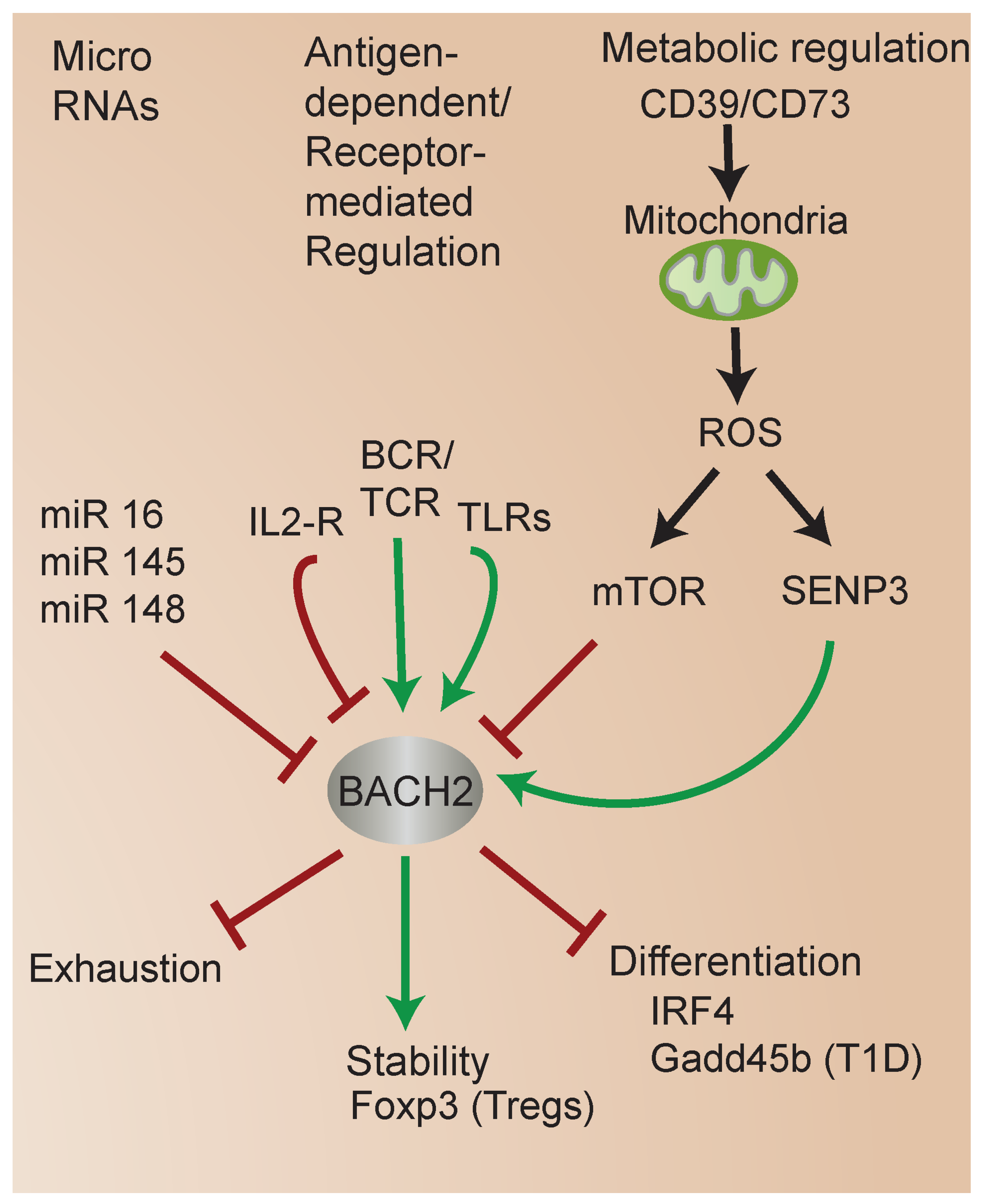
4. Molecular Mechanisms That Regulate BACH2
4.1. BACH2 Regulation by MicroRNAs (miRs)
4.2. Metabolic Regulation of BACH2
4.3. Regulation of BACH2 by SUMOylation and Role in iTreg Stability
5. BACH2-Associated Diseases
5.1. BACH2 in Murine T Cell Inflammatory Disease Models and Autoimmunity
5.2. BACH2 in Human Autoimmune Conditions
5.3. BACH2 in Murine Tumor Models and Human Cancers
6. Prospects for Stabilizing iTreg Phenotype and Function via Targeting BACH2
6.1. Approaches to Modulating iTreg Stability
6.2. Identifying Targets for Pharmacological Modulation of BACH2 Function
7. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muto, A.; Tashiro, S.; Nakajima, O.; Hoshino, H.; Takahashi, S.; Sakoda, E.; Ikebe, D.; Yamamoto, M.; Igarashi, K. The transcriptional programme of antibody class switching involves the repressor Bach2. Nature 2004, 429, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Kometani, K.; Nakagawa, R.; Shinnakasu, R.; Kaji, T.; Rybouchkin, A.; Moriyama, S.; Furukawa, K.; Koseki, H.; Takemori, T.; Kurosaki, T. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity 2013, 39, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhuri, R.; Clever, D.; Li, P.; Wakabayashi, Y.; Quinn, K.M.; Klebanoff, C.A.; Ji, Y.; Sukumar, M.; Eil, R.L.; Yu, Z.; et al. BACH2 regulates CD8+ T cell differentiation by controlling access of AP-1 factors to enhancers. Nat. Immunol. 2016, 17, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Richer, M.J.; Lang, M.L.; Butler, N.S. T Cell Fates Zipped Up: How the Bach2 Basic Leucine Zipper Transcriptional Repressor Directs T Cell Differentiation and Function. J. Immunol. 2016, 197, 1009–1015. [Google Scholar] [CrossRef]
- Roychoudhuri, R.; Hirahara, K.; Mousavi, K.; Clever, D.; Klebanoff, C.A.; Bonelli, M.; Sciume, G.; Zare, H.; Vahedi, G.; Dema, B.; et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature 2013, 498, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Tsukumo, S.; Unno, M.; Muto, A.; Takeuchi, A.; Kometani, K.; Kurosaki, T.; Igarashi, K.; Saito, T. Bach2 maintains T cells in a naive state by suppressing effector memory-related genes. Proc. Natl. Acad. Sci. USA 2013, 110, 10735–10740. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H. Regulatory T cells: Friend or foe in immunity to infection? Nat. Rev. Immunol. 2004, 4, 841–855. [Google Scholar] [CrossRef]
- Lourenco, E.V.; La Cava, A. Natural regulatory T cells in autoimmunity. Autoimmunity 2011, 44, 33–42. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Davidson, T.S.; DiPaolo, R.J.; Andersson, J.; Shevach, E.M. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J. Immunol. 2007, 178, 4022–4026. [Google Scholar] [CrossRef]
- Schmitt, E.G.; Williams, C.B. Generation and function of induced regulatory T cells. Front. Immunol. 2013, 4, 152. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, S.; Zhao, Q.; Sun, Y.; Nie, H. The Critical Role of Bach2 in Shaping the Balance between CD4+ T Cell Subsets in Immune-Mediated Diseases. Mediat. Inflamm. 2019, 2019, 2609737. [Google Scholar] [CrossRef] [PubMed]
- Trujillo-Ochoa, J.L.; Kazemian, M.; Afzali, B. The role of transcription factors in shaping regulatory T cell identity. Nat. Rev. Immunol. 2023, 23, 842–856. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, H.; Zhao, M.; Chang, C.; Lu, Q. The Bach Family of Transcription Factors: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 50, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Klasic, M.; Markulin, D.; Vojta, A.; Samarzija, I.; Birus, I.; Dobrinic, P.; Ventham, N.T.; Trbojevic-Akmacic, I.; Simurina, M.; Stambuk, J.; et al. Promoter methylation of the MGAT3 and BACH2 genes correlates with the composition of the immunoglobulin G glycome in inflammatory bowel disease. Clin. Epigenetics 2018, 10, 75. [Google Scholar] [CrossRef] [PubMed]
- Laffin, M.R.; Fedorak, R.N.; Wine, E.; Dicken, B.; Madsen, K.L. A BACH2 Gene Variant Is Associated with Postoperative Recurrence of Crohn's Disease. J. Am. Coll. Surg. 2018, 226, 902–908. [Google Scholar] [CrossRef]
- Cushing, K.C.; Du, X.; Chen, Y.; Stetson, L.C.; Kuppa, A.; Chen, V.L.; Kahlenberg, J.M.; Gudjonsson, J.E.; Vanderwerff, B.; Higgins, P.D.R.; et al. Inflammatory Bowel Disease Risk Variants Are Associated with an Increased Risk of Skin Cancer. Inflamm. Bowel Dis. 2022, 28, 1667–1676. [Google Scholar] [CrossRef]
- Sasikala, M.; Ravikanth, V.V.; Murali Manohar, K.; Deshpande, N.; Singh, S.; Pavan Kumar, P.; Talukdar, R.; Ghosh, S.; Aslam, M.; Rao, G.V.; et al. Bach2 repression mediates Th17 cell induced inflammation and associates with clinical features of advanced disease in chronic pancreatitis. United Eur. Gastroenterol. J. 2018, 6, 272–282. [Google Scholar] [CrossRef]
- Morris, D.L.; Sheng, Y.; Zhang, Y.; Wang, Y.F.; Zhu, Z.; Tombleson, P.; Chen, L.; Cunninghame Graham, D.S.; Bentham, J.; Roberts, A.L.; et al. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat. Genet. 2016, 48, 940–946. [Google Scholar] [CrossRef]
- Afzali, B.; Gronholm, J.; Vandrovcova, J.; O'Brien, C.; Sun, H.W.; Vanderleyden, I.; Davis, F.P.; Khoder, A.; Zhang, Y.; Hegazy, A.N.; et al. BACH2 immunodeficiency illustrates an association between super-enhancers and haploinsufficiency. Nat. Immunol. 2017, 18, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Sun, G.; Chen, R.; Chen, J.; Fang, S.; Xu, Q.; Tang, W.; Dai, R.; Zhang, Z.; An, Y.; et al. An early-onset SLE patient with a novel paternal inherited BACH2 mutation. J. Clin. Immunol. 2023, 43, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Zhou, X.; Gu, Y.; Han, Q.; Li, J.; Chen, B.; Ge, Q.; Dovat, E.; Payne, J.L.; Sun, T.; et al. Ikaros regulation of the BCL6/BACH2 axis and its clinical relevance in acute lymphoblastic leukemia. Oncotarget 2017, 8, 8022–8034. [Google Scholar] [CrossRef] [PubMed]
- Ciardullo, C.; Szoltysek, K.; Zhou, P.; Pietrowska, M.; Marczak, L.; Willmore, E.; Enshaei, A.; Walaszczyk, A.; Ho, J.Y.; Rand, V.; et al. Low BACH2 Expression Predicts Adverse Outcome in Chronic Lymphocytic Leukaemia. Cancers 2021, 14, 23. [Google Scholar] [CrossRef]
- Ferreira, M.A.; Matheson, M.C.; Duffy, D.L.; Marks, G.B.; Hui, J.; Le Souëf, P.; Danoy, P.; Baltic, S.; Nyholt, D.R.; Jenkins, M.; et al. Identification of IL6R and chromosome 11q13.5 as risk loci for asthma. Lancet 2011, 378, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.D.; Smyth, D.J.; Smiles, A.M.; Plagnol, V.; Walker, N.M.; Allen, J.E.; Downes, K.; Barrett, J.C.; Healy, B.C.; Mychaleckyj, J.C.; et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat. Genet. 2008, 40, 1399–1401. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Santin, I.; Dos Santos, R.S.; Marselli, L.; Marchetti, P.; Eizirik, D.L. BACH2, a candidate risk gene for type 1 diabetes, regulates apoptosis in pancreatic beta-cells via JNK1 modulation and crosstalk with the candidate gene PTPN2. Diabetes 2014, 63, 2516–2527. [Google Scholar] [CrossRef]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Dubois, P.C.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.; Adany, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef]
- Jin, Y.; Birlea, S.A.; Fain, P.R.; Ferrara, T.M.; Ben, S.; Riccardi, S.L.; Cole, J.B.; Gowan, K.; Holland, P.J.; Bennett, D.C.; et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 2012, 44, 676–680. [Google Scholar] [CrossRef]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Ochiai, K.; Itoh-Nakadai, A.; Muto, A. Orchestration of plasma cell differentiation by Bach2 and its gene regulatory network. Immunol. Rev. 2014, 261, 116–125. [Google Scholar] [CrossRef] [PubMed]
- McAllister, K.; Yarwood, A.; Bowes, J.; Orozco, G.; Viatte, S.; Diogo, D.; Hocking, L.J.; Steer, S.; Wordsworth, P.; Wilson, A.G.; et al. Identification of BACH2 and RAD51B as rheumatoid arthritis susceptibility loci in a meta-analysis of genome-wide data. Arthritis Rheumatol. 2013, 65, 3058–3062. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Røyrvik, E.C. GWAS for autoimmune Addison's disease identifies multiple risk loci and highlights AIRE in disease susceptibility. Nat. Commun. 2021, 12, 959. [Google Scholar] [CrossRef] [PubMed]
- Pazderska, A.; Oftedal, B.E.; Napier, C.M.; Ainsworth, H.F.; Husebye, E.S.; Cordell, H.J.; Pearce, S.H.; Mitchell, A.L. A Variant in the BACH2 Gene Is Associated with Susceptibility to Autoimmune Addison’s Disease in Humans. J. Clin. Endocrinol. Metab. 2016, 101, 3865–3869. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Ochiai, K.; Muto, A. Architecture and dynamics of the transcription factor network that regulates B-to-plasma cell differentiation. J. Biochem. 2007, 141, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Ando, R.; Shima, H.; Tamahara, T.; Sato, Y.; Watanabe-Matsui, M.; Kato, H.; Sax, N.; Motohashi, H.; Taguchi, K.; Yamamoto, M.; et al. The Transcription Factor Bach2 Is Phosphorylated at Multiple Sites in Murine B Cells but a Single Site Prevents Its Nuclear Localization. J. Biol. Chem. 2016, 291, 1826–1840. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.R.; Xia, X.; Nguyen, K.; Zhu, F.; Geng, J.; Sialer, D.O.; Hu, H. Bach2 Integrates Cytokine Signals to Arbitrate Differentiation Decisions between T Follicular Helper and Th17 Lineages. J. Immunol. 2023, 211, 1756–1761. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Lou, G.; Sun, H.-W.; Zhu, Z.; Sun, Y.; Chen, Z.; Chauss, D.; Moseman, E.A.; Cheng, J.; D’Antonio, M.A.; et al. BACH2 enforces the transcriptional and epigenetic programs of stem-like CD8+ T cells. Nat. Immunol. 2021, 22, 370–380. [Google Scholar] [CrossRef]
- Russ, B.E.; Barugahare, A.; Dakle, P.; Tsyganov, K.; Quon, S.; Yu, B.; Li, J.; Lee, J.K.C.; Olshansky, M.; He, Z.; et al. Active maintenance of CD8+ T cell naivety through regulation of global genome architecture. Cell Rep. 2023, 42, 113301. [Google Scholar] [CrossRef]
- Sidwell, T.; Liao, Y.; Garnham, A.L.; Vasanthakumar, A.; Gloury, R.; Blume, J.; Teh, P.P.; Chisanga, D.; Thelemann, C.; de Labastida Rivera, F.; et al. Attenuation of TCR-induced transcription by Bach2 controls regulatory T cell differentiation and homeostasis. Nat. Commun. 2020, 11, 252. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Gasper, D.J.; Lee, S.H.; Plisch, E.H.; Svaren, J.; Suresh, M. Bach2 regulates homeostasis of Foxp3+ regulatory T cells and protects against fatal lung disease in mice. J. Immunol. 2014, 192, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, Y.; Kiyokawa, N.; Ochiai, N.; Imadome, K.; Horiuchi, Y.; Onda, K.; Yajima, M.; Nakamura, H.; Katagiri, Y.U.; Okita, H.; et al. Ex vivo expanded cord blood CD4 T lymphocytes exhibit a distinct expression profile of cytokine-related genes from those of peripheral blood origin. Immunology 2009, 128, 405–419. [Google Scholar] [CrossRef]
- Lesniewski, M.L.; Haviernik, P.; Weitzel, R.P.; Kadereit, S.; Kozik, M.M.; Fanning, L.R.; Yang, Y.C.; Hegerfeldt, Y.; Finney, M.R.; Ratajczak, M.Z.; et al. Regulation of IL-2 expression by transcription factor BACH2 in umbilical cord blood CD4+ T cells. Leukemia 2008, 22, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dai, D.; Hu, Q.; Yang, F.; Xue, Y.; Li, F.; Shen, N.; Zhang, M.; Huang, C. Bach2 attenuates IL-2R signaling to control Treg homeostasis and Tfr development. Cell Rep. 2021, 35, 109096. [Google Scholar] [CrossRef]
- Zheng, Y.; Lu, Y.; Huang, X.; Han, L.; Chen, Z.; Zhou, B.; Ma, Y.; Xie, G.; Yang, J.; Bian, B.; et al. BACH2 regulates the function of human CD4+ CD45RA(-) Foxp3(l) degrees cytokine-secreting T cells and promotes B-cell response in systemic lupus erythematosus. Eur. J. Immunol. 2020, 50, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Do, J.S.; Zhong, F.; Huang, A.Y.; Van’t Hof, W.J.; Finney, M.; Laughlin, M.J. Foxp3 expression in induced T regulatory cells derived from human umbilical cord blood vs. adult peripheral blood. Bone Marrow Transplant. 2018, 53, 1568–1577. [Google Scholar] [CrossRef]
- Hogan, P.G. Calcium-NFAT transcriptional signalling in T cell activation and T cell exhaustion. Cell Calcium 2017, 63, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Borde, M.; Heissmeyer, V.; Feuerer, M.; Lapan, A.D.; Stroud, J.C.; Bates, D.L.; Guo, L.; Han, A.; Ziegler, S.F.; et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 2006, 126, 375–387. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers 2019, 11, 1037. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.; Lee, H.R.; Lee, G.H.; Oh, A.R.; Cha, J.Y.; Igarashi, K.; Youn, J. Bach2 represses the AP-1-driven induction of interleukin-2 gene transcription in CD4 T cells. BMB Rep. 2017, 50, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zeng, X.; Zha, X.; Lai, J.; Tan, G.; Chen, S.; Yu, X.; Li, Y.; Xu, L. Correlation of the transcription factors IRF4 and BACH2 with the abnormal NFATC1 expression in T cells from chronic myeloid leukemia patients. Hematology 2022, 27, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, D.T.; Gabriel, S.S.; Chisanga, D.; Gloury, R.; Gubser, P.M.; Vasanthakumar, A.; Shi, W.; Kallies, A. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol. 2020, 21, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Grant, F.M.; Yang, J.; Nasrallah, R.; Clarke, J.; Sadiyah, F.; Whiteside, S.K.; Imianowski, C.J.; Kuo, P.; Vardaka, P.; Todorov, T.; et al. BACH2 drives quiescence and maintenance of resting Treg cells to promote homeostasis and cancer immunosuppression. J. Exp. Med. 2020, 217, e20190711. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, R.P.; Lesniewski, M.L.; Greco, N.J.; Laughlin, M.J. Reduced methyl-CpG protein binding contributing to miR-184 expression in umbilical cord blood CD4+ T-cells. Leukemia 2011, 25, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, R.P.; Lesniewski, M.L.; Haviernik, P.; Kadereit, S.; Leahy, P.; Greco, N.J.; Laughlin, M.J. microRNA 184 regulates expression of NFAT1 in umbilical cord blood CD4+ T cells. Blood 2009, 113, 6648–6657. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Song, Z.; Dong, J.; Shu, R. microRNA-142 is upregulated by tumor necrosis factor-alpha and triggers apoptosis in human gingival epithelial cells by repressing BACH2 expression. Am. J. Transl. Res. 2017, 9, 175–183. [Google Scholar]
- Liu, X.; Su, K.; Kuang, S.; Fu, M.; Zhang, Z. miR-16-5p and miR-145-5p trigger apoptosis in human gingival epithelial cells by down-regulating BACH2. Int. J. Clin. Exp. Pathol. 2020, 13, 901–911. [Google Scholar]
- Porstner, M.; Winkelmann, R.; Daum, P.; Schmid, J.; Pracht, K.; Corte-Real, J.; Schreiber, S.; Haftmann, C.; Brandl, A.; Mashreghi, M.F.; et al. miR-148a promotes plasma cell differentiation and targets the germinal center transcription factors Mitf and Bach2. Eur. J. Immunol. 2015, 45, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, J.; Wang, F.; Guo, R.; Xing, H.; Chen, Y.; Chen, D.; Xie, X.; Wan, D.; Jiang, Z. MiR-150-5p regulate T cell activation in severe aplastic anemia by targeting Bach2. Cell Tissue Res. 2021, 384, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xu, M.; Chen, Z.; Huang, L.; Wu, Z.; Huang, Z.; Liu, L. circ_SPEF2 Regulates the Balance of Treg Cells by Regulating miR-16-5p/BACH2 in Lymphoma and Participates in the Immune Response. Tissue Eng. Regen. Med. 2023, 20, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, H.; Kobayashi, A.; Yoshida, M.; Kudo, N.; Oyake, T.; Motohashi, H.; Hayashi, N.; Yamamoto, M.; Igarashi, K. Oxidative stress abolishes leptomycin B-sensitive nuclear export of transcription repressor Bach2 that counteracts activation of Maf recognition element. J. Biol. Chem. 2000, 275, 15370–15376. [Google Scholar] [CrossRef] [PubMed]
- Muto, A.; Tashiro, S.; Tsuchiya, H.; Kume, A.; Kanno, M.; Ito, E.; Yamamoto, M.; Igarashi, K. Activation of Maf/AP-1 repressor Bach2 by oxidative stress promotes apoptosis and its interaction with promyelocytic leukemia nuclear bodies. J. Biol. Chem. 2002, 277, 20724–20733. [Google Scholar] [CrossRef] [PubMed]
- Tamahara, T.; Ochiai, K.; Muto, A.; Kato, Y.; Sax, N.; Matsumoto, M.; Koseki, T.; Igarashi, K. The mTOR-Bach2 Cascade Controls Cell Cycle and Class Switch Recombination during B Cell Differentiation. Mol. Cell. Biol. 2017, 37, e00418-17. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Yang, K.; Cloer, C.; Neale, G.; Vogel, P.; Chi, H. mTORC1 couples immune signals and metabolic programming to establish Treg-cell function. Nature 2013, 499, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Chi, H. mTOR signaling, Tregs and immune modulation. Immunotherapy 2014, 6, 1295–1311. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Zeng, H.; Nguyen, T.M.; Wang, Y.; Vogel, P.; Dhungana, Y.; Liu, X.; Neale, G.; Locasale, J.W.; Chi, H. mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat. Commun. 2018, 9, 2095. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef]
- Jang, K.J.; Mano, H.; Aoki, K.; Hayashi, T.; Muto, A.; Nambu, Y.; Takahashi, K.; Itoh, K.; Taketani, S.; Nutt, S.L.; et al. Mitochondrial function provides instructive signals for activation-induced B-cell fates. Nat. Commun. 2015, 6, 6750. [Google Scholar] [CrossRef] [PubMed]
- Tsui, C.; Martinez-Martin, N.; Gaya, M.; Maldonado, P.; Llorian, M.; Legrave, N.M.; Rossi, M.; MacRae, J.I.; Cameron, A.J.; Parker, P.J.; et al. Protein Kinase C-beta Dictates B Cell Fate by Regulating Mitochondrial Remodeling, Metabolic Reprogramming, and Heme Biosynthesis. Immunity 2018, 48, 1144–1159.e1145. [Google Scholar] [CrossRef] [PubMed]
- Do, J.S.; Zwick, D.; Kenyon, J.D.; Zhong, F.; Askew, D.; Huang, A.Y.; Van't Hof, W.; Finney, M.; Laughlin, M.J. Mesenchymal stromal cell mitochondrial transfer to human induced T-regulatory cells mediates FOXP3 stability. Sci. Rep. 2021, 11, 10676. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Pompura, S.L.; Wagner, A.; Kitz, A.; LaPerche, J.; Yosef, N.; Dominguez-Villar, M.; Hafler, D.A. Oleic acid restores suppressive defects in tissue-resident FOXP3 Tregs from patients with multiple sclerosis. J. Clin. Investig. 2021, 131, e138519. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.L.; Kuchroo, J.R.; Sage, P.T.; Liang, D.; Francisco, L.M.; Buck, J.; Thaker, Y.R.; Zhang, Q.; McArdel, S.L.; Juneja, V.R.; et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J. Exp. Med. 2021, 218, e20182232. [Google Scholar] [CrossRef] [PubMed]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e1287. [Google Scholar] [CrossRef]
- Chen, Z.; Pittman, E.F.; Romaguera, J.; Fayad, L.; Wang, M.; Neelapu, S.S.; McLaughlin, P.; Kwak, L.; McCarty, N. Nuclear translocation of B-cell-specific transcription factor, BACH2, modulates ROS mediated cytotoxic responses in mantle cell lymphoma. PLoS ONE 2013, 8, e69126. [Google Scholar] [CrossRef]
- Tashiro, S.; Muto, A.; Tanimoto, K.; Tsuchiya, H.; Suzuki, H.; Hoshino, H.; Yoshida, M.; Walter, J.; Igarashi, K. Repression of PML nuclear body-associated transcription by oxidative stress-activated Bach2. Mol. Cell. Biol. 2004, 24, 3473–3484. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lao, Y.; Teng, X.L.; Li, S.; Zhou, Y.; Wang, F.; Guo, X.; Deng, S.; Chang, Y.; Wu, X.; et al. SENP3 maintains the stability and function of regulatory T cells via BACH2 deSUMOylation. Nat. Commun. 2018, 9, 3157. [Google Scholar] [CrossRef] [PubMed]
- Barbi, J.; Pardoll, D.M.; Pan, F. Ubiquitin-dependent regulation of Foxp3 and Treg function. Immunol. Rev. 2015, 266, 27–45. [Google Scholar] [CrossRef]
- Venuprasad, K.; Huang, H.; Harada, Y.; Elly, C.; Subramaniam, M.; Spelsberg, T.; Su, J.; Liu, Y.C. The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat. Immunol. 2008, 9, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wei, P.; Barbi, J.; Huang, Q.; Yang, E.; Bai, Y.; Nie, J.; Gao, Y.; Tao, J.; Lu, Y.; et al. The deubiquitinase USP44 promotes Treg function during inflammation by preventing FOXP3 degradation. EMBO Rep. 2020, 21, e50308. [Google Scholar] [CrossRef]
- Cortez, J.T.; Montauti, E.; Shifrut, E.; Gatchalian, J.; Zhang, Y.; Shaked, O.; Xu, Y.; Roth, T.L.; Simeonov, D.R.; Zhang, Y.; et al. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature 2020, 582, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Loo, C.-S.; Gatchalian, J.; Liang, Y.; Leblanc, M.; Xie, M.; Ho, J.; Venkatraghavan, B.; Hargreaves, D.C.; Zheng, Y. A Genome-wide CRISPR Screen Reveals a Role for the Non-canonical Nucleosome-Remodeling BAF Complex in Foxp3 Expression and Regulatory T Cell Function. Immunity 2020, 53, 143–157.e148. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Michalec, L. The molecular and epigenetic mechanisms of innate lymphoid cell (ILC) memory and its relevance for asthma. J. Exp. Med. 2021, 218, e20201354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hu, Q.; Zhang, M.; Yang, F.; Peng, C.; Zhang, Z.; Huang, C. Bach2 Deficiency Leads to Spontaneous Expansion of IL-4-Producing T Follicular Helper Cells and Autoimmunity. Front. Immunol. 2019, 10, 2050. [Google Scholar] [CrossRef]
- Contreras, A.; Wiesner, D.L.; Kingstad-Bakke, B.; Lee, W.; Svaren, J.P.; Klein, B.S.; Suresh, M. BACH2 in TRegs Limits the Number of Adipose Tissue Regulatory T Cells and Restrains Type 2 Immunity to Fungal Allergens. J. Immunol. Res. 2022, 2022, 6789055. [Google Scholar] [CrossRef]
- Fichna, M.; Żurawek, M.; Słomiński, B.; Sumińska, M.; Czarnywojtek, A.; Rozwadowska, N.; Fichna, P.; Myśliwiec, M.; Ruchała, M. Polymorphism in BACH2 gene is a marker of polyglandular autoimmunity. Endocrine 2021. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes, A. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes. Diabetes Care 2018, 41, S13–S27. [Google Scholar] [CrossRef]
- Kruger, C.; Schallreuter, K.U. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int. J. Dermatol. 2012, 51, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Linker, R.A. Inflammation in multiple sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211007687. [Google Scholar] [CrossRef] [PubMed]
- Perga, S.; Montarolo, F.; Martire, S.; Berchialla, P.; Malucchi, S.; Bertolotto, A. Anti-inflammatory genes associated with multiple sclerosis: A gene expression study. J. Neuroimmunol. 2015, 279, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhuri, R.; Eil, R.L.; Clever, D.; Klebanoff, C.A.; Sukumar, M.; Grant, F.M.; Yu, Z.; Mehta, G.; Liu, H.; Jin, P.; et al. The transcription factor BACH2 promotes tumor immunosuppression. J. Clin. Investig. 2016, 126, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Petanidis, S.; Domvri, K.; Porpodis, K.; Anestakis, D.; Freitag, L.; Hohenforst-Schmidt, W.; Tsavlis, D.; Zarogoulidis, K. Inhibition of kras-derived exosomes downregulates immunosuppressive BACH2/GATA-3 expression via RIP-3 dependent necroptosis and miR-146/miR-210 modulation. Biomed. Pharmacother. 2020, 122, 109461. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Bern, M.D.; Miao, B.; Fan, C.; Xing, X.; Inoue, T.; Piersma, S.J.; Wang, T.; Colonna, M.; Kurosaki, T.; et al. The transcription factor Bach2 negatively regulates murine natural killer cell maturation and function. Elife 2022, 11, e77294. [Google Scholar] [CrossRef]
- Honaker, Y.; Hubbard, N.; Xiang, Y.; Fisher, L.; Hagin, D.; Sommer, K.; Song, Y.; Yang, S.J.; Lopez, C.; Tappen, T.; et al. Gene editing to induce FOXP3 expression in human CD4+ T cells leads to a stable regulatory phenotype and function. Sci. Transl. Med. 2020, 12, eaay6422. [Google Scholar] [CrossRef]
- Lan, Q.; Fan, H.; Quesniaux, V.; Ryffel, B.; Liu, Z.; Zheng, S.G. Induced Foxp3+ regulatory T cells: A potential new weapon to treat autoimmune and inflammatory diseases? J. Mol. Cell Biol. 2012, 4, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Hippen, K.L.; Merkel, S.C.; Schirm, D.K.; Nelson, C.; Tennis, N.C.; Riley, J.L.; June, C.H.; Miller, J.S.; Wagner, J.E.; Blazar, B.R. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2011, 11, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tey, S.K.; Koyama, M.; Kuns, R.D.; Olver, S.D.; Lineburg, K.E.; Lor, M.; Teal, B.E.; Raffelt, N.C.; Raju, J.; et al. Induced regulatory T cells promote tolerance when stabilized by rapamycin and IL-2 in vivo. J. Immunol. 2013, 191, 5291–5303. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, M.; Nakatsukasa, H.; Okada, M.; Lu, Q.; Yoshimura, A. Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol. 2016, 37, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Muto, A.; Shima, H.; Katoh, Y.; Sax, N.; Tajima, S.; Brydun, A.; Ikura, T.; Yoshizawa, N.; Masai, H.; et al. Epigenetic Regulation of the Blimp-1 Gene (Prdm1) in B Cells Involves Bach2 and Histone Deacetylase 3. J. Biol. Chem. 2016, 291, 6316–6330. [Google Scholar] [CrossRef]
- Hu, Q.; Xu, T.; Zhang, W.; Huang, C. Bach2 regulates B cell survival to maintain germinal centers and promote B cell memory. Biochem. Biophys. Res. Commun. 2022, 618, 86–92. [Google Scholar] [CrossRef]
- Son, J.; Ding, H.; Farb, T.B.; Efanov, A.M.; Sun, J.; Gore, J.L.; Syed, S.K.; Lei, Z.; Wang, Q.; Accili, D.; et al. BACH2 inhibition reverses beta cell failure in type 2 diabetes models. J. Clin. Investig. 2021, 131, e153876. [Google Scholar] [CrossRef]
- Xiong, E.; Popp, O.; Salomon, C.; Mertins, P.; Kocks, C.; Rajewsky, K.; Chu, V.T. A CRISPR/Cas9-mediated screen identifies determinants of early plasma cell differentiation. Front. Immunol. 2022, 13, 1083119. [Google Scholar] [CrossRef]
- Vardaka, P.; Lozano, T.; Bot, C.; Ellery, J.; Whiteside, S.K.; Imianowski, C.J.; Farrow, S.; Walker, S.; Okkenhaug, H.; Yang, J.; et al. A cell-based bioluminescence assay reveals dose-dependent and contextual repression of AP-1-driven gene expression by BACH2. Sci. Rep. 2020, 10, 18902. [Google Scholar] [CrossRef]
- Mouri, K.; Guo, M.H.; de Boer, C.G.; Lissner, M.M.; Harten, I.A.; Newby, G.A.; DeBerg, H.A.; Platt, W.F.; Gentili, M.; Liu, D.R.; et al. Prioritization of autoimmune disease-associated genetic variants that perturb regulatory element activity in T cells. Nat. Genet. 2022, 54, 603–612. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | BACH2 Mutation | Role of BACH2 in Disease | Reference |
|---|---|---|---|
| Inflammatory Bowel Syndrome | BACH2 promoter methylation | Affects IgG glycosylation associated with IBS | [16] |
| Crohn’s Disease, IBS and Non-melanoma skin cancer | BACH2 variant, SNP intron variant and genic upstream transcript variant | Associated with post-operative recurrence of Crohn’s Disease | [17,18] |
| Chronic pancreatitis | BACH2 associated with protection | BACH2 repression associated with features of advanced disease | [19] |
| Systemic lupus erythematosus | SNP rs597325 | Associated with SLE | [20] |
| BRIDA | T71C, G2362A G1727T | BACH2 related immunodeficiency | [21,22] |
| Leukemia | BACH2 associated with incidence, genome wide association studies, and SNPs | Low expression associated with poor overall survival | [23,24] |
| Asthma | SNP associated with disease risk | [25] | |
| Type 1 Diabetes (T1D) | [26,27] | ||
| Crohn’s and Celiac’s disease | [28,29] | ||
| Vitiligo | [30] | ||
| MS | [31,32] | ||
| Rhumatoid Arthritis | [33] | ||
| Addison’s Disease | [34,35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zwick, D.; Vo, M.T.; Shim, Y.J.; Reijonen, H.; Do, J.-s. BACH2: The Future of Induced T-Regulatory Cell Therapies. Cells 2024, 13, 891. https://doi.org/10.3390/cells13110891
Zwick D, Vo MT, Shim YJ, Reijonen H, Do J-s. BACH2: The Future of Induced T-Regulatory Cell Therapies. Cells. 2024; 13(11):891. https://doi.org/10.3390/cells13110891
Chicago/Turabian StyleZwick, Daniel, Mai Tram Vo, Young Jun Shim, Helena Reijonen, and Jeong-su Do. 2024. "BACH2: The Future of Induced T-Regulatory Cell Therapies" Cells 13, no. 11: 891. https://doi.org/10.3390/cells13110891