
From Environment to Gene Expression: Epigenetic Methylations and One-Carbon Metabolism in Amyotrophic Lateral Sclerosis

Abstract
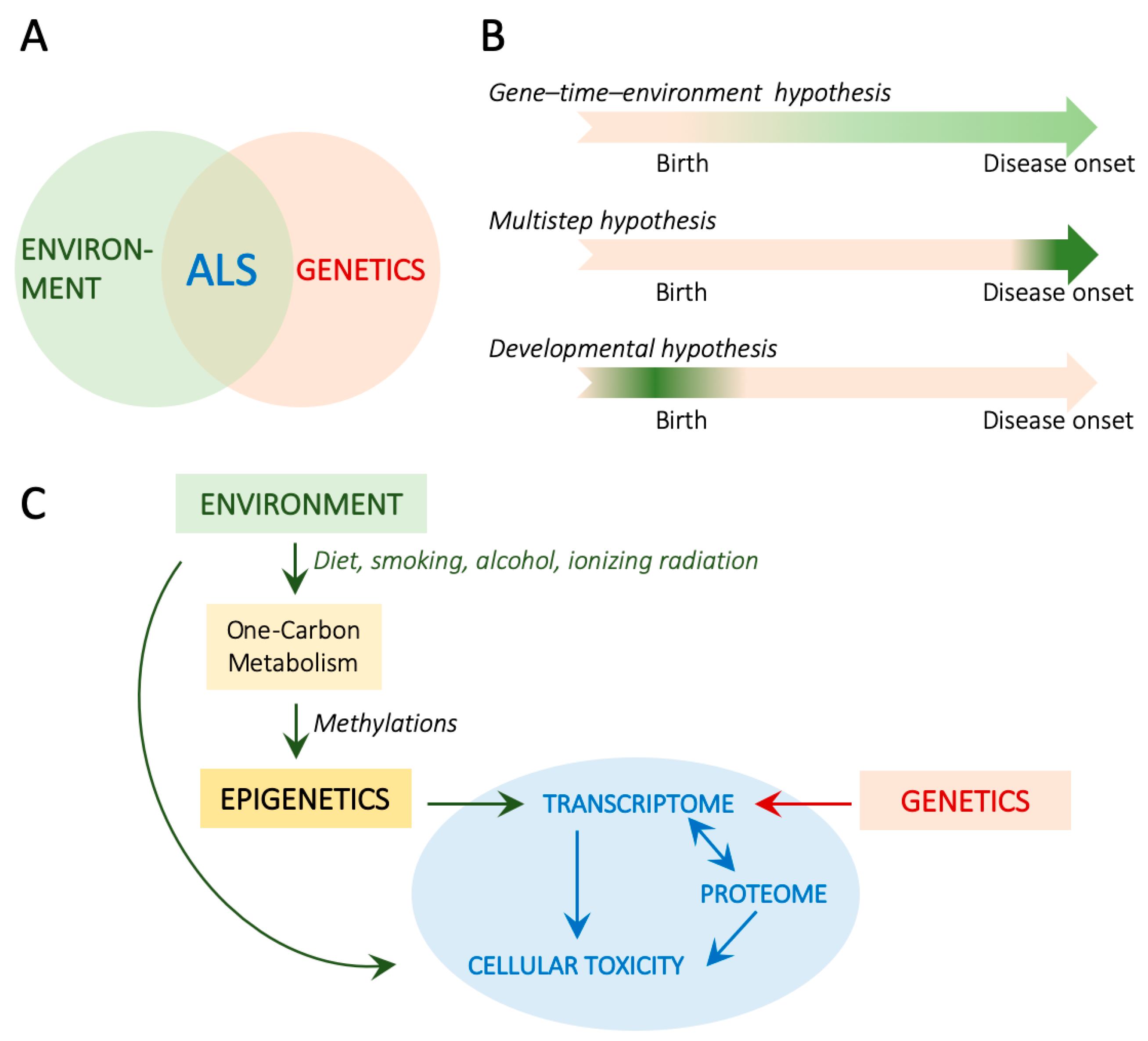
:1. Introduction
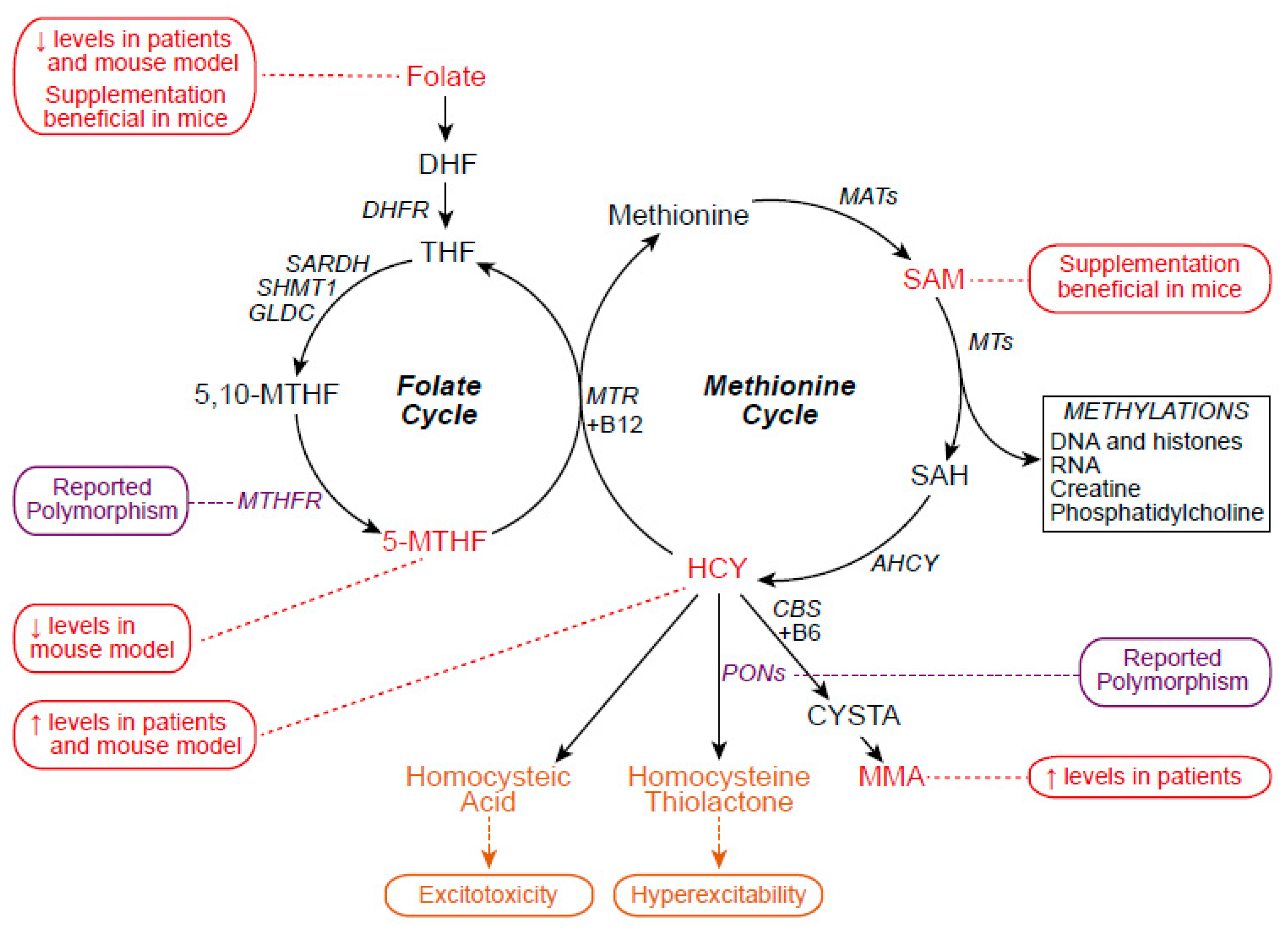
2. One-Carbon Metabolism
2.1. One-Carbon Metabolism in ALS
2.2. One-Carbon Metabolism and Epigenetics
3. DNA Methylation
3.1. Increased Expression of DNMTs and DNAm in ALS Is Associated with Neuronal Death
3.2. DNAm Age Contributes to ALS Onset and Heterogeneity
3.3. DNAm Changes Confirm Transcriptomic Analyses
3.4. DNAm as an Epigenetic Disease Modifier
4. Histone Methylation
4.1. Polycomb Repressive Complex 2 and Repressive H3K27me3 Mark
4.2. Lysine-Specific Demethylase 1 and the H3K4me2 Active Mark
4.3. Protein Arginine Methyltransferase 1 and the Active H4R3me2 Mark
4.4. Complex Patterns of Altered Histone Methylation Characterize C9ORF72 Pathology

{kind=link}
{kind=link}
| Tissue/Model | Observations | References |
|---|---|---|
| Post-mortem frontal cortex and cerebellar tissues of C9-ALS/FTD patients, sALS, and controls |
| [101] |
| Post-mortem brains of C9-ALS/FTD, ALSFTD non-expansion carriers, and controls |
| [100] |
| Post-mortem motor cortex of C9-ALS/FTD patients and controls |
| [99] |
| MN from lumbar spinal cord of SOD1G93A mice |
| [102] |
| NSC34 cells expressing mSod1 |
| |
| SOD1G93A mice |
| |
| MNs from spinal cord of SOD1G93A mice and SH-SY5HY cells expressing SOD1 mutants G93A and H80R |
| [96] |
| Post-mortem spinal cord of sALS patients and controls |
| [105] |
| Primary spinal cord cultures of mutant FUS MN |
| [108] |
| Spinal cord of transgenic FUSR495 mouse |
| |
| Post-mortem cortex of C9-FTD/ALS and GFP-(PR)50 mouse |
| [118] |
| Post-mortem frontal cortex and cerebellum of C9-ALS/FTD patients and controls |
| [115] |
| Cortex of the transgenic C9-BAC mice |
| [116] |
| Primary spinal cord astrocyte cultures and astrocytes and neurons of spinal cord, motor cortex, and hippocampus of transgenic C9-BAC mice |
| [117] |
5. Pharmacological Targeting of Epigenetic Methylation in ALS
6. Environmental and Developmental Impacts on 1C Metabolism
Author Contributions
Funding
Conflicts of Interest
References
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chiò, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1847–1850. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M.; et al. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology 2018, 91, e635–e642. [Google Scholar] [CrossRef]
- Schor, N.F.; Bianchi, D.W. Neurodevelopmental Clues to Neurodegeneration. Pediatr. Neurol. 2021, 123, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Tartaglione, A.M.; Venerosi, A.; Calamandrei, G. Early-Life Toxic Insults and Onset of Sporadic Neurodegenerative Diseases-an Overview of Experimental Studies. Curr. Top. Behav. Neurosci. 2016, 29, 231–264. [Google Scholar]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Gregory, P.D.; Wagner, K.; Hörz, W. Histone acetylation and chromatin remodeling. Exp. Cell Res. 2001, 265, 195–202. [Google Scholar] [CrossRef]
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in amyotrophic lateral sclerosis: A role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 2019, 204, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Bankole, O.; Scambi, I.; Parrella, E.; Muccilli, M.; Bonafede, R.; Turano, E.; Pizzi, M.; Mariotti, R. Beneficial and Sexually Dimorphic Response to Combined HDAC Inhibitor Valproate and AMPK/SIRT1 Pathway Activator Resveratrol in the Treatment of ALS Mice. Int. J. Mol. Sci. 2022, 23, 1047. [Google Scholar] [CrossRef] [PubMed]
- Rossaert, E.; Pollari, E.; Jaspers, T.; Van Helleputte, L.; Jarpe, M.; Van Damme, P.; De Bock, K.; Moisse, M.; Van Den Bosch, L. Restoration of histone acetylation ameliorates disease and metabolic abnormalities in a FUS mouse model. Acta Neuropathol. Commun. 2019, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Rouaux, C.; Panteleeva, I.; Rene, F.; Gonzalez de Aguilar, J.-L.; Echaniz-Laguna, A.; Dupuis, L.; Menger, Y.; Boutillier, A.-L.; Loeffler, J.-P. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J. Neurosci. 2007, 27, 5535–5545. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.-E.; Ko, C.-P. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2011, 231, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Quintana, M.; Sherman, A.V.; Vestrucci, M.; Wu, Y.; Timmons, J.; Cudkowicz, M.; Pooled Resource Open-Access ALS Clinical Trials Consortium. Analysis of sodium phenylbutyrate and taurursodiol survival effect in ALS using external controls. Ann. Clin. Transl. Neurol. 2023, 10, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Burg, T.; Rossaert, E.; Moisse, M.; Van Damme, P.; Van Den Bosch, L. Histone Deacetylase Inhibition Regulates Lipid Homeostasis in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 11224. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef]
- Parrella, E.; Porrini, V.; Scambi, I.; Gennari, M.M.; Gussago, C.; Bankole, O.; Benarese, M.; Mariotti, R.; Pizzi, M. Synergistic association of resveratrol and histone deacetylase inhibitors as treatment in amyotrophic lateral sclerosis. Front. Pharmacol. 2022, 13, 1017364. [Google Scholar] [CrossRef]
- Gruber, A.J.; Zavolan, M. Modulation of epigenetic regulators and cell fate decisions by miRNAs. Epigenomics 2013, 5, 671–683. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, F.; Guan, Y.; Meng, F.; Zhao, Z.; Su, Q.; Bao, W.; Wang, X.; Zhao, J.; Huo, Z.; et al. The Biogenesis of miRNAs and Their Role in the Development of Amyotrophic Lateral Sclerosis. Cells 2022, 11, 572. [Google Scholar] [CrossRef] [PubMed]
- Koike, Y.; Onodera, O. Implications of miRNAs dysregulation in amyotrophic lateral sclerosis: Challenging for clinical applications. Front. Neurosci. 2023, 17, 1131758. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Schübeler, D. Genetics and epigenetics: Stability and plasticity during cellular differentiation. Trends Genet. 2009, 25, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Cordaro, M.; Siracusa, R.; Fusco, R.; Cuzzocrea, S.; Di Paola, R.; Impellizzeri, D. Involvements of Hyperhomocysteinemia in Neurological Disorders. Metabolites 2021, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Zoccolella, S.; Bendotti, C.; Beghi, E.; Logroscino, G. Homocysteine levels and amyotrophic lateral sclerosis: A possible link. Amyotroph. Lateral Scler. 2010, 11, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Kühnlein, P.; Jung, H.; Farkas, M.; Keskitalo, S.; Ineichen, B.; Jelcic, I.; Petersen, J.; Semmler, A.; Weller, M.; Ludolph, A.C.; et al. The thermolabile variant of 5,10-methylenetetrahydrofolate reductase is a possible risk factor for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2011, 12, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Sazci, A.; Ozel, M.D.; Emel, E.; Idrisoglu, H.A. Gender-specific association of methylenetetrahydrofolate reductase gene polymorphisms with sporadic amyotrophic lateral sclerosis. Genet. Test. Mol. Biomark. 2012, 16, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Reichert, C.O.; Levy, D.; Bydlowski, S.P. Paraoxonase Role in Human Neurodegenerative Diseases. Antioxidants 2020, 10, 11. [Google Scholar] [CrossRef]
- Zoccolella, S.; Simone, I.L.; Lamberti, P.; Samarelli, V.; Tortelli, R.; Serlenga, L.; Logroscino, G. Elevated plasma homocysteine levels in patients with amyotrophic lateral sclerosis. Neurology 2008, 70, 222–225. [Google Scholar] [CrossRef]
- Levin, J.; Bötzel, K.; Giese, A.; Vogeser, M.; Lorenzl, S. Elevated levels of methylmalonate and homocysteine in Parkinson’s disease, progressive supranuclear palsy and amyotrophic lateral sclerosis. Dement. Geriatr. Cogn. Disord. 2010, 29, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Valentino, F.; Bivona, G.; Butera, D.; Paladino, P.; Fazzari, M.; Piccoli, T.; Ciaccio, M.; La Bella, V. Elevated cerebrospinal fluid and plasma homocysteine levels in ALS. Eur. J. Neurol. 2010, 17, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, S.; Li, L.; Wang, Q.; Le, W. Decreased level of 5-methyltetrahydrofolate: A potential biomarker for pre-symptomatic amyotrophic lateral sclerosis. J. Neurol. Sci. 2010, 293, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yang, Q.; Liu, Z.; Zhang, R.; Yu, H.; Wang, M.; Chen, S.; Xu, G.; Shao, Y.; Le, W. Integrative analysis of metabolomics and proteomics unravels purine metabolism dysregulation in the SOD1G93A mouse model of amyotrophic lateral scelrosis. Neurobiol. Dis. 2023, 181, 106110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, S.; Li, L.; Wang, Q.; Le, W. Folic acid protects motor neurons against the increased homocysteine, inflammation and apoptosis in SOD1 G93A transgenic mice. Neuropharmacology 2008, 54, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Suchy, J.; Lee, S.; Ahmed, A.; Shea, T.B. Dietary supplementation with S-adenosyl methionine delays the onset of motor neuron pathology in a murine model of amyotrophic lateral sclerosis. Neuromol. Med. 2010, 12, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Adalbert, R.; Engelhardt, J.I.; Siklos, L. DL-Homocysteic acid application disrupts calcium homeostasis and induces degeneration of spinal motor neurons in vivo. Acta Neuropathol. 2002, 103, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Deep, S.N.; Mitra, S.; Rajagopal, S.; Paul, S.; Poddar, R. GluN2A-NMDA receptor-mediated sustained Ca2+ influx leads to homocysteine-induced neuronal cell death. J. Biol. Chem. 2019, 294, 11154–11165. [Google Scholar] [CrossRef] [PubMed]
- Mareš, P.; Folbergrová, J.; Haugvicová, R.; Kubová, H. Do stereoisomers of homocysteic acid exhibit different convulsant action in immature rats? Physiol. Res. 2019, 68, S361–S366. [Google Scholar] [CrossRef]
- Rasić-Marković, A.; Stanojlović, O.; Hrncić, D.; Krstić, D.; Colović, M.; Susić, V.; Radosavljević, T.; Djuric, D. The activity of erythrocyte and brain Na+/K+ and Mg2+-ATPases in rats subjected to acute homocysteine and homocysteine thiolactone administration. Mol. Cell. Biochem. 2009, 327, 39–45. [Google Scholar] [CrossRef]
- Gabbi, P.; Ribeiro, L.R.; Jessié Martins, G.; Cardoso, A.S.; Haupental, F.; Rodrigues, F.S.; Machado, A.K.; Sperotto Brum, J.; Medeiros Frescura Duarte, M.M.; Schetinger, M.R.C.; et al. Methylmalonate Induces Inflammatory and Apoptotic Potential: A Link to Glial Activation and Neurological Dysfunction. J. Neuropathol. Exp. Neurol. 2017, 76, 160–178. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Ward, R.L. Folate and One-Carbon Metabolism and Its Impact on Aberrant DNA Methylation in Cancer, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2010; Volume 71, pp. 79–121. [Google Scholar]
- Lionaki, E.; Ploumi, C.; Tavernarakis, N. One-Carbon Metabolism: Pulling the Strings behind Aging and Neurodegeneration. Cells 2022, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- McGee, M.; Bainbridge, S.; Fontaine-Bisson, B. A crucial role for maternal dietary methyl donor intake in epigenetic programming and fetal growth outcomes. Nutr. Rev. 2018, 76, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Serefidou, M.; Venkatasubramani, A.V.; Imhof, A. The Impact of One Carbon Metabolism on Histone Methylation. Front. Genet. 2019, 10, 764. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Xiahou, Z.; Zhao, H.; Peng, B.; Zhao, H.; Xu, X. MTHFR promotes heterochromatin maintenance. Biochem. Biophys. Res. Commun. 2014, 447, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Perła-Kaján, J.; Jakubowski, H. Dysregulation of Epigenetic Mechanisms of Gene Expression in the Pathologies of Hyperhomocysteinemia. Int. J. Mol. Sci. 2019, 20, 3140. [Google Scholar] [CrossRef]
- Xie, J.; Xie, L.; Wei, H.; Li, X.-J.; Lin, L. Dynamic Regulation of DNA Methylation and Brain Functions. Biology 2023, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Yen, T.J.; Bellacosa, A. Active DNA demethylation-The epigenetic gatekeeper of development, immunity, and cancer. Adv. Genet. 2021, 2, e10033. [Google Scholar] [CrossRef]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef]
- Starr, J.M. Ageing and epigenetics: Linking neurodevelopmental and neurodegenerative disorders. Dev. Med. Child Neurol. 2019, 61, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Seal, S.; Ruark, E.; Harmer, J.; Ramsay, E.; Del Vecchio Duarte, S.; Zachariou, A.; Hanks, S.; O’Brien, E.; Aksglaede, L.; et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 2014, 46, 385–388. [Google Scholar] [CrossRef]
- Tatton-Brown, K.; Zachariou, A.; Loveday, C.; Renwick, A.; Mahamdallie, S.; Aksglaede, L.; Baralle, D.; Barge-Schaapveld, D.; Blyth, M.; Bouma, M.; et al. The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res. 2018, 3, 46. [Google Scholar] [CrossRef]
- Klein, C.J.; Botuyan, M.-V.; Wu, Y.; Ward, C.J.; Nicholson, G.A.; Hammans, S.; Hojo, K.; Yamanishi, H.; Karpf, A.R.; Wallace, D.C.; et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 2011, 43, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Winkelmann, J.; Lin, L.; Schormair, B.; Kornum, B.R.; Faraco, J.; Plazzi, G.; Melberg, A.; Cornelio, F.; Urban, A.E.; Pizza, F.; et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum. Mol. Genet. 2012, 21, 2205–2210. [Google Scholar] [CrossRef]
- Martin, L.J.; Adams, D.A.; Niedzwiecki, M.V.; Wong, M. Aberrant DNA and RNA Methylation Occur in Spinal Cord and Skeletal Muscle of Human SOD1 Mouse Models of ALS and in Human ALS: Targeting DNA Methylation Is Therapeutic. Cells 2022, 11, 3448. [Google Scholar] [CrossRef]
- Appleby-Mallinder, C.; Schaber, E.; Kirby, J.; Shaw, P.J.; Cooper-Knock, J.; Heath, P.R.; Highley, J.R. TDP43 proteinopathy is associated with aberrant DNA methylation in human amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2021, 47, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Hartung, T.; Rhein, M.; Kalmbach, N.; Thau-Habermann, N.; Naujock, M.; Müschen, L.; Frieling, H.; Sterneckert, J.; Hermann, A.; Wegner, F.; et al. Methylation and Expression of Mutant FUS in Motor Neurons Differentiated From Induced Pluripotent Stem Cells From ALS Patients. Front. Cell Dev. Biol. 2021, 9, 774751. [Google Scholar] [CrossRef]
- Li, J.; Jaiswal, M.K.; Chien, J.-F.; Kozlenkov, A.; Jung, J.; Zhou, P.; Gardashli, M.; Pregent, L.J.; Engelberg-Cook, E.; Dickson, D.W.; et al. Divergent single cell transcriptome and epigenome alterations in ALS and FTD patients with C9orf72 mutation. Nat. Commun. 2023, 14, 5714. [Google Scholar] [CrossRef]
- Rasmi, Y.; Shokati, A.; Hassan, A.; Aziz, S.G.-G.; Bastani, S.; Jalali, L.; Moradi, F.; Alipour, S. The role of DNA methylation in progression of neurological disorders and neurodegenerative diseases as well as the prospect of using DNA methylation inhibitors as therapeutic agents for such disorders. IBRO Neurosci. Rep. 2023, 14, 28–37. [Google Scholar] [CrossRef]
- Niccoli, T.; Partridge, L.; Isaacs, A.M. Ageing as a risk factor for ALS/FTD. Hum. Mol. Genet. 2017, 26, R105–R113. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Tremolizzo, L.; Messina, P.; Conti, E.; Sala, G.; Cecchi, M.; Airoldi, L.; Pastorelli, R.; Pupillo, E.; Bandettini Di Poggio, M.; Filosto, M.; et al. EURALS Consortium Whole-blood global DNA methylation is increased in amyotrophic lateral sclerosis independently of age of onset. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; McKeever, P.M.; Xi, Z.; Moreno, D.; Sato, C.; Bergsma, T.; McGoldrick, P.; Keith, J.; Robertson, J.; Zinman, L.; et al. DNA methylation age acceleration is associated with ALS age of onset and survival. Acta Neuropathol. 2020, 139, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Tartaglia, M.C.; Moreno, D.; Sato, C.; McKeever, P.; Weichert, A.; Keith, J.; Robertson, J.; Zinman, L.; Rogaeva, E. DNA methylation age-acceleration is associated with disease duration and age at onset in C9orf72 patients. Acta Neuropathol. 2017, 134, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Morahan, J.M.; Yu, B.; Trent, R.J.; Pamphlett, R. A genome-wide analysis of brain DNA methylation identifies new candidate genes for sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Fang, F.; Hanby, M.F.; Leigh, P.N.; Shaw, C.E.; Ye, W.; Rijsdijk, F. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1324–1326. [Google Scholar] [CrossRef]
- Young, P.E.; Kum Jew, S.; Buckland, M.E.; Pamphlett, R.; Suter, C.M. Epigenetic differences between monozygotic twins discordant for amyotrophic lateral sclerosis (ALS) provide clues to disease pathogenesis. PLoS ONE 2017, 12, e0182638. [Google Scholar] [CrossRef]
- Tarr, I.; McCann, E.P.; Benyamin, B.; Peters, T.J.; Twine, N.A.; Zhang, K.Y.; Zhao, Q.; Zhang, Z.-H.; Rowe, D.B.; Nicholson, G.A.; et al. Monozygotic twins and tripletsdiscordant for amyotrophic lateral. Sci. Rep. 2019, 9, 8254. [Google Scholar] [CrossRef]
- Zhang, M.; Xi, Z.; Ghani, M.; Jia, P.; Pal, M.; Werynska, K.; Moreno, D.; Sato, C.; Liang, Y.; Robertson, J.; et al. Genetic and epigenetic study of ALS-discordant identical twins with double mutations in SOD1 and ARHGEF28. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1268–1270. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef] [PubMed]
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2019, 9, 419229. [Google Scholar] [CrossRef] [PubMed]
- Ebbert, M.T.W.; Ross, C.A.; Pregent, L.J.; Lank, R.J.; Zhang, C.; Katzman, R.B.; Jansen-West, K.; Song, Y.; Rocha, E.L.; Palmucci, C.; et al. Conserved DNA methylation combined with differential frontal cortex and cerebellar expression distinguishes C9orf72-associated and sporadic ALS, and implicates SERPINA1 in disease. Acta Neuropathol. 2017, 134, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.; Koczulla, A.-R.; Mengel, D.; Koepke, J.; Dodel, R.; Dontcheva, G.; Habib, P.; Bach, J.-P. Reduction of glutamate-induced excitotoxicity in murine primary neurons involving calpain inhibtion. J. Neurol. Sci. 2015, 359, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Welch, G.; Tsai, L.-H. Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep. 2022, 23, e54217. [Google Scholar] [CrossRef] [PubMed]
- Ronen, A.; Glickman, B.W. Human DNA repair genes. Environ. Mol. Mutagen. 2001, 37, 241–283. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Commun. 2020, 8, 7. [Google Scholar] [CrossRef]
- Oates, N.; Pamphlett, R. An epigenetic analysis of SOD1 and VEGF in ALS. Amyotroph. Lateral Scler. 2007, 8, 83–86. [Google Scholar] [CrossRef]
- Coppedè, F.; Stoccoro, A.; Mosca, L.; Gallo, R.; Tarlarini, C.; Lunetta, C.; Marocchi, A.; Migliore, L.; Penco, S. Increase in DNA methylation in patients with amyotrophic lateral sclerosis carriers of not fully penetrant SOD1 mutations. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 93–101. [Google Scholar] [CrossRef]
- Jackson, J.L.; Finch, N.A.; Baker, M.C.; Kachergus, J.M.; DeJesus-Hernandez, M.; Pereira, K.; Christopher, E.; Prudencio, M.; Heckman, M.G.; Thompson, E.A.; et al. Elevated methylation levels, reduced expression levels, and frequent contractions in a clinical cohort of C9orf72 expansion carriers. Mol. Neurodegener. 2020, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Zinman, L.; Moreno, D.; Schymick, J.; Liang, Y.; Sato, C.; Zheng, Y.; Ghani, M.; Dib, S.; Keith, J.; et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9ORF72 expansion. Am. J. Hum. Genet. 2013, 92, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.Y.; Russ, J.; Wu, K.; Neal, D.; Suh, E.; McNally, A.G.; Irwin, D.J.; Van Deerlin, V.M.; Lee, E.B. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. 2014, 128, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Russ, J.; Liu, E.Y.; Wu, K.; Neal, D.; Suh, E.; Irwin, D.J.; McMillan, C.T.; Harms, M.B.; Cairns, N.J.; Wood, E.M.; et al. Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol. 2015, 129, 39–52. [Google Scholar] [CrossRef] [PubMed]
- McMillan, C.T.; Russ, J.; Wood, E.M.; Irwin, D.J.; Grossman, M.; McCluskey, L.; Elman, L.; Van Deerlin, V.; Lee, E.B. C9orf72 promoter hypermethylation is neuroprotective: Neuroimaging and neuropathologic evidence. Neurology 2015, 84, 1622–1630. [Google Scholar] [CrossRef] [PubMed]
- Koike, Y.; Sugai, A.; Hara, N.; Ito, J.; Yokoseki, A.; Ishihara, T.; Yamagishi, T.; Tsuboguchi, S.; Tada, M.; Ikeuchi, T.; et al. Age-related demethylation of the TDP-43 autoregulatory region in the human motor cortex. Commun. Biol. 2021, 4, 1107. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Mazzio, E.A.; Soliman, K.F.A. Basic concepts of epigenetics: Impact of environmental signals on gene expression. Epigenetics 2012, 7, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Déjardin, J. The chromatin signatures of enhancers and their dynamic regulation. Nucleus 2023, 14, 2160551. [Google Scholar] [CrossRef]
- Ito, S.; Das, N.D.; Umehara, T.; Koseki, H. Factors and Mechanisms That Influence Chromatin-Mediated Enhancer-Promoter Interactions and Transcriptional Regulation. Cancers 2022, 14, 5404. [Google Scholar] [CrossRef]
- Park, J.; Lee, K.; Kim, K.; Yi, S.-J. The role of histone modifications: From neurodevelopment to neurodiseases. Signal Transduct. Target. Ther. 2022, 7, 217. [Google Scholar] [CrossRef] [PubMed]
- Swahari, V.; West, A.E. Histone demethylases in neuronal differentiation, plasticity, and disease. Curr. Opin. Neurobiol. 2019, 59, 9–15. [Google Scholar] [CrossRef]
- Cobos, S.N.; Bennett, S.A.; Torrente, M.P. The impact of histone post-translational modifications in neurodegenerative diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kundu, S.; Singh, A.; Singh, S. Understanding the Role of Histone Deacetylase and their Inhibitors in Neurodegenerative Disorders: Current Targets and Future Perspective. Curr. Neuropharmacol. 2022, 20, 158–178. [Google Scholar] [CrossRef]
- Masala, A.; Sanna, S.; Esposito, S.; Rassu, M.; Galioto, M.; Zinellu, A.; Carru, C.; Carrì, M.T.; Iaccarino, C.; Crosio, C. Epigenetic Changes Associated with the Expression of Amyotrophic Lateral Sclerosis (ALS) Causing Genes. Neuroscience 2018, 390, 1–11. [Google Scholar] [CrossRef]
- von Schimmelmann, M.; Feinberg, P.A.; Sullivan, J.M.; Ku, S.M.; Badimon, A.; Duff, M.K.; Wang, Z.; Lachmann, A.; Dewell, S.; Ma’ayan, A.; et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 2016, 19, 1321–1330. [Google Scholar] [CrossRef]
- Jackson, W.S.; Bauer, S.; Kaczmarczyk, L.; Magadi, S.S. Selective Vulnerability to Neurodegenerative Disease: Insights from Cell Type-Specific Translatome Studies. Biology 2024, 13, 67. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Goodrich, K.J.; Conlon, E.G.; Gao, J.; Erbse, A.H.; Manley, J.L.; Cech, T.R. C9orf72 and triplet repeat disorder RNAs: G-quadruplex formation, binding to PRC2 and implications for disease mechanisms. RNA 2019, 25, 935–947. [Google Scholar] [CrossRef]
- Finch, N.A.; Wang, X.; Baker, M.C.; Heckman, M.G.; Gendron, T.F.; Bieniek, K.F.; Wuu, J.; DeJesus-Hernandez, M.; Brown, P.H.; Chew, J.; et al. Abnormal expression of homeobox genes and transthyretin in C9ORF72 expansion carriers. Neurol. Genet. 2017, 3, e161. [Google Scholar] [CrossRef]
- Prudencio, M.; Belzil, V.V.; Batra, R.; Ross, C.A.; Gendron, T.F.; Pregent, L.J.; Murray, M.E.; Overstreet, K.K.; Piazza-Johnston, A.E.; Desaro, P.; et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 2015, 18, 1175–1182. [Google Scholar] [CrossRef]
- Choi, S.-H.; Yousefian-Jazi, A.; Hyeon, S.J.; Nguyen, P.T.-T.; Chu, J.; Kim, S.; Kim, S.; Ryu, H.L.; Kowall, N.W.; Ryu, H.; et al. Modulation of histone H3K4 dimethylationby spermidine ameliorates motor neuron survival and neuropathology in a mouse model of ALS. J. Biomed. Sci. 2022, 29, 106. [Google Scholar] [CrossRef] [PubMed]
- Christopher, M.A.; Myrick, D.A.; Barwick, B.G.; Engstrom, A.K.; Porter-Stransky, K.A.; Boss, J.M.; Weinshenker, D.; Levey, A.I.; Katz, D.J. LSD1 protects against hippocampal and cortical neurodegeneration. Nat. Commun. 2017, 8, 805. [Google Scholar] [CrossRef] [PubMed]
- Pineda, S.S.; Lee, H.; Ulloa-Navas, M.J.; Linville, R.M.; Garcia, F.J.; Galani, K.; Engelberg-Cook, E.; Castanedes, M.C.; Fitzwalter, B.E.; Pregent, L.J.; et al. Single-cell dissection of the human motor and prefrontal cortices in ALS and FTLD. Cell 2024, 187, 1971–1989.e16. [Google Scholar] [CrossRef]
- Ikenaka, K.; Atsuta, N.; Maeda, Y.; Hotta, Y.; Nakamura, R.; Kawai, K.; Yokoi, D.; Hirakawa, A.; Taniguchi, A.; Morita, M.; et al. Increase of arginine dimethylation correlates with the progression and prognosis of ALS. Neurology 2019, 92, e1868–e1877. [Google Scholar] [CrossRef] [PubMed]
- Ikenaka, K.; Miyata, S.; Mori, Y.; Koyama, Y.; Taneda, T.; Okuda, H.; Kousaka, A.; Tohyama, M. Immunohistochemical and western analyses of protein arginine N-methyltransferase 3 in the mouse brain. Neuroscience 2006, 141, 1971–1982. [Google Scholar] [CrossRef]
- So, H.-K.; Kim, H.; Lee, J.; You, C.-L.; Yun, C.-E.; Jeong, H.-J.; Jin, E.-J.; Jo, Y.; Ryu, D.; Bae, G.-U.; et al. Protein Arginine Methyltransferase 1 Ablation in Motor Neurons Causes Mitochondrial Dysfunction Leading to Age-related Motor Neuron Degeneration with Muscle Loss. Research 2023, 6, 0158. [Google Scholar] [CrossRef]
- Tibshirani, M.; Tradewell, M.L.; Mattina, K.R.; Minotti, S.; Yang, W.; Zhou, H.; Strong, M.J.; Hayward, L.J.; Durham, H.D. Cytoplasmic sequestration of FUS/TLS associated with ALS alters histone marks through loss of nuclear protein arginine methyltransferase 1. Hum. Mol. Genet. 2015, 24, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Madl, T.; Valori, C.F.; Bentmann, E.; Tahirovic, S.; Abou-Ajram, C.; Kremmer, E.; Ansorge, O.; Mackenzie, I.R.A.; Neumann, M.; et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012, 31, 4258–4275. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Arai, S.; Kawamura, T.; Matsushita, A.; Kurokawa, R. TLS and PRMT1 synergistically coactivate transcription at the survivin promoter through TLS arginine methylation. Biochem. Biophys. Res. Commun. 2011, 404, 991–996. [Google Scholar] [CrossRef]
- Scaramuzzino, C.; Monaghan, J.; Milioto, C.; Lanson, N.A.; Maltare, A.; Aggarwal, T.; Casci, I.; Fackelmayer, F.O.; Pennuto, M.; Pandey, U.B. Protein arginine methyltransferase 1 and 8 interact with FUS to modify its sub-cellular distribution and toxicity in vitro and in vivo. PLoS ONE 2013, 8, e61576. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Kitajo, K. The effect of PRMT1-mediated arginine methylation on the subcellular localization, stress granules, and detergent-insoluble aggregates of FUS/TLS. PLoS ONE 2012, 7, e49267. [Google Scholar] [CrossRef] [PubMed]
- Premasiri, A.S.; Gill, A.L.; Vieira, F.G. Type I PRMT Inhibition Protects Against C9ORF72 Arginine-Rich Dipeptide Repeat Toxicity. Front. Pharmacol. 2020, 11, 569661. [Google Scholar] [CrossRef] [PubMed]
- Dane, T.L.; Gill, A.L.; Vieira, F.G.; Denton, K.R. Reduced C9orf72 expression exacerbates polyGR toxicity in patient iPSC-derived motor neurons and a Type I protein arginine methyltransferase inhibitor reduces that toxicity. Front. Cell. Neurosci. 2023, 17, 1134090. [Google Scholar] [CrossRef] [PubMed]
- Belzil, V.V.; Bauer, P.O.; Prudencio, M.; Gendron, T.F.; Stetler, C.T.; Yan, I.K.; Pregent, L.; Daughrity, L.; Baker, M.C.; Rademakers, R.; et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013, 126, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Esanov, R.; Cabrera, G.T.; Andrade, N.S.; Gendron, T.F.; Brown, R.H.; Benatar, M.; Wahlestedt, C.; Mueller, C.; Zeier, Z. A C9ORF72 BAC mouse model recapitulates key epigenetic perturbations of ALS/FTD. Mol. Neurodegener. 2017, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Jury, N.; Abarzua, S.; Diaz, I.; Guerra, M.V.; Ampuero, E.; Cubillos, P.; Martinez, P.; Herrera-Soto, A.; Arredondo, C.; Rojas, F.; et al. Widespread loss of the silencing epigenetic mark H3K9me3 in astrocytes and neurons along with hippocampal-dependent cognitive impairment in C9orf72 BAC transgenic mice. Clin. Epigenet. 2020, 12, 32. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Guo, L.; Gonzales, P.K.; Gendron, T.F.; Wu, Y.; Jansen-West, K.; O’Raw, A.D.; Pickles, S.R.; Prudencio, M.; Carlomagno, Y.; et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 2019, 363, eaav2606. [Google Scholar] [CrossRef]
- Zhang, Y.; Amaral, M.L.; Zhu, C.; Grieco, S.F.; Hou, X.; Lin, L.; Buchanan, J.; Tong, L.; Preissl, S.; Xu, X.; et al. Single-cell epigenome analysis reveals age-associated decay of heterochromatin domains in excitatory neurons in the mouse brain. Cell Res. 2022, 32, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Benjamin, D.I.; Kim, S.; Salvi, J.S.; Dhaliwal, G.; Lam, R.; Goshayeshi, A.; Brett, J.O.; Liu, L.; Rando, T.A. Depletion of SAM leading to loss of heterochromatin drives muscle stem cell ageing. Nat. Metab. 2024, 6, 153–168. [Google Scholar] [CrossRef]
- Lee, J.; An, S.; Lee, S.-J.; Kang, J.-S. Protein Arginine Methyltransferases in Neuromuscular Function and Diseases. Cells 2022, 11, 364. [Google Scholar] [CrossRef]
- Chen, K.; Bennett, S.A.; Rana, N.; Yousuf, H.; Said, M.; Taaseen, S.; Mendo, N.; Meltser, S.M.; Torrente, M.P. Neurodegenerative Disease Proteinopathies Are Connected to Distinct Histone Post-translational Modification Landscapes. ACS Chem. Neurosci. 2018, 9, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Czuppa, M.; Dhingra, A.; Zhou, Q.; Schludi, C.; König, L.; Scharf, E.; Farny, D.; Dalmia, A.; Täger, J.; Castillo-Lizardo, M.; et al. Drug screen in iPSC-Neurons identifies nucleoside analogs as inhibitors of (G4C2)n expression in C9orf72 ALS/FTD. Cell Rep. 2022, 39, 110913. [Google Scholar] [CrossRef] [PubMed]
- Ramic, M.; Andrade, N.S.; Rybin, M.J.; Esanov, R.; Wahlestedt, C.; Benatar, M.; Zeier, Z. Epigenetic Small Molecules Rescue Nucleocytoplasmic Transport and DNA Damage Phenotypes in C9ORF72 ALS/FTD. Brain Sci. 2021, 11, 1543. [Google Scholar] [CrossRef] [PubMed]
- Green, K.M.; Sheth, U.J.; Flores, B.N.; Wright, S.E.; Sutter, A.B.; Kearse, M.G.; Barmada, S.J.; Ivanova, M.I.; Todd, P.K. High-throughput screening yields several small-molecule inhibitors of repeat-associated non-AUG translation. J. Biol. Chem. 2019, 294, 18624–18638. [Google Scholar] [CrossRef] [PubMed]
- Montalvo-Casimiro, M.; González-Barrios, R.; Meraz-Rodriguez, M.A.; Juárez-González, V.T.; Arriaga-Canon, C.; Herrera, L.A. Epidrug Repurposing: Discovering New Faces of Old Acquaintances in Cancer Therapy. Front. Oncol. 2020, 10, 605386. [Google Scholar] [CrossRef] [PubMed]
- Yahn, G.B.; Abato, J.E.; Jadavji, N.M. Role of vitamin B12 deficiency in ischemic stroke risk and outcome. Neural Regen. Res. 2021, 16, 470–474. [Google Scholar] [PubMed]
- Zeisel, S.H. Diet-gene interactions underlie metabolic individuality and influence brain development: Implications for clinical practice derived from studies on choline metabolism. Ann. Nutr. Metab. 2012, 60, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.H. Causes of vitamin B12 and folate deficiency. Food Nutr. Bull. 2008, 29 (Suppl. S1), S20–S34. [Google Scholar] [CrossRef]
- Barros, A.N.d.A.B.; Felipe, M.L.D.N.; Barbosa, I.R.; Leite-Lais, L.; Pedrosa, L.F.C. Dietary Intake of Micronutrients and Disease Severity in Patients with Amyotrophic Lateral Sclerosis. Metabolites 2023, 13, 696. [Google Scholar] [CrossRef]
- Fragou, D.; Pakkidi, E.; Aschner, M.; Samanidou, V.; Kovatsi, L. Smoking and DNA methylation: Correlation of methylation with smoking behavior and association with diseases and fœtus development following prenatal exposure. Food Chem. Toxicol. 2019, 129, 312–327. [Google Scholar] [CrossRef]
- Kruman, I.I.; Fowler, A.-K. Impaired one carbon metabolism and DNA methylation in alcohol toxicity. J. Neurochem. 2014, 129, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Miousse, I.R.; Tobacyk, J.; Melnyk, S.; James, S.J.; Cheema, A.K.; Boerma, M.; Hauer-Jensen, M.; Koturbash, I. One-carbon metabolism and ionizing radiation: A multifaceted interaction. Biomol. Concepts 2017, 8, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Fawal, M.-A.; Jungas, T.; Kischel, A.; Audouard, C.; Iacovoni, J.S.; Davy, A. Cross Talk between One-Carbon Metabolism, Eph Signaling, and Histone Methylation Promotes Neural Stem Cell Differentiation. Cell Rep. 2018, 23, 2864–2873.e7. [Google Scholar] [CrossRef]
- Korsmo, H.W.; Jiang, X. One carbon metabolism and early development: A diet-dependent destiny. Trends Endocrinol. Metab. 2021, 32, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Jungas, T.T.; Ohayon, D.; Audouard, C.; Ye, T.; Fawal, M.-A.; Davy, A. Dihydrofolate reductase activity controls neurogenic transitions in the developing neocortex. Development 2023, 150, dev201696. [Google Scholar] [CrossRef]
- Hendricks, E.; Quihuis, A.M.; Hung, S.-T.; Chang, J.; Dorjsuren, N.; Der, B.; Staats, K.A.; Shi, Y.; Maria, N.S.S.; Jacobs, R.E.; et al. The C9ORF72 repeat expansion alters neurodevelopment. Cell Rep. 2023, 42, 112983. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.C.; Schuck, T.; Wheeler, J.M.; Robinson, L.C.; Trojanowski, J.Q.; Lee, V.M.Y.; Schellenberg, G.D. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010, 119, 409–419. [Google Scholar] [CrossRef]
- Scekic-Zahirovic, J.; Sendschied, O.; El Oussini, H.; Jambeau, M.; Sun, Y.; Mersmann, S.; Wagner, M.; Dieterlé, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 2016, 35, 1077–1097. [Google Scholar] [CrossRef]
| Tissue/Model | Observations | References |
|---|---|---|
| Post-mortem motor cortex of sALS and controls |
| [49] |
| Post-mortem LMN fom sALS and controls |
| |
| NSC34 cell line |
| |
| Post-mortem LMN from sALS and C9-ALS and controls |
| [58] |
| Post-mortem upper and lower cortical neurons from C9-ALS patients and controls |
| [60] |
| FUS iPSC-derived LMN |
| [59] |
| Transgenic hSOD1G93A and hSOD1G37R mouse spinal cord and skeletal muscle |
| [57] |
| Transgenic hSOD1G93A |
|
| Targets and Drugs | Effects | References |
|---|---|---|
| DNMT inhibition | ||
|
| [123] |
|
| [57] [123] |
| KMT inhibition | ||
|
| [124] |
|
| [125] |
| PRMT inhibition | ||
|
| [113] [114] |
| KDM inhibition | ||
|
| [102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernan-Godoy, M.; Rouaux, C. From Environment to Gene Expression: Epigenetic Methylations and One-Carbon Metabolism in Amyotrophic Lateral Sclerosis. Cells 2024, 13, 967. https://doi.org/10.3390/cells13110967
Hernan-Godoy M, Rouaux C. From Environment to Gene Expression: Epigenetic Methylations and One-Carbon Metabolism in Amyotrophic Lateral Sclerosis. Cells. 2024; 13(11):967. https://doi.org/10.3390/cells13110967
Chicago/Turabian StyleHernan-Godoy, Marina, and Caroline Rouaux. 2024. "From Environment to Gene Expression: Epigenetic Methylations and One-Carbon Metabolism in Amyotrophic Lateral Sclerosis" Cells 13, no. 11: 967. https://doi.org/10.3390/cells13110967