
WISP-1 Regulates Cardiac Fibrosis by Promoting Cardiac Fibroblasts’ Activation and Collagen Processing



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. In Vitro Sample Collection and Preparation
2.3. Western Blotting
2.4. Quantitative Polymerase Chain Reaction
2.5. Silencing RNA
2.6. Immunocytochemistry
2.7. Kinetic Cell Motility Assay
2.8. Animals
2.9. Infusion of Angiotensin II
2.10. Immunohistochemistry
2.11. Enzyme-Linked Immunosorbent Assay (ELISA)
2.12. Statistical Analysis
3. Results
3.1. WISP-1 Protein Induced Type I Collagen Processing
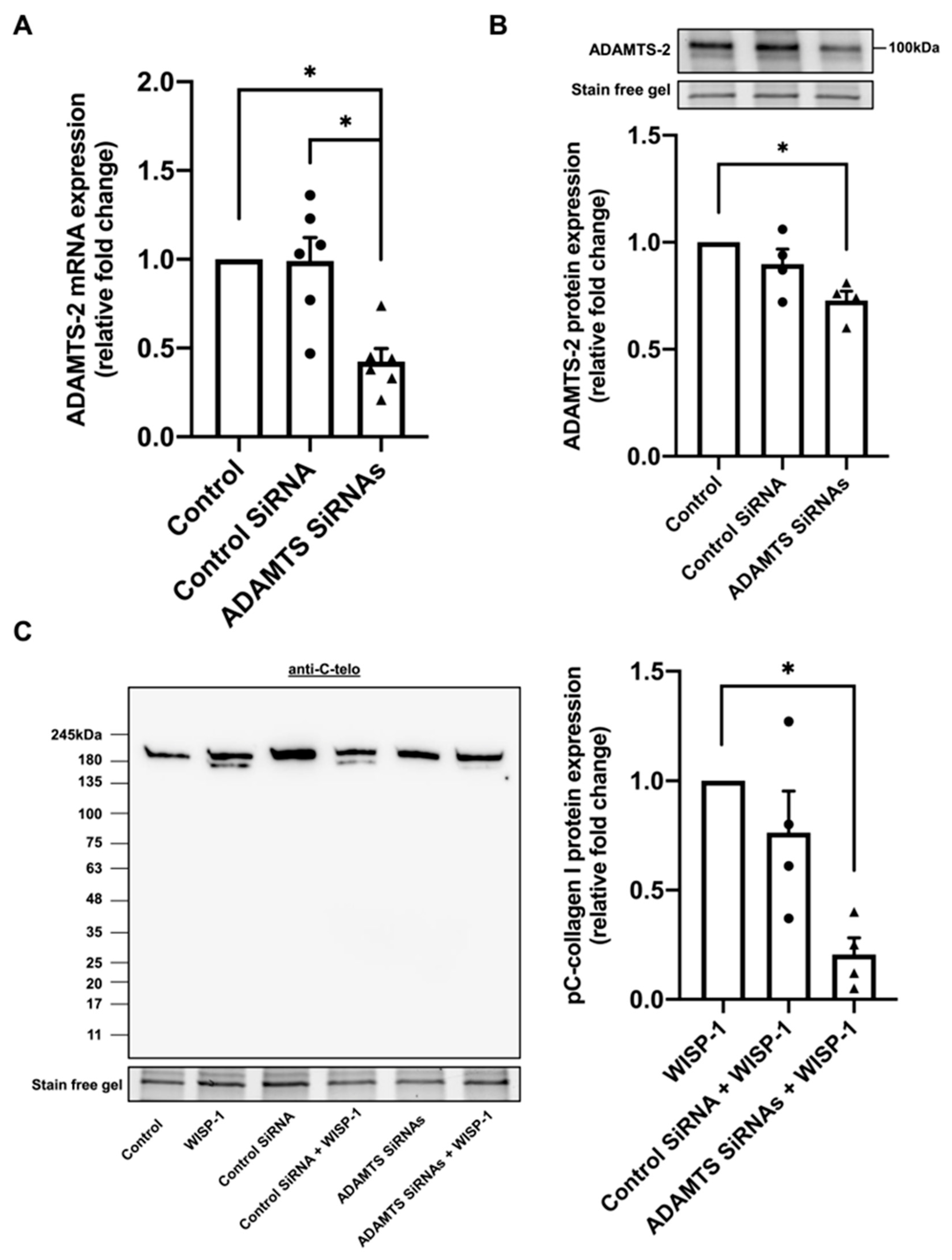
3.2. Silencing of ADAMTS-2 Inhibited WISP-1 Protein-Induced Type I Collagen Processing
3.3. WISP-1 Protein-Induced Type I Collagen Processing Is Independent of Collagenase MMPs
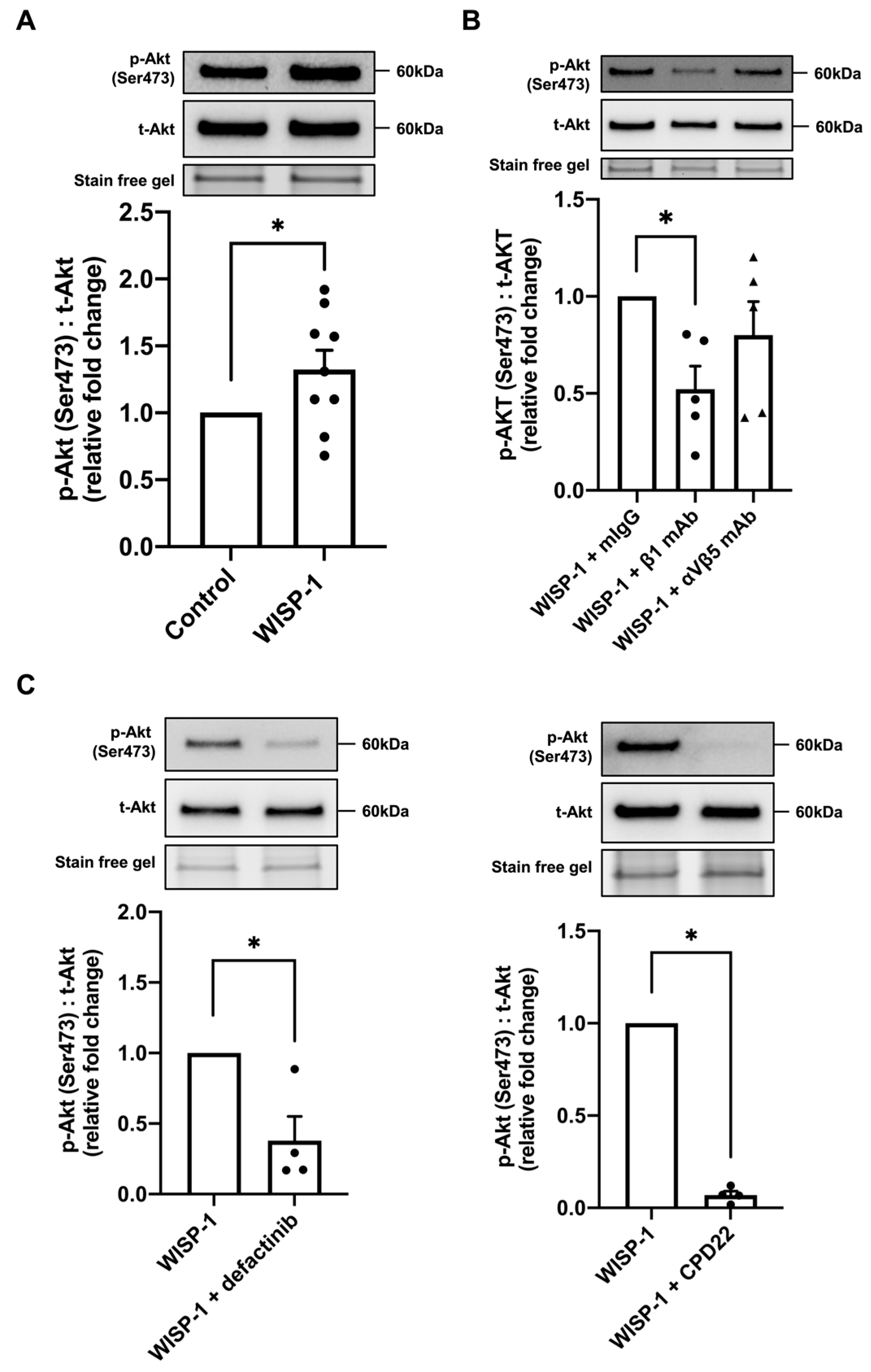
3.4. WISP-1 Protein Induced Akt Phosphorylation via Integrin β1/Focal Adhesion Kinase (FAK)/Integrin-Linked Kinase (ILK)
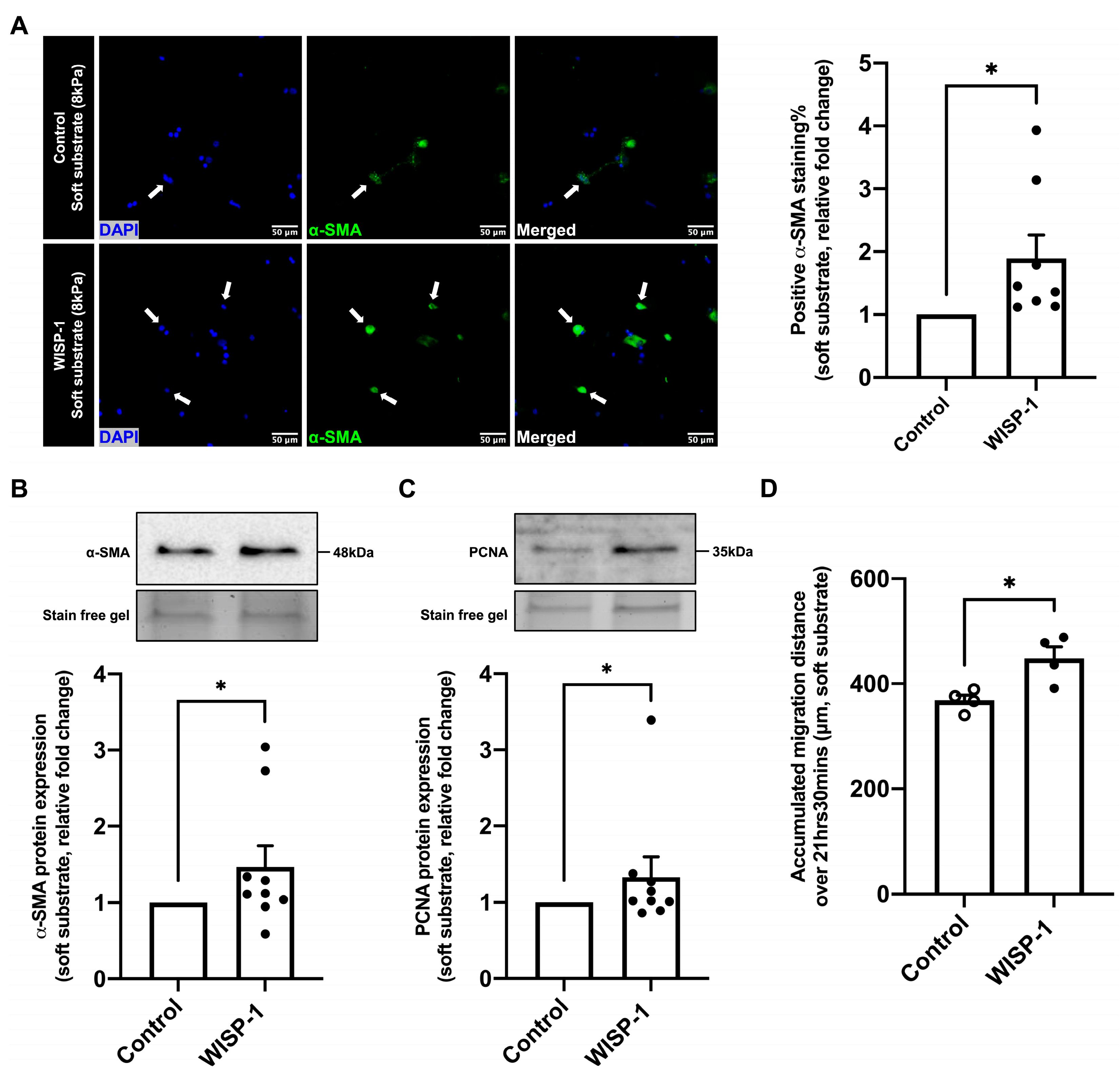
3.5. WISP-1 Protein Promoted HCFs Activation
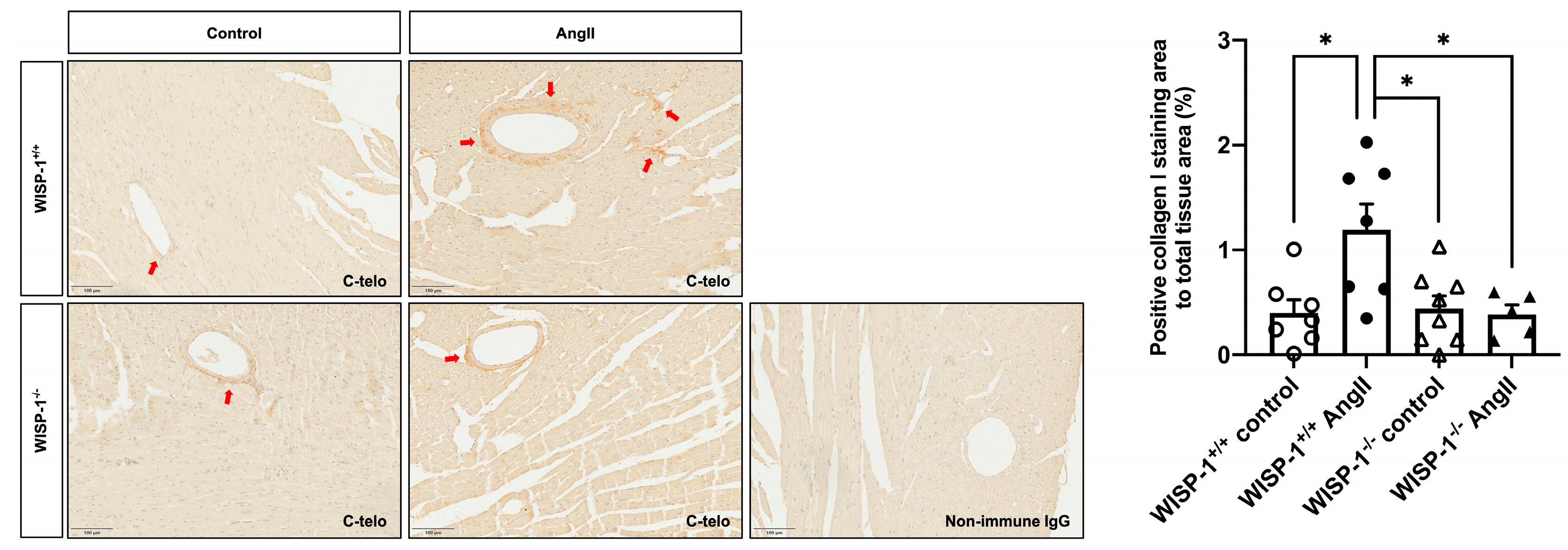
3.6. WISP-1 Deficiency Mice Attenuated AngII-Induced Coronary Artery Perivascular Collagen Deposition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation. Circ. Res. 2020, 127, 427–447. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schellings, M.W.; Vanhoutte, D.; van Almen, G.C.; Swinnen, M.; Leenders, J.J.; Kubben, N.; van Leeuwen, R.E.; Hofstra, L.; Heymans, S.; Pinto, Y.M. Syndecan-1 amplifies angiotensin II-induced cardiac fibrosis. Hypertension 2010, 55, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Bagnost, T.; Ma, L.; da Silva, R.F.; Rezakhaniha, R.; Houdayer, C.; Stergiopulos, N.; Andre, C.; Guillaume, Y.; Berthelot, A.; Demougeot, C. Cardiovascular effects of arginase inhibition in spontaneously hypertensive rats with fully developed hypertension. Cardiovasc. Res. 2010, 87, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Schwartzkopff, B.; Motz, W.; Frenzel, H.; Vogt, M.; Knauer, S.; Strauer, B.E. Structural and functional alterations of the intramyocardial coronary arterioles in patients with arterial hypertension. Circulation 1993, 88, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Aoki, T.; Fukumoto, Y.; Shimokawa, H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J. Cardiol. 2012, 60, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Boutouyrie, P. The structural factor of hypertension: Large and small artery alterations. Circ. Res. 2015, 116, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, D.; Yildirim, I.; Ciftci, O.; Ozer, I.; Caliskan, M.; Gullu, H.; Muderrisoglu, H. Effects of normal blood pressure, prehypertension, and hypertension on coronary microvascular function. Circulation 2007, 115, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Kelshiker, M.A.; Seligman, H.; Howard, J.P.; Rahman, H.; Foley, M.; Nowbar, A.N.; Rajkumar, C.A.; Shun-Shin, M.J.; Ahmad, Y.; Sen, S.; et al. Coronary flow reserve and cardiovascular outcomes: A systematic review and meta-analysis. Eur. Heart J. 2022, 43, 1582–1593. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, F.D.; Zhou, J.; Chen, E.Q. Molecular Mechanisms and Potential New Therapeutic Drugs for Liver Fibrosis. Front. Pharmacol. 2022, 13, 787748. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Koudelka, A.; Cechova, V.; Rojas, M.; Mitash, N.; Bondonese, A.; St Croix, C.; Ross, M.A.; Freeman, B.A. Fatty acid nitroalkene reversal of established lung fibrosis. Redox Biol. 2022, 50, 102226. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ruiz-Ortega, M.; Lamas, S.; Ortiz, A. Antifibrotic Agents for the Management of CKD: A Review. Am. J. Kidney Dis. 2022, 80, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Sprangers, S.; Everts, V. Molecular pathways of cell-mediated degradation of fibrillar collagen. Matrix Biol. 2019, 75–76, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Weber, K.T. Cardiac interstitium in health and disease: The fibrillar collagen network. J. Am. Coll. Cardiol. 1989, 13, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Canty, E.G.; Kadler, K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005, 118 Pt 7, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Trackman, P.C. Diverse biological functions of extracellular collagen processing enzymes. J. Cell. Biochem. 2005, 96, 927–937. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bekhouche, M.; Colige, A. The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biol. 2015, 44–46, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Kessler, E.; Takahara, K.; Biniaminov, L.; Brusel, M.; Greenspan, D.S. Bone morphogenetic protein-1: The type I procollagen C-proteinase. Science 1996, 271, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shi, Y.; O’Brien, J.E.; Fard, A.; Zalewski, A. Transforming growth factor-beta 1 expression and myofibroblast formation during arterial repair. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Pennica, D.; Swanson, T.A.; Welsh, J.W.; Roy, M.A.; Lawrence, D.A.; Lee, J.; Brush, J.; Taneyhill, L.A.; Deuel, B.; Lew, M.; et al. WISP genes are members of the connective tissue growth factor family that are up-regulated in wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc. Natl. Acad. Sci. USA 1998, 95, 14717–14722. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, L.; Corcoran, R.B.; Welsh, J.W.; Pennica, D.; Levine, A.J. WISP-1 is a Wnt-1- and beta-catenin-responsive oncogene. Genes Dev. 2000, 14, 585–595. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Colston, J.T.; de la Rosa, S.D.; Koehler, M.; Gonzales, K.; Mestril, R.; Freeman, G.L.; Bailey, S.R.; Chandrasekar, B. Wnt-induced secreted protein-1 is a prohypertrophic and profibrotic growth factor. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1839–H1846. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; LaCanna, R.; Ma, H.Y.; N’Diaye, E.N.; Gierke, S.; Caplazi, P.; Sagolla, M.; Huang, Z.; Lucio, L.; Arlantico, A.; et al. A WISP1 antibody inhibits MRTF signaling to prevent the progression of established liver fibrosis. Cell Metab. 2022, 34, 1377–1393.e8. [Google Scholar] [CrossRef] [PubMed]
- Königshoff, M.; Kramer, M.; Balsara, N.; Wilhelm, J.; Amarie, O.V.; Jahn, A.; Rose, F.; Fink, L.; Seeger, W.; Schaefer, L.; et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J. Clin. Investig. 2009, 119, 772–787. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhong, X.; Tu, Y.J.; Li, Y.; Zhang, P.; Wang, W.; Chen, S.S.; Li, L.; Chung, A.C.; Lan, H.Y.; Chen, H.Y.; et al. Serum levels of WNT1-inducible signaling pathway protein-1 (WISP-1): A noninvasive biomarker of renal fibrosis in subjects with chronic kidney disease. Am. J. Transl. Res. 2017, 9, 2920–2932. [Google Scholar] [PubMed] [PubMed Central]
- Soon, L.L.; Yie, T.A.; Shvarts, A.; Levine, A.J.; Su, F.; Tchou-Wong, K.M. Overexpression of WISP-1 down-regulated motility and invasion of lung cancer cells through inhibition of Rac activation. J. Biol. Chem. 2003, 278, 11465–11470. [Google Scholar] [CrossRef] [PubMed]
- Stephens, S.; Palmer, J.; Konstantinova, I.; Pearce, A.; Jarai, G.; Day, E. A functional analysis of Wnt inducible signalling pathway protein -1 (WISP-1/CCN4). J. Cell Commun. Signal. 2015, 9, 63–72. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Deng, W.; Fernandez, A.; McLaughlin, S.L.; Klinke, D.J. WNT1-inducible signaling pathway protein 1 (WISP1/CCN4) stimulates melanoma invasion and metastasis by promoting the epithelial-mesenchymal transition. J. Biol. Chem. 2019, 294, 5261–5280. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, C.L.; Tsai, H.C.; Chen, Z.W.; Wu, C.M.; Li, T.M.; Fong, Y.C.; Tang, C.H. Ras activation mediates WISP-1-induced increases in cell motility and matrix metalloproteinase expression in human osteosarcoma. Cell Signal. 2013, 25, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Tzeng, H.E.; Huang, C.Y.; Huang, Y.L.; Tsai, C.H.; Wang, S.W.; Wang, P.C.; Chang, A.C.; Fong, Y.C.; Tang, C.H. WISP-1 positively regulates angiogenesis by controlling VEGF-A expression in human osteosarcoma. Cell Death Dis. 2017, 8, e2750. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Williams, H.; Mill, C.A.; Monk, B.A.; Hulin-Curtis, S.; Johnson, J.L.; George, S.J. Wnt2 and WISP-1/CCN4 Induce Intimal Thickening via Promotion of Smooth Muscle Cell Migration. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.; Wadey, K.S.; Frankow, A.; Blythe, H.C.; Forbes, T.; Johnson, J.L.; George, S.J. Aneurysm severity is suppressed by deletion of CCN4. J. Cell Commun. Signal. 2021, 15, 421–432. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. BMJ Open Sci. 2020, 4, e100115. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lyon, C.A.; Williams, H.; Bianco, R.; Simmonds, S.J.; Brown, B.A.; Wadey, K.S.; Smith, F.C.T.; Johnson, J.L.; George, S.J. Aneurysm Severity is Increased by Combined Mmp-7 Deletion and N-cadherin Mimetic (EC4-Fc) Over-Expression. Sci. Rep. 2017, 7, 17342. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hsu, H.S.; Liu, C.C.; Lin, J.H.; Hsu, T.W.; Hsu, J.W.; Su, K.; Hung, S.C. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci. Rep. 2017, 7, 14272. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, G.; Li, Y.Y.; Sun, J.E.; Lin, W.H.; Zhou, R.X. ILK-PI3K/AKT pathway participates in cutaneous wound contraction by regulating fibroblast migration and differentiation to myofibroblast. Lab. Investig. 2016, 96, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Bujor, A.M.; Pannu, J.; Bu, S.; Smith, E.A.; Muise-Helmericks, R.C.; Trojanowska, M. Akt blockade downregulates collagen and upregulates MMP1 in human dermal fibroblasts. J. Investig. Dermatol. 2008, 128, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Ettehad, D.; Emdin, C.A.; Kiran, A.; Anderson, S.G.; Callender, T.; Emberson, J.; Chalmers, J.; Rodgers, A.; Rahimi, K. Blood pressure lowering for prevention of cardiovascular disease and death: A systematic review and meta-analysis. Lancet 2016, 387, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Rapsomaniki, E.; Timmis, A.; George, J.; Pujades-Rodriguez, M.; Shah, A.D.; Denaxas, S.; White, I.R.; Caulfield, M.J.; Deanfield, J.E.; Smeeth, L.; et al. Blood pressure and incidence of twelve cardiovascular diseases: Lifetime risks, healthy life-years lost, and age-specific associations in 1·25 million people. Lancet 2014, 383, 1899–1911. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, 1269–1324. [Google Scholar] [CrossRef] [PubMed]
- Brilla, C.G.; Pick, R.; Tan, L.B.; Janicki, J.S.; Weber, K.T. Remodeling of the rat right and left ventricles in experimental hypertension. Circ. Res. 1990, 67, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Brilla, C.G.; Janicki, J.S.; Weber, K.T. Impaired diastolic function and coronary reserve in genetic hypertension. Role of interstitial fibrosis and medial thickening of intramyocardial coronary arteries. Circ. Res. 1991, 69, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Broder, C.; Arnold, P.; Vadon-Le Goff, S.; Konerding, M.A.; Bahr, K.; Müller, S.; Overall, C.M.; Bond, J.S.; Koudelka, T.; Tholey, A.; et al. Metalloproteases meprin α and meprin β are C- and N-procollagen proteinases important for collagen assembly and tensile strength. Proc. Natl. Acad. Sci. USA 2013, 110, 14219–14224. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Z.; Li, W.; Chen, S.; Tang, X.X. Role of ADAM and ADAMTS proteases in pathological tissue remodeling. Cell Death Discov. 2023, 9, 447. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kilic, T.; Okuno, K.; Eguchi, S.; Kassiri, Z. Disintegrin and Metalloproteinases (ADAMs [A Disintegrin and Metalloproteinase] and ADAMTSs [ADAMs With a Thrombospondin Motif]) in Aortic Aneurysm. Hypertension 2022, 79, 1327–1338. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fernandes, R.J.; Hirohata, S.; Engle, J.M.; Colige, A.; Cohn, D.H.; Eyre, D.R.; Apte, S.S. Procollagen II amino propeptide processing by ADAMTS-3. Insights on dermatosparaxis. J. Biol. Chem. 2001, 276, 31502–31509. [Google Scholar] [CrossRef] [PubMed]
- Bode, W. A helping hand for collagenases: The haemopexin-like domain. Structure 1995, 3, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Laronha, H.; Caldeira, J. Structure and Function of Human Matrix Metalloproteinases. Cells 2020, 9, 1076. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Colige, A.; Vandenberghe, I.; Thiry, M.; Lambert, C.A.; Van Beeumen, J.; Li, S.W.; Prockop, D.J.; Lapiere, C.M.; Nusgens, B.V. Cloning and characterization of ADAMTS-14, a novel ADAMTS displaying high homology with ADAMTS-2 and ADAMTS-3. J. Biol. Chem. 2002, 277, 5756–5766. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova-Ramich, O.; Loske, J.; Hornemann, S.; Markova, M.; Seebeck, N.; Rosenthal, A.; Klauschen, F.; Castro, J.P.; Buschow, R.; Grune, T.; et al. Hepatic Wnt1 Inducible Signaling Pathway Protein 1 (WISP-1/CCN4) Associates with Markers of Liver Fibrosis in Severe Obesity. Cells 2021, 10, 1048. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Klee, S.; Lehmann, M.; Wagner, D.E.; Baarsma, H.A.; Königshoff, M. WISP1 mediates IL-6-dependent proliferation in primary human lung fibroblasts. Sci. Rep. 2016, 6, 20547. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lu, S.; Liu, H.; Lu, L.; Wan, H.; Lin, Z.; Qian, K.; Yao, X.; Chen, Q.; Liu, W.; Yan, J.; et al. WISP1 overexpression promotes proliferation and migration of human vascular smooth muscle cells via AKT signaling pathway. Eur. J. Pharmacol. 2016, 788, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Overholtzer, M.; Besser, D.; Levine, A.J. WISP-1 attenuates p53-mediated apoptosis in response to DNA damage through activation of the Akt kinase. Genes Dev. 2002, 16, 46–57. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hou, C.H.; Chiang, Y.C.; Fong, Y.C.; Tang, C.H. WISP-1 increases MMP-2 expression and cell motility in human chondrosarcoma cells. Biochem. Pharmacol. 2011, 81, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Chen, P.C.; Lein, M.Y.; Tsao, C.W.; Huang, C.C.; Wang, S.W.; Tang, C.H.; Tung, K.C. WISP-1 promotes VEGF-C-dependent lymphangiogenesis by inhibiting miR-300 in human oral squamous cell carcinoma cells. Oncotarget 2016, 7, 9993–10005. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Casar, B.; Rimann, I.; Kato, H.; Shattil, S.J.; Quigley, J.P.; Deryugina, E.I. In vivo cleaved CDCP1 promotes early tumor dissemination via complexing with activated β1 integrin and induction of FAK/PI3K/Akt motility signaling. Oncogene 2014, 33, 255–268. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Persad, S.; Attwell, S.; Gray, V.; Delcommenne, M.; Troussard, A.; Sanghera, J.; Dedhar, S. Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3207–3212. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Delcommenne, M.; Tan, C.; Gray, V.; Rue, L.; Woodgett, J.; Dedhar, S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 11211–11216. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nho, R.S.; Xia, H.; Kahm, J.; Kleidon, J.; Diebold, D.; Henke, C.A. Role of integrin-linked kinase in regulating phosphorylation of Akt and fibroblast survival in type I collagen matrices through a beta1 integrin viability signaling pathway. J. Biol. Chem. 2005, 280, 26630–26639. [Google Scholar] [CrossRef] [PubMed]
- Snider, P.; Standley, K.N.; Wang, J.; Azhar, M.; Doetschman, T.; Conway, S.J. Origin of cardiac fibroblasts and the role of periostin. Circ. Res. 2009, 105, 934–947. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex Relationship Between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J. Am. Heart Assoc. 2021, 10, e019338. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Baudino, T.A.; Carver, W.; Giles, W.; Borg, T.K. Cardiac fibroblasts: Friend or foe? Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1015–H1026. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Choppe, J.; Kumar, A.; Engler, A.J.; McCulloch, A.D. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol. Biol. Cell. 2017, 28, 1871–1882. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; J Lin, S.C.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brown, J.P.; Don-Wauchope, A.; Douville, P.; Albert, C.; Vasikaran, S.D. Current use of bone turnover markers in the management of osteoporosis. Clin. Biochem. 2022, 109–110, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bhattoa, H.P.; Cavalier, E.; Eastell, R.; Heijboer, A.C.; Jørgensen, N.R.; Makris, K.; Ulmer, C.Z.; Kanis, J.A.; Cooper, C.; Silverman, S.L.; et al. Analytical considerations and plans to standardize or harmonize assays for the reference bone turnover markers PINP and β-CTX in blood. Clin. Chim. Acta 2021, 515, 16–20. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kaye, D.M.; Khammy, O.; Mariani, J.; Maeder, M.T. Relationship of circulating matrix biomarkers to myocardial matrix metabolism in advanced heart failure. Eur. J. Heart Fail. 2013, 15, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.F.; Nigri, M.; Higuchi, M.L.; Pomerantzeff, P.M.; Spina, G.S.; Sampaio, R.O.; Tarasoutchi, F.; Grinberg, M.; Rochitte, C.E. Prognostic significance of myocardial fibrosis quantification by histopathology and magnetic resonance imaging in patients with severe aortic valve disease. J. Am. Coll. Cardiol. 2010, 56, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Manhenke, C.; Ueland, T.; Jugdutt, B.I.; Godang, K.; Aukrust, P.; Dickstein, K.; Ørn, S. The relationship between markers of extracellular cardiac matrix turnover: Infarct healing and left ventricular remodelling following primary PCI in patients with first-time STEMI. Eur. Heart J. 2014, 35, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Iraqi, W.; Rossignol, P.; Angioi, M.; Fay, R.; Nuée, J.; Ketelslegers, J.M.; Vincent, J.; Pitt, B.; Zannad, F. Extracellular cardiac matrix biomarkers in patients with acute myocardial infarction complicated by left ventricular dysfunction and heart failure: Insights from the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) study. Circulation 2009, 119, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.; Ueland, T.; Aukrust, P.; Nilsen, D.W.T.; Grundt, H.; Staines, H.; Pönitz, V.; Kontny, F. Procollagen type 1 N-terminal propeptide is associated with adverse outcome in acute chest pain of suspected coronary origin. Front. Cardiovasc. Med. 2023, 10, 1191055. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Williams, H.; Jackson, M.L.; Johnson, J.L.; George, S.J. WISP-1 Regulates Cardiac Fibrosis by Promoting Cardiac Fibroblasts’ Activation and Collagen Processing. Cells 2024, 13, 989. https://doi.org/10.3390/cells13110989
Li Z, Williams H, Jackson ML, Johnson JL, George SJ. WISP-1 Regulates Cardiac Fibrosis by Promoting Cardiac Fibroblasts’ Activation and Collagen Processing. Cells. 2024; 13(11):989. https://doi.org/10.3390/cells13110989
Chicago/Turabian StyleLi, Ze, Helen Williams, Molly L. Jackson, Jason L. Johnson, and Sarah J. George. 2024. "WISP-1 Regulates Cardiac Fibrosis by Promoting Cardiac Fibroblasts’ Activation and Collagen Processing" Cells 13, no. 11: 989. https://doi.org/10.3390/cells13110989
APA StyleLi, Z., Williams, H., Jackson, M. L., Johnson, J. L., & George, S. J. (2024). WISP-1 Regulates Cardiac Fibrosis by Promoting Cardiac Fibroblasts’ Activation and Collagen Processing. Cells, 13(11), 989. https://doi.org/10.3390/cells13110989