
Primary Ciliary Dyskinesia Associated Disease-Causing Variants in CCDC39 and CCDC40 Cause Axonemal Absence of Inner Dynein Arm Heavy Chains DNAH1, DNAH6, and DNAH7


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Samples and Ethics Statement
2.2. Immunofluorescence (IF) Staining of Human Respiratory Epithelium
2.3. Western Blotting
3. Results
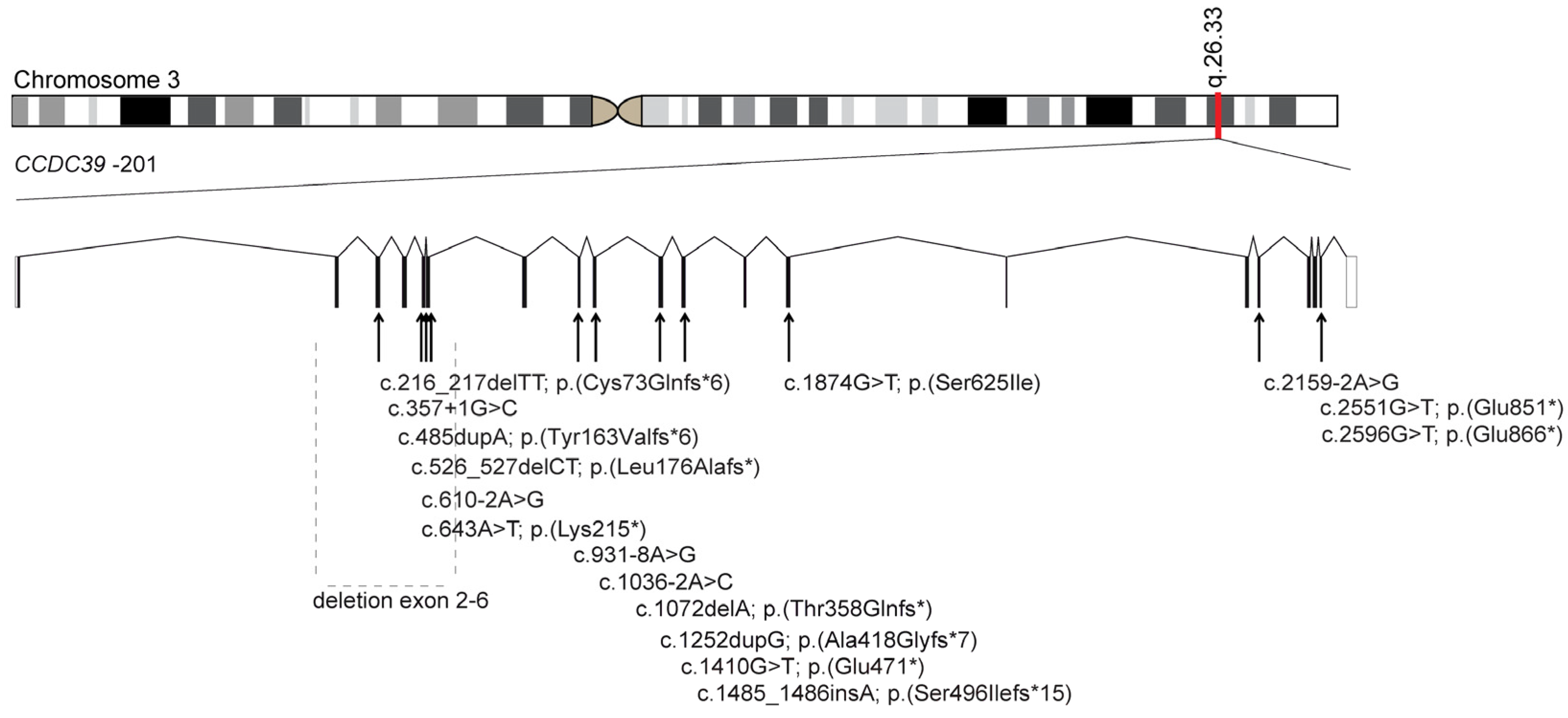
3.1. Identification of Bi-Allelic Disease-Causing CCDC39 Variants in PCD Individuals
3.2. Examination of Pathogenicity of the Novel Disease-Causing CCDC39 Variants
3.3. Identification of Bi-Allelic Disease-Causing CCDC40 Variants in PCD Individuals
3.4. Examination of Pathogenicity for the Novel Disease-Causing CCDC40 Variants
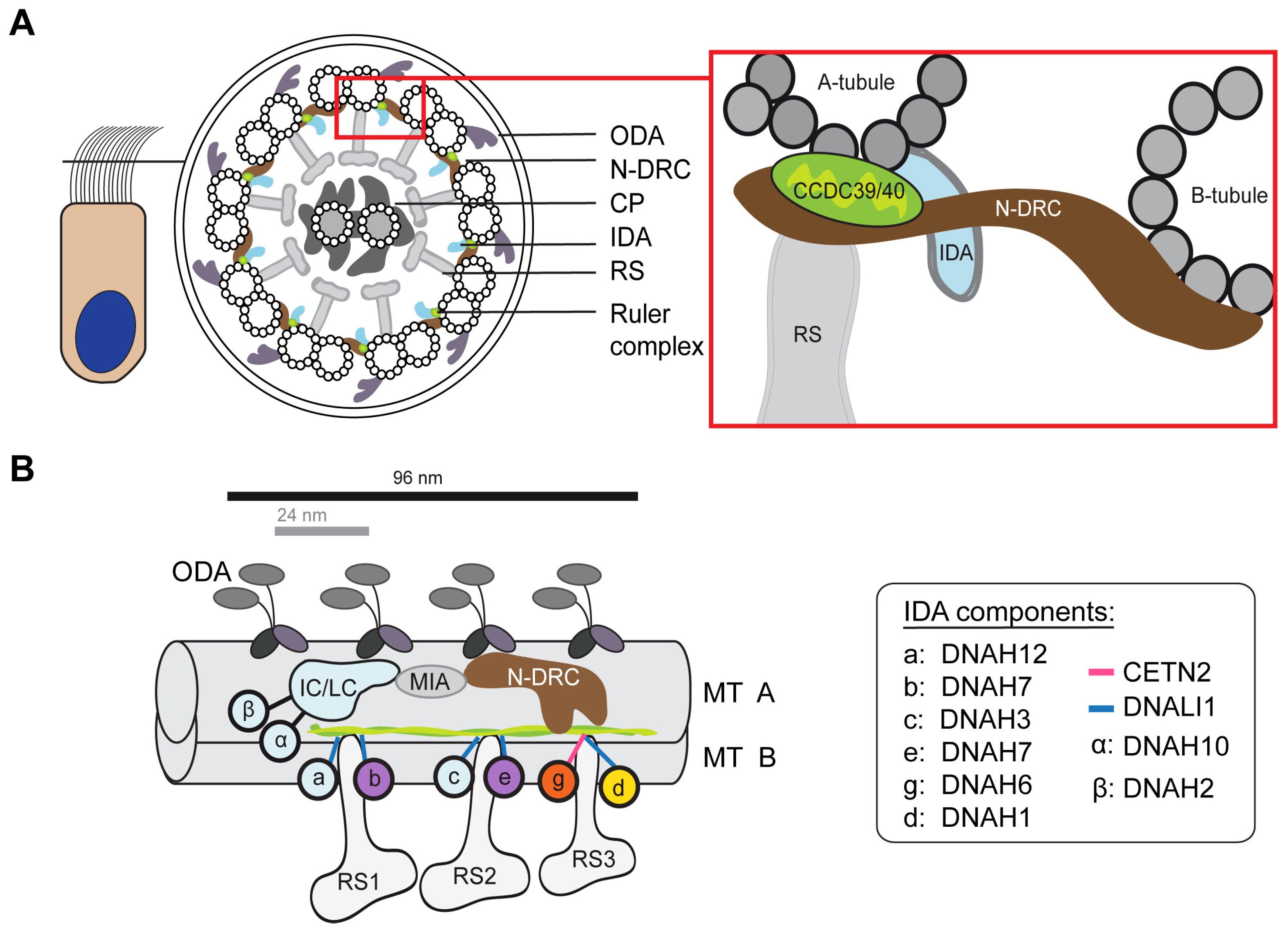
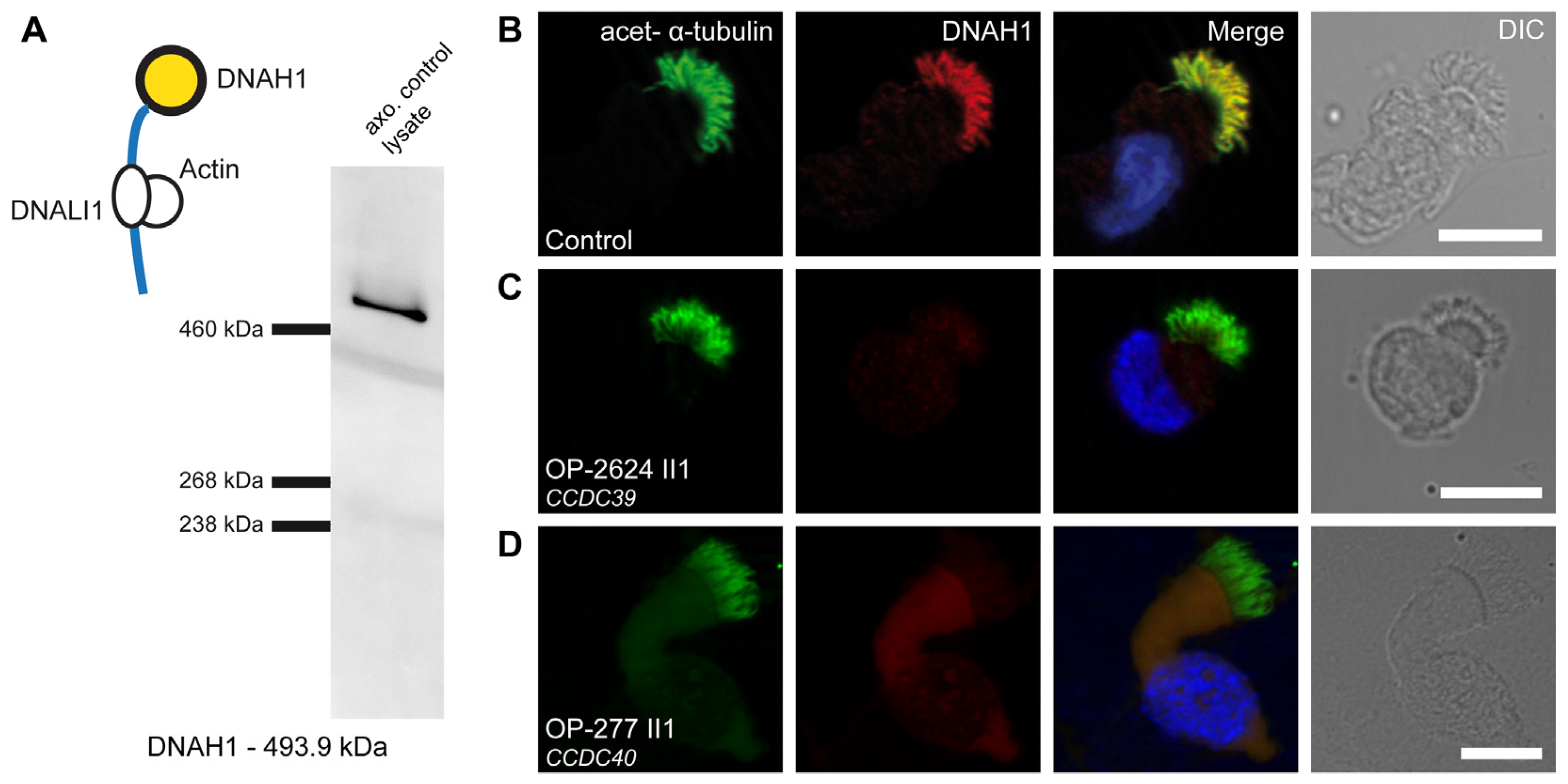
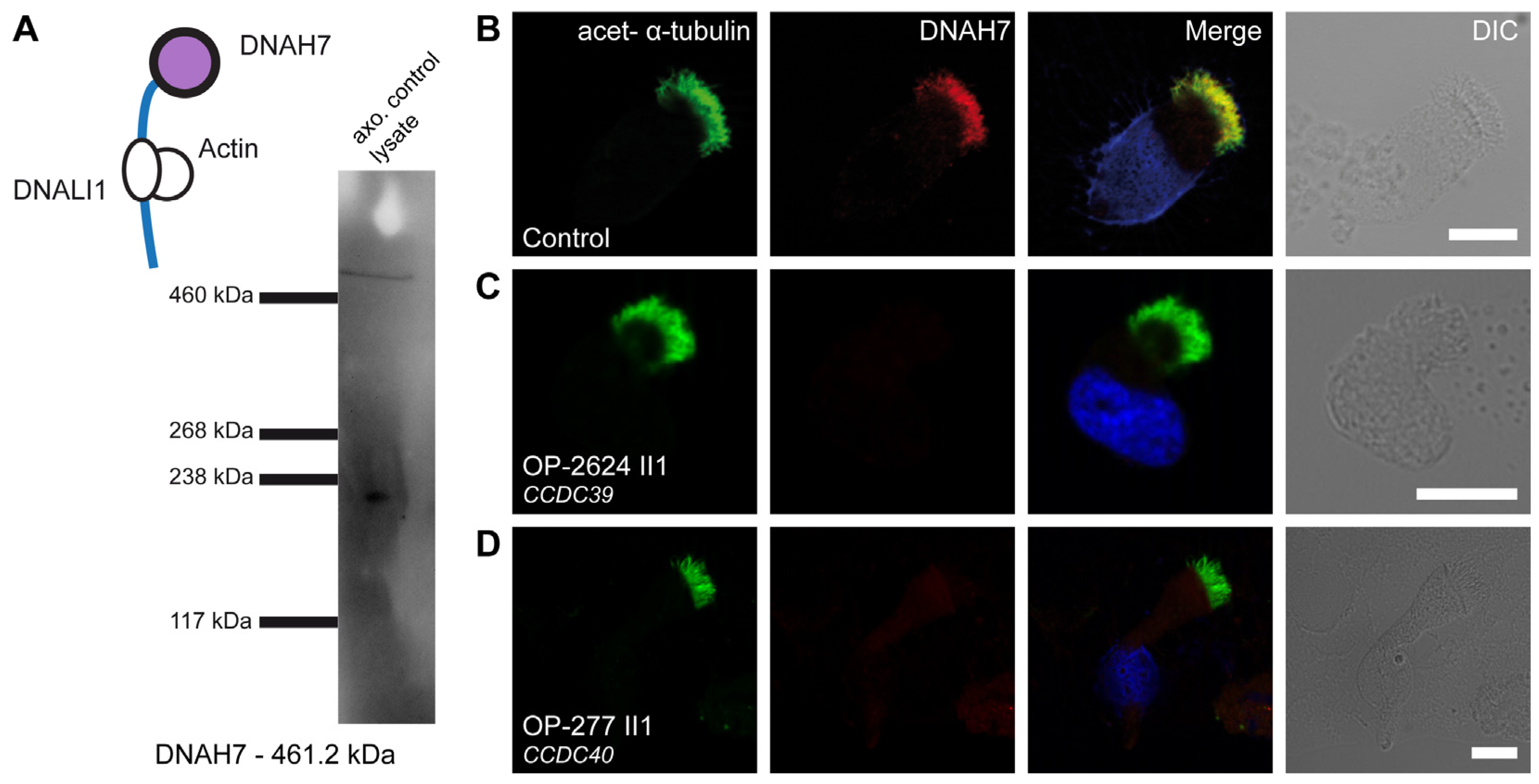
3.5. Defects of the 96 nm Axonemal Ruler Cause Deficiency of IDA Heavy Chains DNAH1, DNAH6, and DNAH7
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallmeier, J.; Nielsen, K.G.; Kuehni, C.E.; Lucas, J.S.; Leigh, M.W.; Zariwala, M.A.; Omran, H. Motile ciliopathies. Nat. Rev. Dis. Primers 2020, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Raidt, J.; Riepenhausen, S.; Pennekamp, P.; Olbrich, O.; Amirav, I.; Athanazio, R.A.; Aviram, M.; Balinotti, J.E.; Bar-On, O.; Bode, S.F.N.; et al. Analyses of 1,236 genotyped primary ciliary dyskinesia individuals identify regional clusters of distinct DNA variants and significant genotype-phenotype correlations. Eur. Respir. J. 2024. submitted; in revision. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Zariwala, M.; Leigh, M. Primary Ciliary Dyskinesia. Clin. Chest Med. 2016, 37, 449–461. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sironen, A.; Shoemark, A.; Patel, M.; Loebinger, M.R.; Mitchison, H.M. Sperm defects in primary ciliary dyskinesia and related causes of male infertility. Cell. Mol. Life Sci. 2020, 77, 2029–2048. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yamamoto, R.; Hwang, J.; Ishikawa, T.; Kon, T.; Sale, W.S. Composition and function of ciliary inner-dynein-arm subunits studied in Chlamydomonas reinhardtii. Cytoskeleton 2021, 78, 77–96. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Oda, T.; Yanagisawa, H.; Kamiya, R.; Kikkawa, M. A molecular ruler determines the repeat length in eukaryotic cilia and flagella. Science 2014, 346, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Walton, T.; Gui, M.; Velkova, S.; Fassad, M.R.; Hirst, R.A.; Haarman, E.; O’Callaghan, C.; Bottier, M.; Burgoyne, T.; Mitchison, H.M.; et al. Axonemal structures reveal mechanoregulatory and disease mechanisms. Nature 2023, 618, 625–633. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Merveille, A.C.; Davis, E.E.; Becker-Heck, A.; Legendre, M.; Amirav, I.; Bataille, G.; Belmont, J.; Beydon, N.; Billen, F.; Clément, A.; et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011, 43, 72–78. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Becker-Heck, A.; Zohn, I.E.; Okabe, N.; Pollock, A.; Lenhart, K.B.; Sullivan-Brown, J.; McSheene, J.; Loges, N.T.; Olbrich, H.; Haeffner, K.; et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011, 43, 79–84. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blanchon, S.; Legendre, M.; Copin, B.; Duquesnoy, P.; Montantin, G.; Kott, E.; Dastot, F.; Jeanson, L.; Cachanado, M.; Rousseau, A.; et al. Delineation of CCDC39/CCDC40 mutation spectrum and associated phenotypes in primary ciliary dyskinesia. J. Med. Genet. 2012, 49, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Antony, D.; Becker-Heck, A.; Zariwala, M.A.; Schmidts, M.; Onoufriadis, A.; Forouhan, M.; Wilson, R.; Taylor-Cox, T.; Dewar, A.; Jackson, C.; et al. Mutations in CCDC39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Hum. Mutat. 2013, 34, 462–472. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Raidt, J.; Wallmeier, J.; Hjeij, R.; Onnebrink, J.G.; Pennekamp, P.; Loges, N.T.; Olbrich, H.; Häffner, K.; Dougherty, G.W.; Omran, H.; et al. Ciliary beat pattern and frequency in genetic variants of primary ciliary dyskinesia. Eur. Respir. J. 2014, 44, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.D.; Ferkol, T.W.; Rosenfeld, M.; Lee, H.S.; Dell, S.D.; Sagel, S.D.; Milla, C.; Zariwala, M.A.; Pittman, J.E.; Shapiro, A.J.; et al. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am. J. Respir. Crit. Care Med. 2015, 191, 316–324. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, D.; Liang, Y.; Li, J.; Zhang, X.; Zheng, R.; Wang, X.; Zhang, H.; Shen, Y. A novel CCDC39 mutation causes multiple morphological abnormalities of the flagella in a primary ciliary dyskinesia patient. Reprod. Biomed. Online 2021, 43, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, B.; Lei, C.; Yang, D.; Ding, S.; Lu, C.; Wang, L.; Guo, T.; Wang, R.; Luo, H. Novel Compound Heterozygous Variants in CCDC40 Associated with Primary Ciliary Dyskinesia and Multiple Morphological Abnormalities of the Sperm Flagella. Pharmacogenomics Pers. Med. 2022, 15, 341–350. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aprea, I.; Wilken, A.; Krallmann, C.; Nöthe-Menchen, T.; Olbrich, H.; Loges, N.T.; Dougherty, G.W.; Bracht, D.; Brenker, C.; Kliesch, S.; et al. Pathogenic gene variants in CCDC39, CCDC40, RSPH1, RSPH9, HYDIN, and SPEF2 cause defects of sperm flagella composition and male infertility. Front. Genet. 2023, 14, 1117821. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Olbrich, H.; Cremers, C.; Loges, N.T.; Werner, C.; Nielsen, K.G.; Marthin, J.K.; Philipsen, M.; Wallmeier, J.; Pennekamp, P.; Menchen, T.; et al. Loss-of-Function GAS8 Mutations Cause Primary Ciliary Dyskinesia and Disrupt the Nexin-Dynein Regulatory Complex. Am. J. Hum. Genet. 2015, 97, 546–554. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Genome Aggregation Database (gnomAD). Available online: http://gnomad.broadinstitute.org (accessed on 1 February 2024).
- SpliceAI Lookup. Available online: https://broadinstitute.github.io/SpliceAI-lookup-dev/index.html (accessed on 1 February 2024).
- ClustalOmega Alignment Analysis. Available online: https://www.ebi.ac.uk/jdispatcher/msa/clustalo (accessed on 1 June 2023).
- Protein Variation Effect Analyzer (Provean). Available online: http://provean.jcvi.org (accessed on 1 December 2023).
- Polymorphism Phenotyping v2 (PolyPhen-2). Available online: http://genetics.bwh.harvard.edu/pph2/ (accessed on 1 March 2023).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Yamamoto, R.; Obbineni, J.M.; Alford, L.M.; Ide, T.; Owa, M.; Hwang, J.; Kon, T.; Inaba, K.; James, N.; King, S.M.; et al. Chlamydomonas DYX1C1/PF23 is essential for axonemal assembly and proper morphology of inner dynein arms. PLoS Genet. 2017, 13, e1006996, Erratum in PLoS Genet. 2017, 13, e1007063. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brody, S.L.; Pan, J.; Huang, T.; Xu, J.; Xu, H.; Koenitizer, J.; Brennan, S.K.; Nanjundappa, R.; Saba, T.G.; Berical, A.; et al. Loss of an extensive ciliary connectome induces proteostasis and cell fate switching in a severe motile ciliopathy. bioRxiv 2024. [Google Scholar] [CrossRef]
- Khelifa, M.; Coutton, C.; Zouari, R.; Karaouzène, T.; Rendu, J.; Bidart, M.; Yassine, S.; Pierre, V.; Delaroche, J.; Hennebicq, S.; et al. Mutations in DNAH1, which encodes an inner arm heavy chain dynein, lead to male infertility from multiple morphological abnormalities of the sperm flagella. Am. J. Hum. Genet. 2014, 94, 95–104. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tu, C.; Nie, H.; Meng, L.; Yuan, S.; He, W.; Luo, A.; Li, H.; Li, W.; Du, J.; Lu, G.; et al. Identification of DNAH6 mutations in infertile men with multiple morphological abnormalities of the sperm flagella. Sci. Rep. 2019, 9, 15864. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gao, Y.; Liu, L.; Shen, Q.; Fu, F.; Xu, C.; Geng, H.; Lv, M.; Li, K.; Tang, D.; Song, B.; et al. Loss of function mutation in DNAH7 induces male infertility associated with abnormalities of the sperm flagella and mitochondria in human. Clin. Genet. 2022, 102, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Keiser, N.W.; Cant, E.; Sitaraman, S.; Shoemark, A.; Limberis, M.P. Restoring Ciliary Function: Gene Therapeutics for Primary Ciliary Dyskinesia. Hum. Gene Ther. 2023, 34, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Fliegauf, M.; Olbrich, H.; Horvath, J.; Wildhaber, J.H.; Zariwala, M.A.; Kennedy, M.; Knowles, M.R.; Omran, H. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 2005, 171, 1343–1349. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Loges, N.T.; Antony, D.; Maver, A.; Deardorff, M.A.; Güleç, E.Y.; Gezdirici, A.; Nöthe-Menchen, T.; Höben, I.M.; Jelten, L.; Frank, D.; et al. Recessive DNAH9 Loss-of-Function Mutations Cause Laterality Defects and Subtle Respiratory Ciliary-Beating Defects. Am. J. Hum. Genet. 2018, 103, 995–1008. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shoemark, A.; Burgoyne, T.; Kwan, R.; Dixon, M.; Patel, M.P.; Rogers, A.V.; Onoufriadis, A.; Scully, J.; Daudvohra, F.; Cullup, T.; et al. Primary ciliary dyskinesia with normal ultrastructure: Three-dimensional tomography detects absence of DNAH11. Eur. Respir. J. 2018, 51, 1701809. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilken, A.; Höben, I.M.; Wolter, A.; Loges, N.T.; Olbrich, H.; Aprea, I.; Dworniczak, B.; Raidt, J.; Omran, H. Primary Ciliary Dyskinesia Associated Disease-Causing Variants in CCDC39 and CCDC40 Cause Axonemal Absence of Inner Dynein Arm Heavy Chains DNAH1, DNAH6, and DNAH7. Cells 2024, 13, 1200. https://doi.org/10.3390/cells13141200
Wilken A, Höben IM, Wolter A, Loges NT, Olbrich H, Aprea I, Dworniczak B, Raidt J, Omran H. Primary Ciliary Dyskinesia Associated Disease-Causing Variants in CCDC39 and CCDC40 Cause Axonemal Absence of Inner Dynein Arm Heavy Chains DNAH1, DNAH6, and DNAH7. Cells. 2024; 13(14):1200. https://doi.org/10.3390/cells13141200
Chicago/Turabian StyleWilken, Alina, Inga Marlena Höben, Alexander Wolter, Niki Tomas Loges, Heike Olbrich, Isabella Aprea, Bernd Dworniczak, Johanna Raidt, and Heymut Omran. 2024. "Primary Ciliary Dyskinesia Associated Disease-Causing Variants in CCDC39 and CCDC40 Cause Axonemal Absence of Inner Dynein Arm Heavy Chains DNAH1, DNAH6, and DNAH7" Cells 13, no. 14: 1200. https://doi.org/10.3390/cells13141200