
Visual Evidence for the Recruitment of Four Enzymes with RNase Activity to the Bacillus subtilis Replication Forks

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Strain Construction
2.2. Growth Conditions
2.3. Western Blot
2.4. Fluorescence Microscopy
2.5. Single-Molecule Tracking (SMT)
3. Results
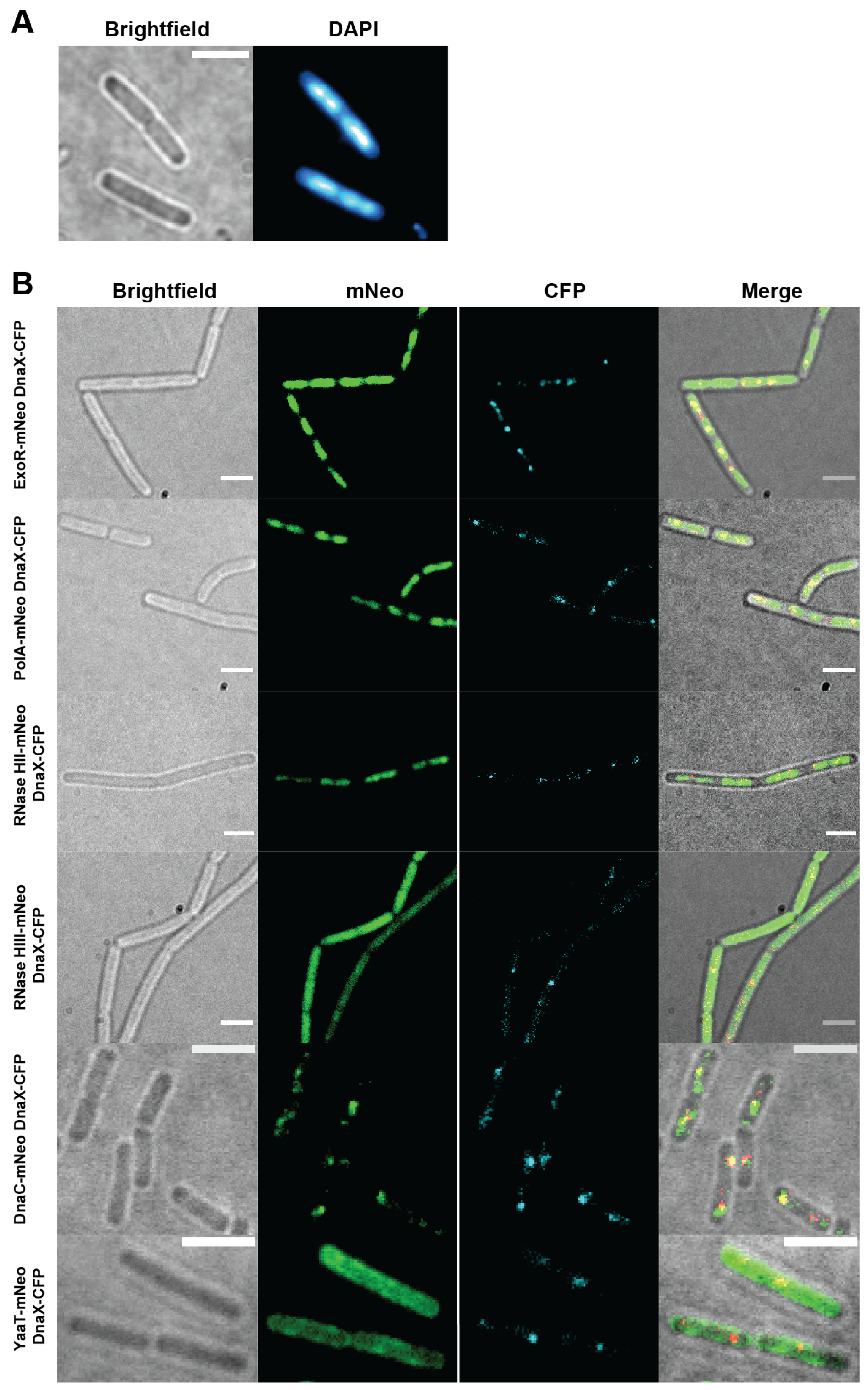
3.1. PolA, ExoR, and RNase HII Show Nucleoid Localization
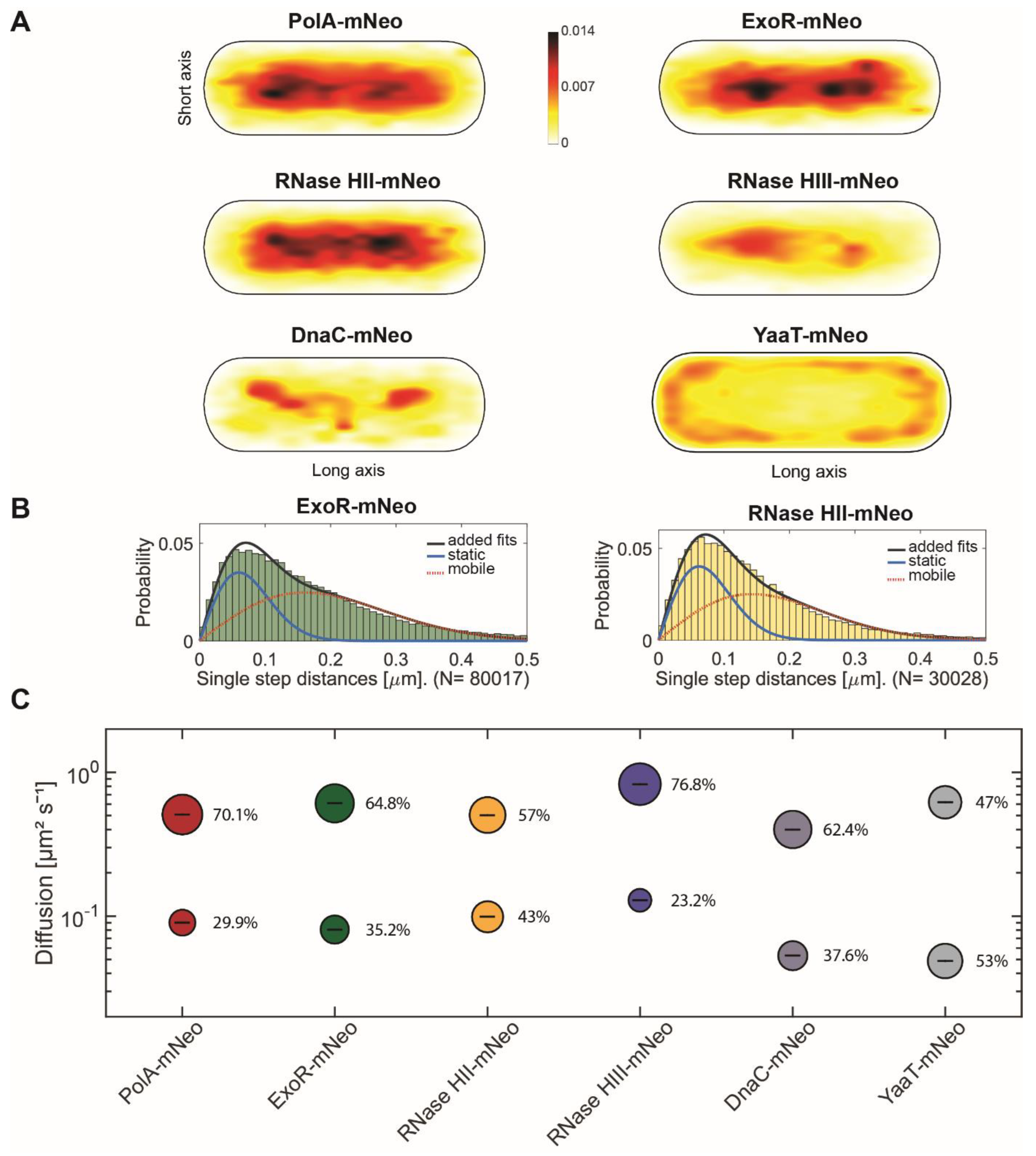
3.2. Single-Molecule Tracking Reveals Distinctive Patterns of Motion for RNases
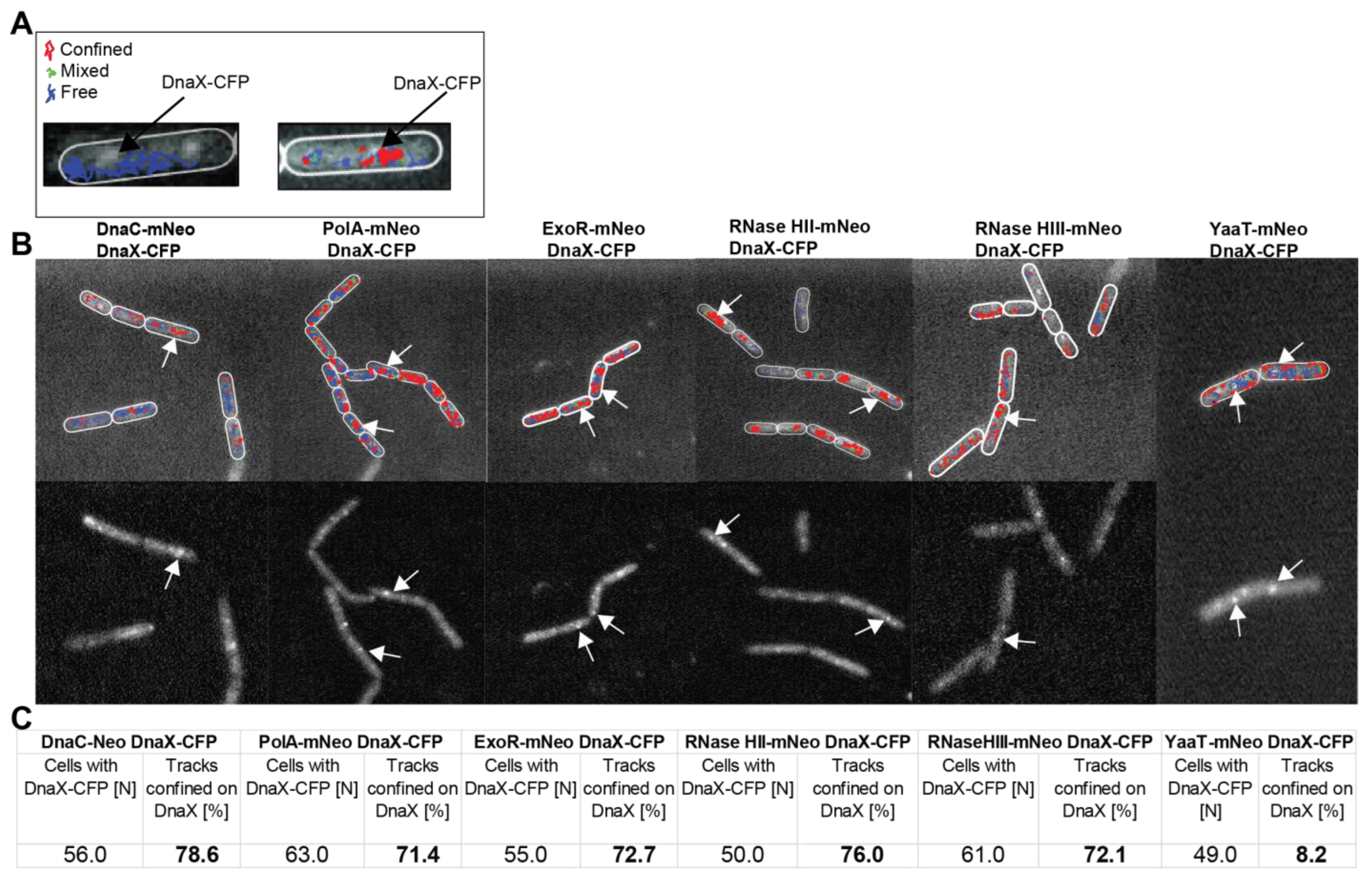
3.3. All Four Enzymes Showing RNase Activity Feature Close Spatial Proximity of Motion and Frequent Arrests at Replication Forks
3.4. PolA, but Not RNase HIII or HII, Shows a Change in Mobility in Response to UV Light-Induced DNA Damage
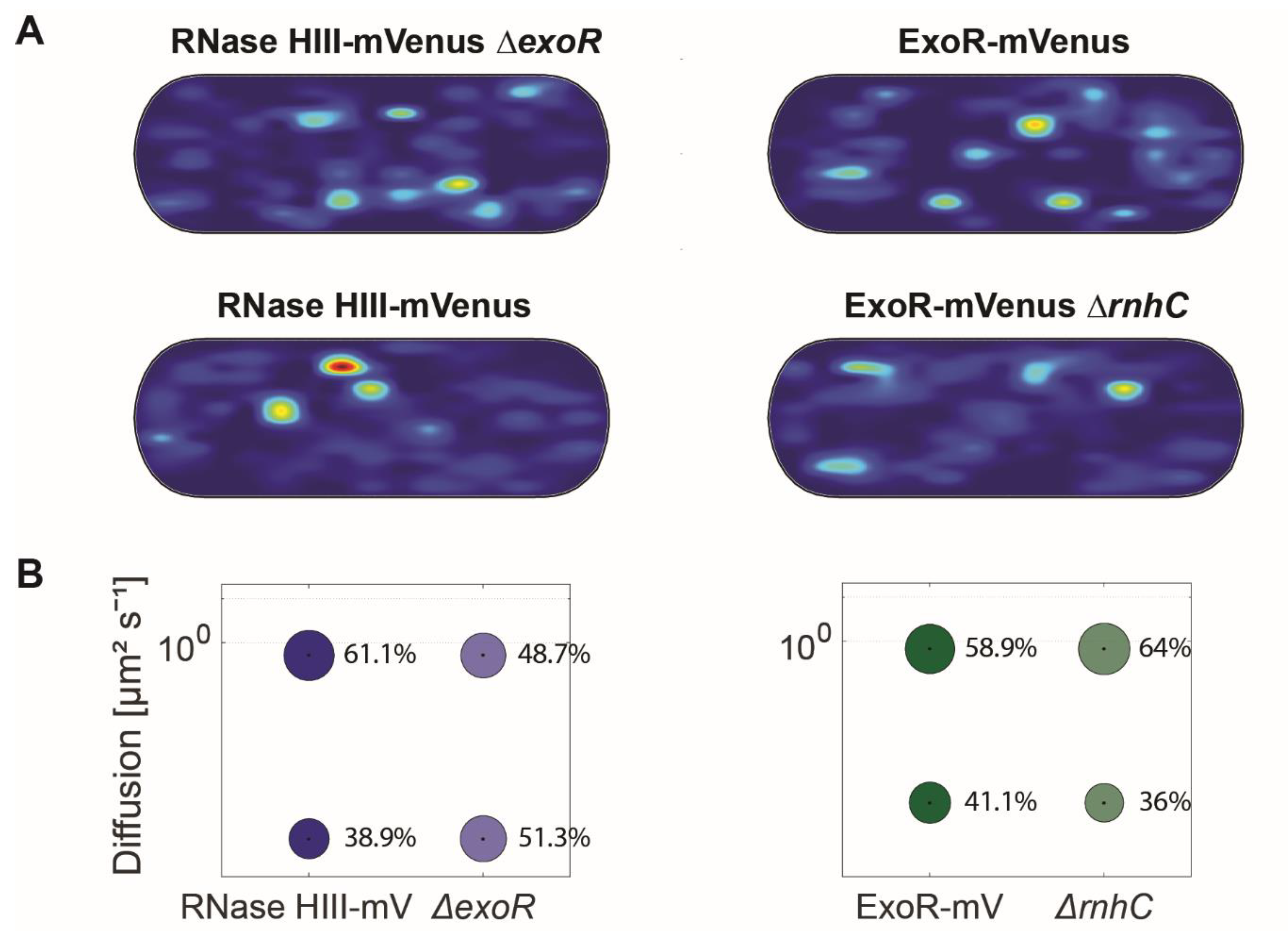
3.5. Lack of ExoR Affects RNase HIII Dynamics
3.6. Inhibiting PolC Activity Leads to a Strong Effect on the Localization of RNase HII and HIII
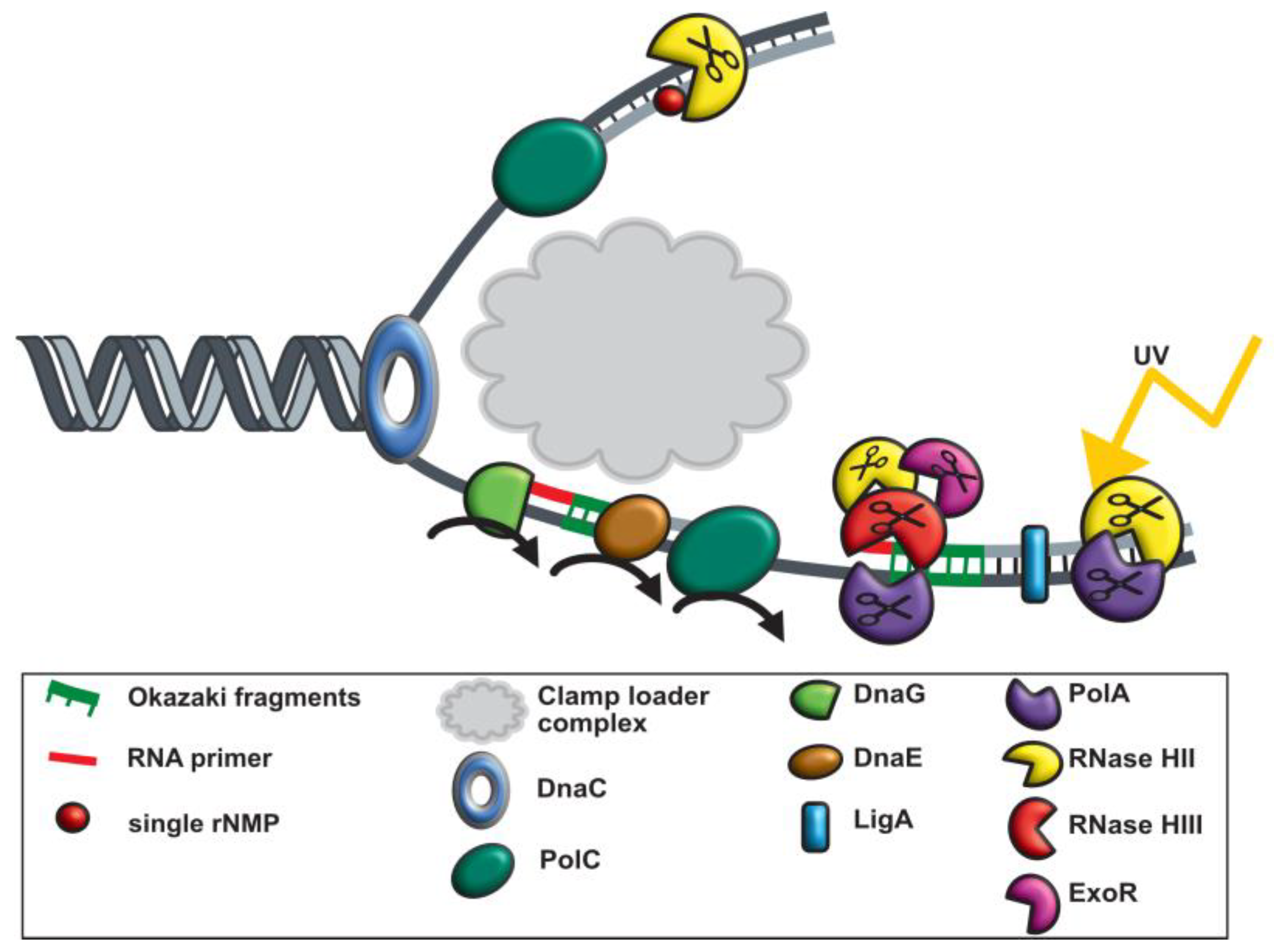
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silva, I.J.; Saramago, M.; Dressaire, C.; Domingues, S.; Viegas, S.C.; Arraiano, C.M. Importance and key events of prokaryotic RNA decay: The ultimate fate of an RNA molecule. Wiley Interdiscip. Rev. RNA 2011, 2, 818–836. [Google Scholar] [CrossRef] [PubMed]
- Jester, B.C.; Romby, P.; Lioliou, E. When ribonucleases come into play in pathogens: A survey of gram-positive bacteria. Int. J. Microbiol. 2012, 2012, 592196. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, S.; Storz, G. Bacterial small RNA regulators: Versatile roles and rapidly evolving variations. Cold Spring Harb. Perspect. Biol. 2011, 3, a003798. [Google Scholar] [CrossRef] [PubMed]
- Arraiano, C.M.; Andrade, J.M.; Domingues, S.; Guinote, I.B.; Malecki, M.; Matos, R.G.; Moreira, R.N.; Pobre, V.; Reis, F.P.; Saramago, M.; et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol. Rev. 2010, 34, 883–923. [Google Scholar] [CrossRef] [PubMed]
- García-Muse, T.; Aguilera, A. R loops: From physiological to pathological roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gomez-Gonzalez, B. DNA–RNA hybrids: The risks of DNA breakage during transcription. Nat. Struct. Mol. Biol. 2017, 24, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.Y.; Schroeder, J.W.; Yurieva, O.; Simmons, L.A.; O’Donnell, M.E. Cost of rNTP/dNTP pool imbalance at the replication fork. Proc. Natl. Acad. Sci. USA 2013, 110, 12942–12947. [Google Scholar] [CrossRef] [PubMed]
- Bechhofer, D.H.; Deutscher, M.P. Bacterial ribonucleases and their roles in RNA metabolism. Crit. Rev. Biochem. Mol. 2019, 54, 242–300. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Deutscher, M.P. Exoribonucleases and Endoribonucleases. EcoSal Plus 2004, 1, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Duigou, S.; Ehrlich, S.D.; Noirot, P.; Noirot-Gros, M.F. DNA polymerase I acts in translesion synthesis mediated by the Y-polymerases in Bacillus subtilis. Mol. Microbiol. 2005, 57, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Okazaki, T.J.M. Function of RNase H in DNA replication revealed by RNase H defective mutants of Escherichia coli. Mol. Gen. Genet. 1984, 193, 231–237. [Google Scholar] [CrossRef]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The enzymes in eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef]
- Ohtani, N.; Haruki, M.; Morikawa, M.; Kanaya, S. Molecular diversities of RNases H. J. Biosci. Bioeng. 1999, 88, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.B.; Kunkel, T.A. The importance of being DNA. Cell Cycle 2010, 9, 4422–4424. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Haruki, M.; Morikawa, M.; Crouch, R.J.; Itaya, M.; Kanaya, S.J.B. Identification of the genes encoding Mn2+-dependent RNase HII and Mg2+-dependent RNase HIII from Bacillus subtilis: Classification of RNases H into three families. Biochemistry 1999, 38, 605–618. [Google Scholar] [CrossRef]
- Kochiwa, H.; Tomita, M.; Kanai, A. Evolution of ribonuclease H genes in prokaryotes to avoid inheritance of redundant genes. BMC Evol. Biol. 2007, 7, 128. [Google Scholar] [CrossRef]
- Sanders, G.M.; Dallmann, H.G.; McHenry, C.S. Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol. Cell 2010, 37, 273–281. [Google Scholar] [CrossRef]
- Dervyn, E.; Suski, C.; Daniel, R.; Bruand, C.; Chapuis, J.; Errington, J.; Janniere, L.; Ehrlich, S.D. Two essential DNA polymerases at the bacterial replication fork. Science 2001, 294, 1716–1719. [Google Scholar] [CrossRef]
- Seco, E.M.; Ayora, S. Bacillus subtilis DNA polymerases, PolC and DnaE, are required for both leading and lagging strand synthesis in SPP1 origin-dependent DNA replication. Nucleic Acids Res. 2017, 45, 8302–8313. [Google Scholar] [CrossRef]
- Ogawa, T.; Okazaki, T. Discontinuous DNA replication. Annu. Rev. Biochem. 1980, 49, 421–457. [Google Scholar] [CrossRef]
- Su’etsugu, M.; Errington, J. The replicase sliding clamp dynamically accumulates behind progressing replication forks in Bacillus subtilis cells. Mol. Cell 2011, 41, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.S.; Kunkel, T.A. Ribonucleotides in DNA: Origins, repair and consequences. DNA Repair. 2014, 19, 27–37. [Google Scholar] [CrossRef]
- López de Saro, F.J.; O’Donnell, M. Interaction of the β sliding clamp with MutS, ligase, and DNA polymerase I. Proc. Natl. Acad. Sci. USA 2001, 98, 8376–8380. [Google Scholar] [CrossRef]
- Randall, J.R.; Nye, T.M.; Wozniak, K.J.; Simmons, L.A. RNase HIII Is Important for Okazaki Fragment Processing in Bacillus subtilis. J. Bacteriol. 2019, 201, e00686-18. [Google Scholar] [CrossRef]
- Schroeder, J.W.; Randall, J.R.; Hirst, W.G.; O’Donnell, M.E.; Simmons, L.A. Mutagenic cost of ribonucleotides in bacterial DNA. Proc. Natl. Acad. Sci. USA 2017, 114, 11733–11738. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Tamayo, R.; Oviedo-Bocanegra, L.M.; Fritz, G.; Graumann, P.L. Symmetric activity of DNA polymerases at and recruitment of exonuclease ExoR and of PolA to the Bacillus subtilis replication forks. Nucleic Acids Res. 2019, 47, 8521–8536. [Google Scholar] [CrossRef] [PubMed]
- Patlán, A.G.; Ayala-García, V.M.; Valenzuela-García, L.I.; Meneses-Plascencia, J.; Vargas-Arias, P.L.; Barraza-Salas, M.; Setlow, P.; Brieba, L.G.; Pedraza-Reyes, M. YwqL (EndoV), ExoA and PolA act in a novel alternative excision pathway to repair deaminated DNA bases in Bacillus subtilis. PLoS ONE 2019, 14, e0211653. [Google Scholar] [CrossRef]
- Lucena, D.; Mauri, M.; Schmidt, F.; Eckhardt, B.; Graumann, P.L. Microdomain formation is a general property of bacterial membrane proteins and induces heterogeneity of diffusion patterns. BMC Biol. 2018, 16, 97. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Koo, B.M.; Kritikos, G.; Farelli, J.D.; Todor, H.; Tong, K.; Kimsey, H.; Wapinski, I.; Galardini, M.; Cabal, A.; Peters, J.M.; et al. Construction and Analysis of Two Genome-Scale Deletion Libraries for Bacillus subtilis. Cell Syst. 2017, 4, 291–305.e7. [Google Scholar] [CrossRef]
- Jaacks, K.J.; Healy, J.; Losick, R.; Grossman, A.D. Identification and characterization of genes controlled by the sporulation-regulatory gene spo0H in Bacillus subtilis. J. Bacteriol. 1989, 171, 4121–4129. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Oviedo-Bocanegra, L.M.; Hinrichs, R.; Rotter, D.A.O.; Dersch, S.; Graumann, P.L. Single molecule/particle tracking analysis program SMTracker 2.0 reveals different dynamics of proteins within the RNA degradosome complex in Bacillus subtilis. Nucleic Acids Res. 2021, 49, e112. [Google Scholar] [CrossRef] [PubMed]
- Paintdakhi, A.; Parry, B.; Campos, M.; Irnov, I.; Elf, J.; Surovtsev, I.; Jacobs-Wagner, C. Oufti: An integrated software package for high-accuracy, high-throughput quantitative microscopy analysis. Mol. Microbiol. 2016, 99, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Jaqaman, K.; Loerke, D.; Mettlen, M.; Kuwata, H.; Grinstein, S.; Schmid, S.L.; Danuser, G. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 2008, 5, 695–702. [Google Scholar] [CrossRef]
- Dersch, S.; Mehl, J.; Stuckenschneider, L.; Mayer, B.; Roth, J.; Rohrbach, A.; Graumann, P.L. Super-Resolution Microscopy and Single-Molecule Tracking Reveal Distinct Adaptive Dynamics of MreB and of Cell Wall-Synthesis Enzymes. Front. Microbiol. 2020, 11, 1946. [Google Scholar] [CrossRef]
- Pediaditakis, M.; Kaufenstein, M.; Graumann, P.L. Bacillus subtilis hlpB Encodes a Conserved Stand-Alone HNH Nuclease-Like Protein That Is Essential for Viability Unless the hlpB Deletion Is Accompanied by the Deletion of Genes Encoding the AddAB DNA Repair Complex. J. Bacteriol. 2012, 194, 6184–6194. [Google Scholar] [CrossRef] [PubMed]
- Bin, L.; Eliason, W.K.; Steitz, T.A. Structure of a helicase-helicase loader complex reveals insights into the mechanism of bacterial primosome assembly. Nat. Commun. 2013, 4, 2495. [Google Scholar] [CrossRef]
- Adusei-Danso, F.; Khaja, F.T.; DeSantis, M.; Jeffrey, P.D.; Dubnau, E.; Demeler, B.; Neiditch, M.B.; Dubnau, D. Structure-Function Studies of the Bacillus subtilis Ric Proteins Identify the Fe-S Cluster-Ligating Residues and Their Roles in Development and RNA Processing. mBio 2019, 10, e01841-19. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, R.; Pozhydaieva, N.; Hofer, K.; Graumann, P.L. Y-Complex Proteins Show RNA-Dependent Binding Events at the Cell Membrane and Distinct Single-Molecule Dynamics. Cells 2022, 11, 933. [Google Scholar] [CrossRef] [PubMed]
- Monahan, L.G.; Liew, A.T.; Bottomley, A.L.; Harry, E.J. Division site positioning in bacteria: One size does not fit all. Front. Microbiol. 2014, 5, 19. [Google Scholar] [CrossRef]
- Brown, N.C. 6-(p-hydroxyphenylazo)-uracil: A selective inhibitor of host DNA replication in phage-infected Bacillus subtilis. Proc. Natl. Acad. Sci. USA 1970, 67, 1454–1461. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Tamayo, R.; Schmitz, H.; Graumann, P.L. Single-Molecule Dynamics at a Bacterial Replication Fork after Nutritional Downshift or Chemically Induced Block in Replication. mSphere 2021, 6, e00948-20. [Google Scholar] [CrossRef]
- Liu, B.; Hu, J.; Wang, J.; Kong, D. Direct Visualization of RNA-DNA Primer Removal from Okazaki Fragments Provides Support for Flap Cleavage and Exonucleolytic Pathways in Eukaryotic Cells. J. Biol. Chem. 2017, 292, 4777–4788. [Google Scholar] [CrossRef]
- Randall, J.R.; Hirst, W.G.; Simmons, L.A. Substrate Specificity for Bacterial RNases HII and HIII Is Influenced by Metal Availability. J. Bacteriol. 2018, 200, e00401-17. [Google Scholar] [CrossRef]
- Rotter, D.A.O.; Heger, C.; Oviedo-Bocanegra, L.M.; Graumann, P.L. Transcription-dependent confined diffusion of enzymes within subcellular spaces of the bacterial cytoplasm. BMC Biol. 2021, 19, 183. [Google Scholar] [CrossRef] [PubMed]
- Stracy, M.; Schweizer, J.; Sherratt, D.J.; Kapanidis, A.N.; Uphoff, S.; Lesterlin, C. Transient non-specific DNA binding dominates the target search of bacterial DNA-binding proteins. Mol. Cell 2021, 81, 1499–1514. [Google Scholar] [CrossRef]
- Negri, A.; Werbowy, O.; Wons, E.; Dersch, S.; Hinrichs, R.; Graumann, P.L.; Mruk, I. Regulator-dependent temporal dynamics of a restriction-modification system’s gene expression upon entering new host cells: Single-cell and population studies. Nucleic Acids Res. 2021, 49, 3826–3840. [Google Scholar] [CrossRef]
- Gan, W.J.; Guan, Z.S.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X.L. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef]
- Lin, Y.L.; Pasero, P. Interference Between DNA Replication and Transcription as a Cause of Genomic Instability. Curr. Genom. 2012, 13, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.S.; Hall, A.N.; Merrikh, C.N.; Ragheb, M.; Tabakh, H.; Pollock, A.J.; Woodward, J.J.; Dreifus, J.E.; Merrikh, H. Replication-Transcription Conflicts Generate R-Loops that Orchestrate Bacterial Stress Survival and Pathogenesis. Cell 2017, 170, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Astatke, M.; Ng, K.; Grindley, N.D.; Joyce, C.M. A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proc. Natl. Acad. Sci. USA 1998, 95, 3402–3407. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Orlova, M.; Georgiadis, M.M.; Hendrickson, W.A.; Goff, S.P. Conferring RNA polymerase activity to a DNA polymerase: A single residue in reverse transcriptase controls substrate selection. Proc. Natl. Acad. Sci. USA 1997, 94, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Joyce, C.M. Choosing the right sugar: How polymerases select a nucleotide substrate. Proc. Natl. Acad. Sci. USA 1997, 94, 1619–1622. [Google Scholar] [CrossRef]
- Nick McElhinny, S.A.; Watts, B.E.; Kumar, D.; Watt, D.L.; Lundstrom, E.B.; Burgers, P.M.; Johansson, E.; Chabes, A.; Kunkel, T.A. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl. Acad. Sci. USA 2010, 107, 4949–4954. [Google Scholar] [CrossRef]
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef]
- Thomaides, H.B.; Davison, E.J.; Burston, L.; Johnson, H.; Brown, D.R.; Hunt, A.C.; Errington, J.; Czaplewski, L. Essential bacterial functions encoded by gene pairs. J. Bacteriol. 2007, 189, 591–602. [Google Scholar] [CrossRef]
- Jameson, K.H.; Wilkinson, A.J. Control of Initiation of DNA Replication in Bacillus subtilis and Escherichia coli. Genes 2017, 8, 22. [Google Scholar] [CrossRef]
- Lenhart, J.S.; Schroeder, J.W.; Walsh, B.W.; Simmons, L.A. DNA repair and genome maintenance in Bacillus subtilis. Microbiol. Mol. Biol. Rev. 2012, 76, 530–564. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Figure 2 | ||||||||
| PolA-mNeo | ExoR-mNeo | RNase HII-mNeo | RNase HIII-mNeo | DnaC-mNeo | YaaT-mNeo | |||
| D1 (static) µm2 s−1 | 0.09 ± 0.0 | 0.09 ± 0.0 | 0.10 ± 0.0 | 0.13 ± 0.001 | 0.05 ± 0.001 | 0.05 ± 0.001 | ||
| D3 (mobile) µm2 s−1 | 0.51 ± 0.001 | 0.63 ± 0.001 | 0.50 ± 0.001 | 0.82 ± 0.001 | 0.4 ± 0.002 | 0.62 ± 0.002 | ||
| Static % | 29.9 ± 0.001 | 35.2 ± 0.001 | 43.0 ± 0.001 | 23.2 ± 0.001 | 37.6 ± 0.001 | 53.0 ± 0.00 | ||
| Mobile % | 70.1 ± 0.001 | 64.8 ± 0.001 | 57.0 ± 0.001 | 76.8 ± 0.001 | 62.4 ± 0.001 | 47.0 ± 0.00 | ||
| R2 | 1 | 0.999 | 1 | 1 | 0.999 | 0.999 | ||
| Figure 5 | ||||||||
| PolA-mNeo | UV | ExoR-mNeo | UV | RNase HII-mNeo | UV | RNase HIII-mNeo | UV | |
| D1 (static) µm2 s−1 | 0.09 ± 0.0 | 0.09 ± 0.0 | 0.09 ± 0.0 | 0.09 ± 0.0 | 0.10 ± 0.0 | 0.10 ± 0.0 | 0.10 ± 0.003 | 0.10 ± 0.003 |
| D3 (mobile) µm2 s−1 | 0.51 ± 0.001 | 0.51 ± 0.001 | 0.63 ± 0.001 | 0.63 ± 0.001 | 0.51 ± 0.002 | 0.51 ± 0.002 | 0.84 ± 0.004 | 0.84 ± 0.004 |
| Static % | 30.2 ± 0.001 | 43.2 ± 0.001 | 37 ± 0.001 | 33.2 ± 0.001 | 42.9 ± 0.002 | 41.3 ± 0.002 | 21.6 ± 0.003 | 23.4 ± 0.003 |
| Mobile % | 69.8 ± 0.001 | 56.8 ± 0.001 | 63 ± 0.001 | 66.8 ± 0.001 | 57.1 ± 0.002 | 58.7 ± 0.002 | 78.4 ± 0.003 | 76.6 ± 0.003 |
| R2 | 1 | 0.999 | 1 | 1 | 1 | 1 | 0.999 | 0.996 |
| Figure 6 | ||||||||
| ExoR-mV | ExoR-mV ΔrnhC | RNaseHIII-mV | RNaseHIII-mV ΔexoR | |||||
| D1 (static) µm2 s−1 | 0.09 ± 0.001 | 0.09 ± 0.001 | 0.05 ± 0.001 | 0.05 ± 0.001 | ||||
| D3 (mobile) µm2 s−1 | 0.85 ± 0.005 | 0.85 ± 0.005 | 0.84 ± 0.001 | 0.84 ± 0.001 | ||||
| Static % | 41.1 ± 0.267 | 36 ± 0.178 | 38.9 ± 0.181 | 48.7 ± 0.264 | ||||
| Mobile % | 58.9 ± 0.267 | 64 ± 0.178 | 61.1 ± 0.181 | 51.3 ± 0.264 | ||||
| R2 | 0.995 | 0.999 | 0.994 | 0.992 | ||||
| Figure 7 | ||||||||
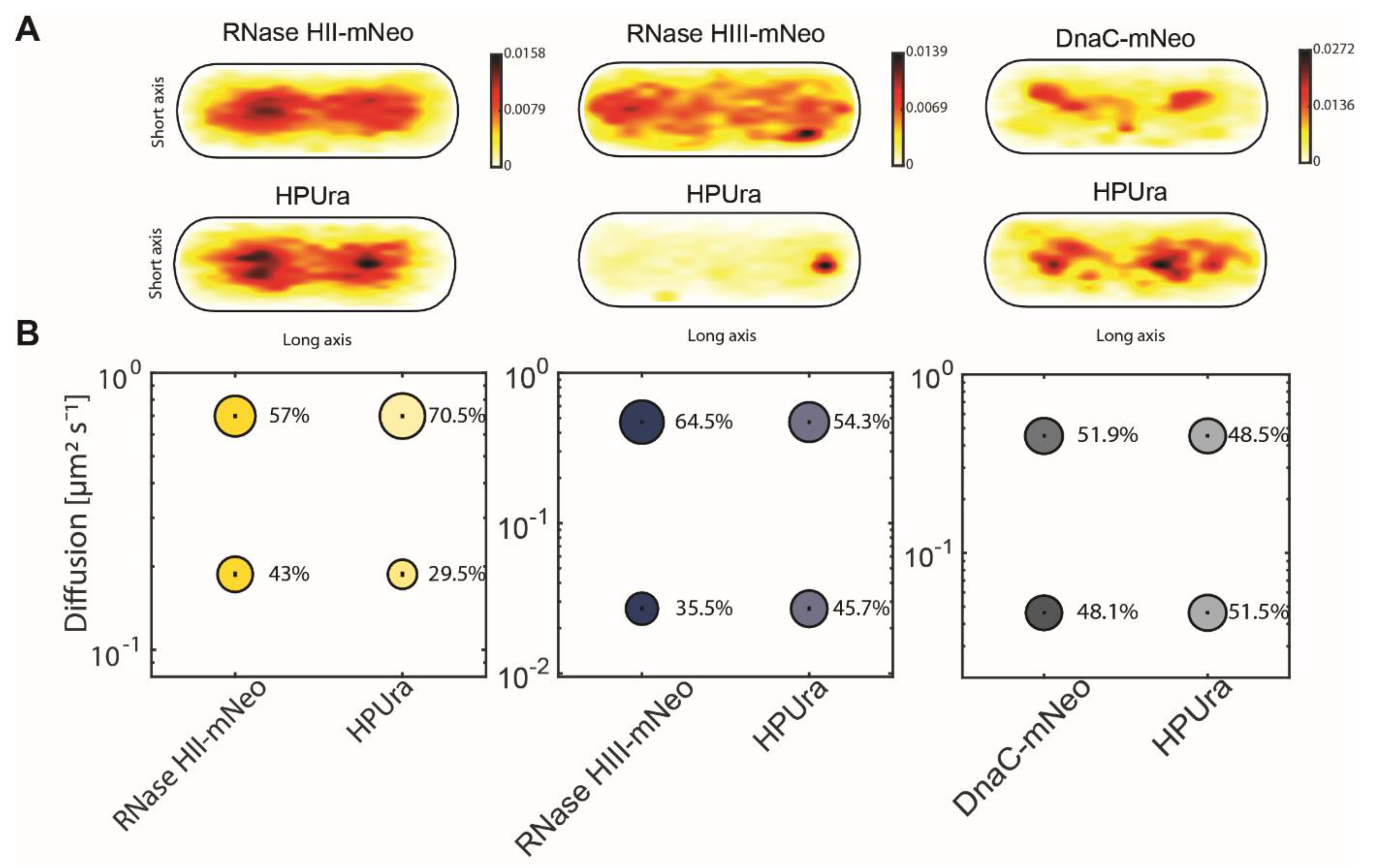
| RNaseHII-mNeo | HPUra | RNaseHIII-mNeo | HPUra | DnaC-mNeo | HPUra | |||
| D1 (static) µm2 s−1 | 0.19 ± 0.002 | 0.19 ± 0.002 | 0.03 ± 0.001 | 0.03 ± 0.0 | 0.05 ± 0.0 | 0.05 ± 0.0 | ||
| D3 (mobile) µm2 s−1 | 0.7 ± 0.005 | 0.7 ± 0.005 | 0.47 ± 0.004 | 0.47 ± 0.004 | 0.45 ± 0.003 | 0.45 ± 0.003 | ||
| Static % | 43 ± 0.006 | 29.5 ± 0.006 | 35.5 ± 0.003 | 45.7 ± 0.003 | 48.1 ± 0.002 | 51.5 ± 0.002 | ||
| Mobile % | 57 ± 0.006 | 70.5 ± 0.006 | 64.5 ± 0.004 | 54.3 ± 0.004 | 51.9 ± 0.002 | 48.5 ± 0.002 | ||
| R2 | 0.999 | 0.999 | 0.972 | 0.972 | 0.999 | 0.999 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinrichs, R.; Graumann, P.L. Visual Evidence for the Recruitment of Four Enzymes with RNase Activity to the Bacillus subtilis Replication Forks. Cells 2024, 13, 1381. https://doi.org/10.3390/cells13161381
Hinrichs R, Graumann PL. Visual Evidence for the Recruitment of Four Enzymes with RNase Activity to the Bacillus subtilis Replication Forks. Cells. 2024; 13(16):1381. https://doi.org/10.3390/cells13161381
Chicago/Turabian StyleHinrichs, Rebecca, and Peter L. Graumann. 2024. "Visual Evidence for the Recruitment of Four Enzymes with RNase Activity to the Bacillus subtilis Replication Forks" Cells 13, no. 16: 1381. https://doi.org/10.3390/cells13161381