
Cell-Specific Effects of Insulin in a Murine Model of Restenosis Under Insulin-Sensitive and Insulin-Resistant Conditions
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model and Animal Experiments
2.2. Diet Treatment
2.3. Generation of Cell-Specific IR-Deficient Mice
2.4. Insulin Pellet Implantation and Surgical Procedure
2.5. Blood Glucose Assessment and Blood Sample Collection
2.6. Insulin Tolerance Test
2.7. Fixed Vessel Collection and Morphological Analysis
2.8. Immunofluorescence
2.9. Statistical Analysis
3. Results
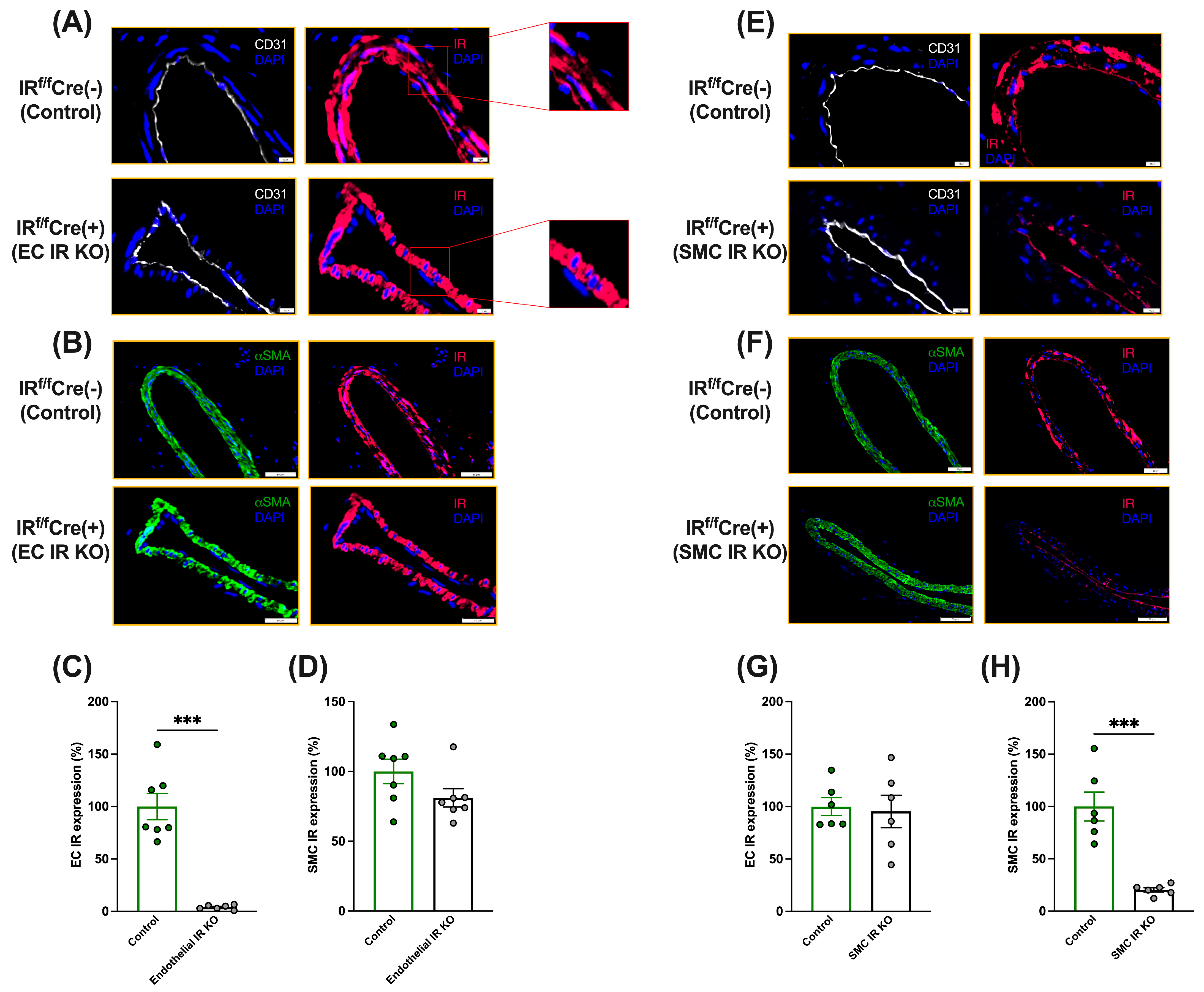
3.1. Generation of Cell-Specific IR-Deficient Mice
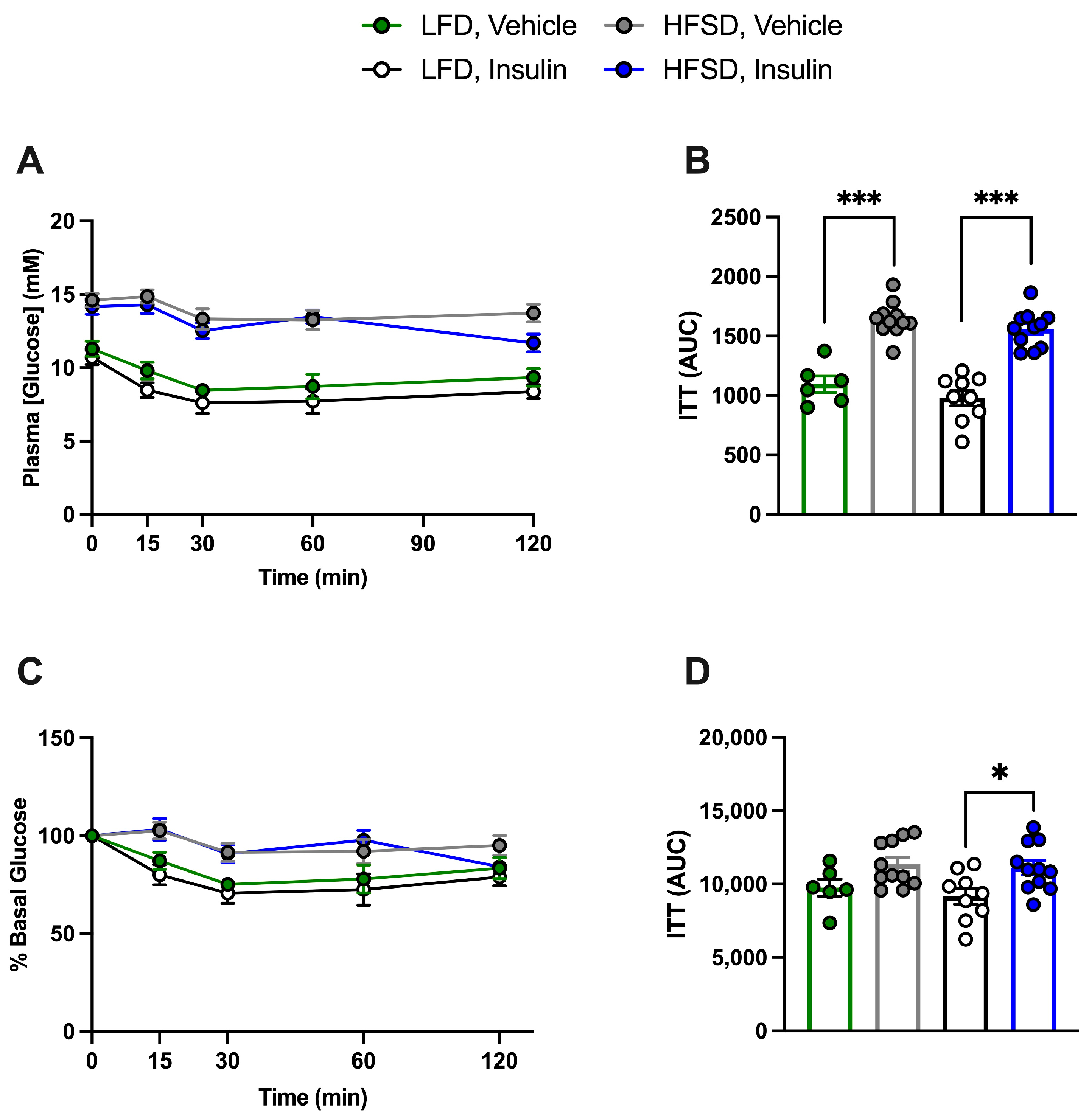
3.2. Metabolic Parameters
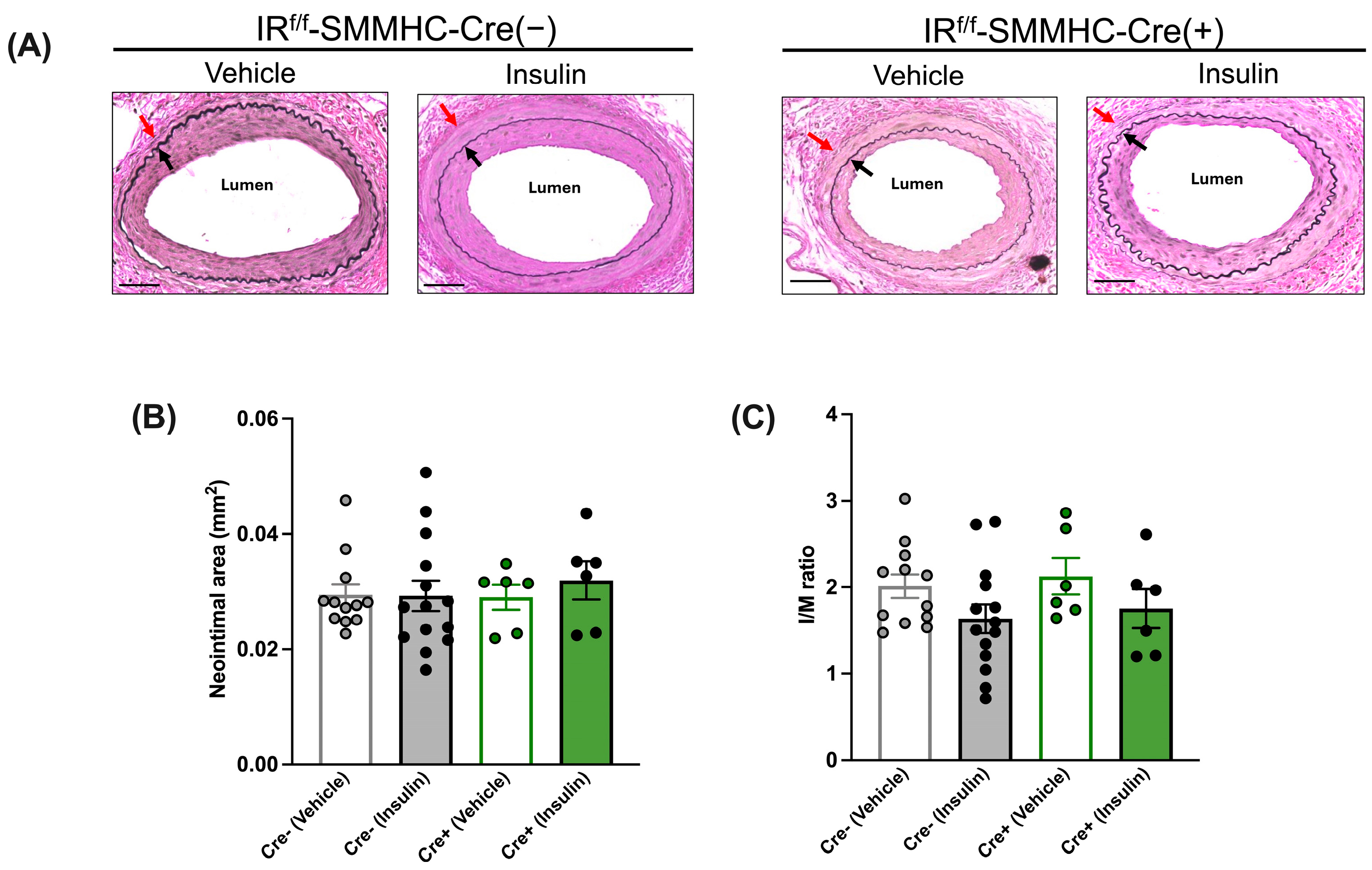
3.3. Cell-Specific Effects of Insulin on Neointimal Growth under Insulin-Sensitive Conditions
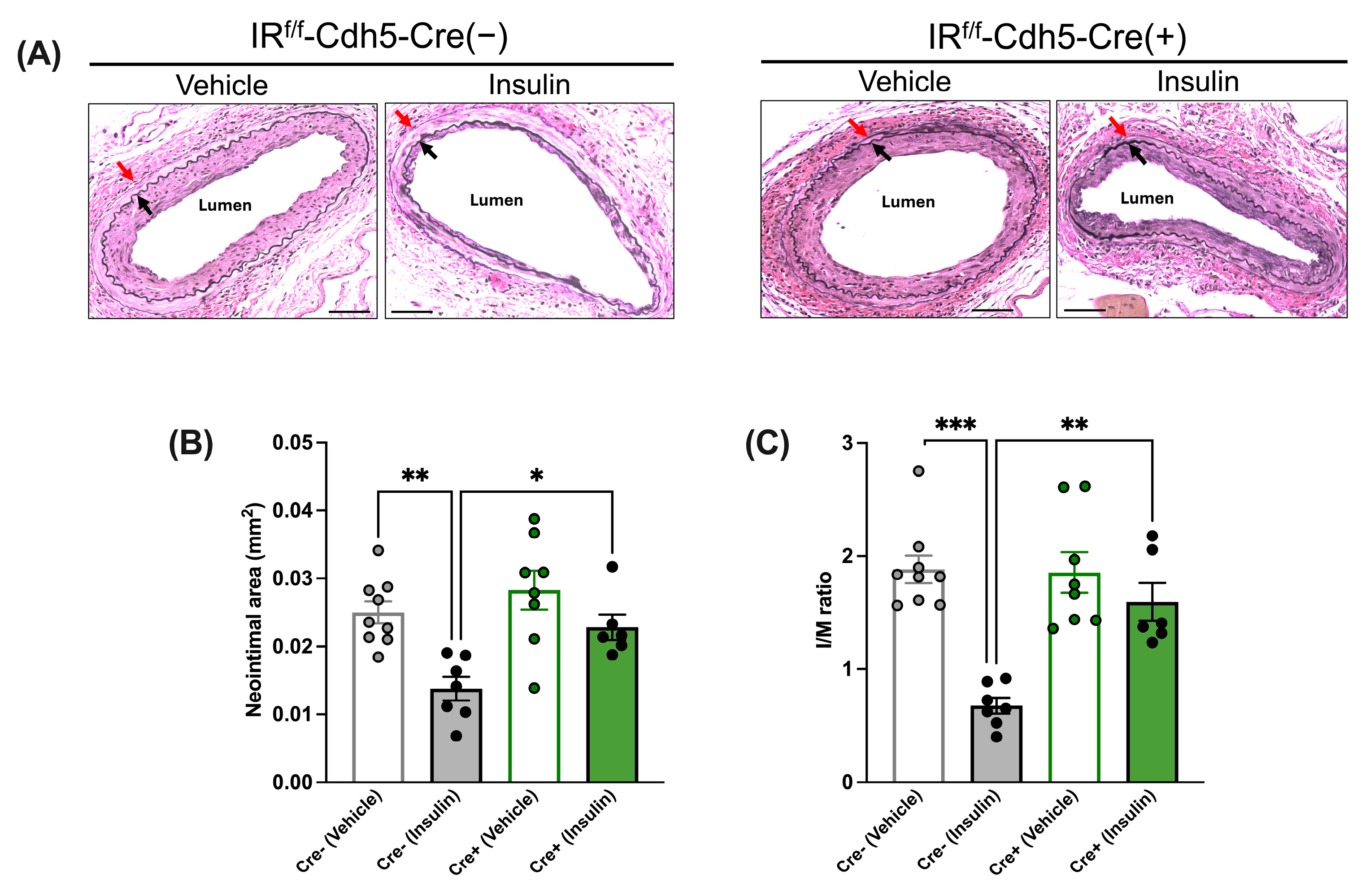
3.3.1. Insulin’s Anti-Restenotic Effect Is Abolished in LFD-Fed EC-Specific IR-Deficient Mice
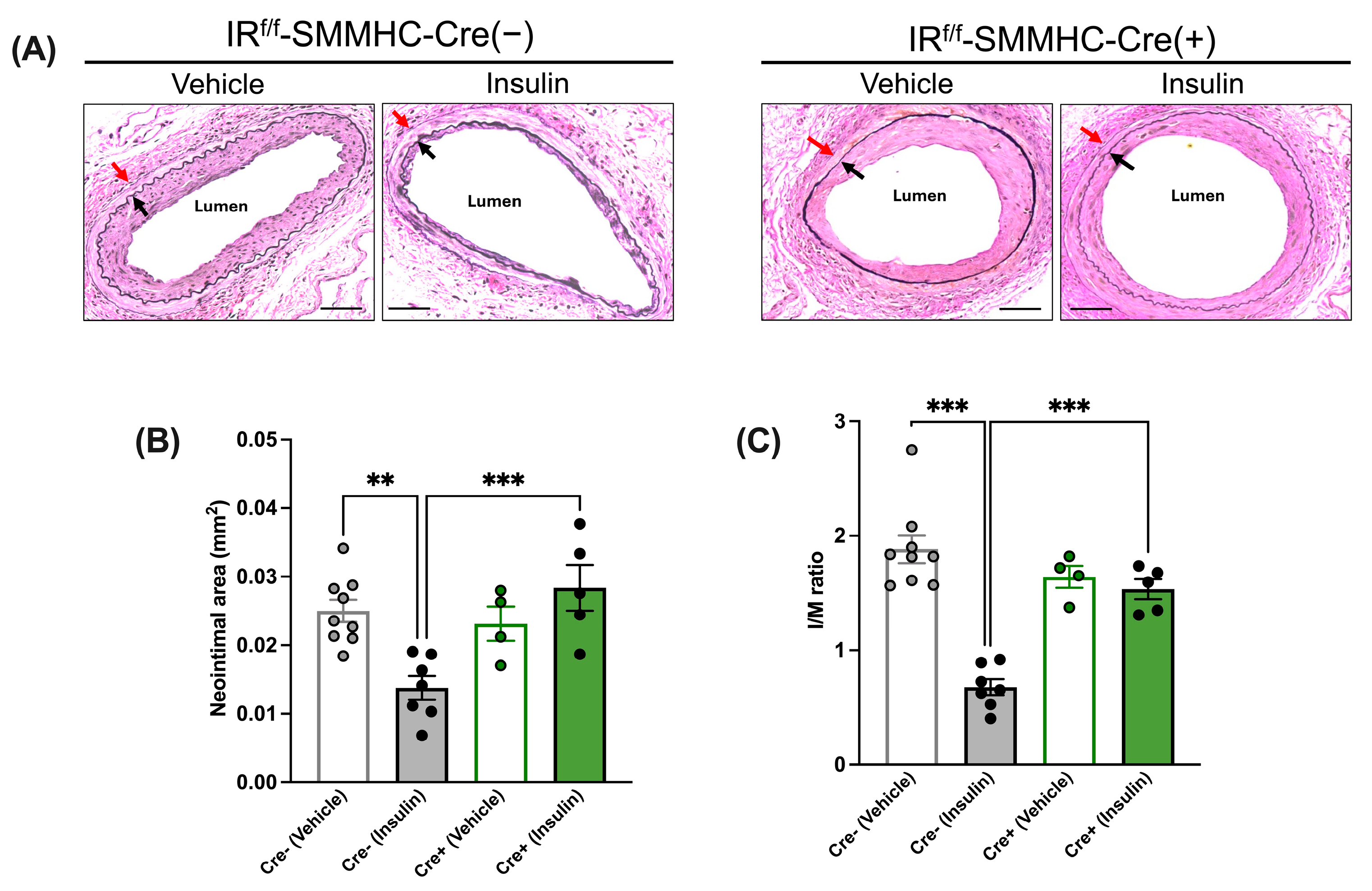
3.3.2. Insulin’s Anti-Restenotic Effect Is Abolished in LFD-Fed SMC-Specific IR-Deficient Mice
3.3.3. Insulin Treatment Reduces Neointimal Growth in LFD-Fed IRW/W-Cre(+) Control Mice
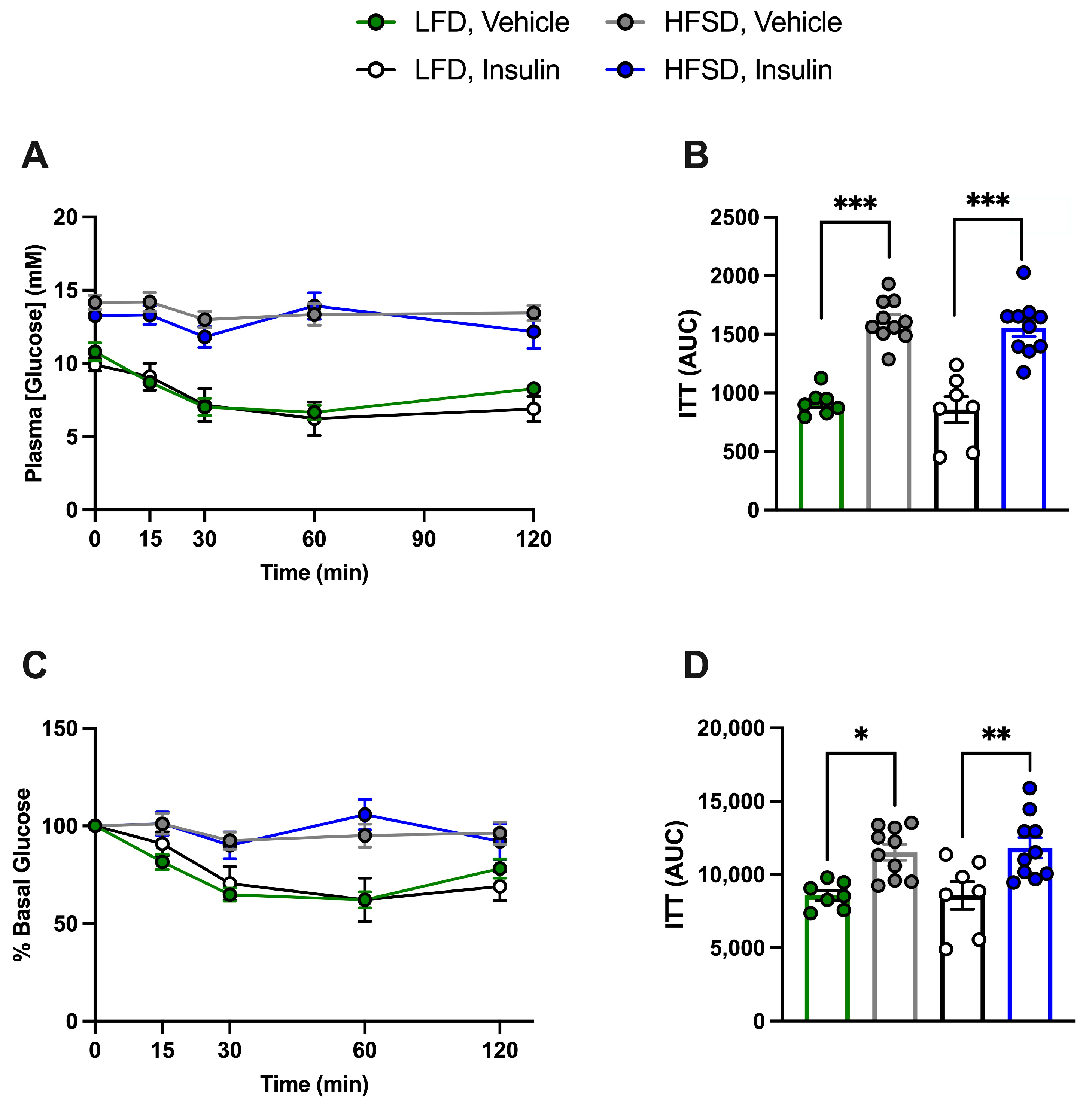
3.4. HFSD Induces Insulin Resistance in Control and Cell-Specific IR-Deficient Mice
3.5. Cell-Specific Effects of Insulin on Neointimal Growth under Insulin-Resistant Conditions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barquera, S.; Pedroza-Tobías, A.; Medina, C.; Hernández-Barrera, L.; Bibbins-Domingo, K.; Lozano, R.; Moran, A.E. Global Overview of the Epidemiology of Atherosclerotic Cardiovascular Disease. Arch. Med. Sci. 2015, 46, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Ferns, G.A.; Avades, T.Y. The Mechanisms of Coronary Restenosis: Insights from Experimental Models. Int. J. Exp. Pathol. 2000, 81, 63–88. [Google Scholar] [CrossRef]
- Morice, M.-C.; Serruys, P.W.; Sousa, J.E.; Fajadet, J.; Ban Hayashi, E.; Perin, M.; Colombo, A.; Schuler, G.; Barragan, P.; Guagliumi, G.; et al. A Randomized Comparison of a Sirolimus-Eluting Stent with a Standard Stent for Coronary Revascularization. N. Engl. J. Med. 2002, 346, 1773–1780. [Google Scholar] [CrossRef]
- Schmucker, J.; Fach, A.; Osteresch, R.; Mata Marin, L.A.; Ruehle, S.; Retzlaff, T.; Garstka, D.; Eitel, I.; Hambrecht, R.; Wienbergen, H. Efficacy of Drug-Eluting Stents in Diabetic Patients Admitted with ST-Elevation Myocardial Infarctions Treated with Primary Percutaneous Coronary Intervention. J. Cardiovasc. Dev. Dis. 2021, 8, 83. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, C.; Ma, Y.; He, Y.; Zhang, X.; Wu, J. Association between Diabetes Mellitus and Primary Restenosis Following Endovascular Treatment: A Comprehensive Meta-Analysis of Randomized Controlled Trials. Cardiovasc. Diabetol. 2024, 23, 132. [Google Scholar] [CrossRef]
- Muniyappa, R.; Montagnani, M.; Koh, K.K.; Quon, M.J. Cardiovascular Actions of Insulin. Endocr. Rev. 2007, 28, 463–491. [Google Scholar] [CrossRef]
- Cubbon, R.M.; Rajwani, A.; Wheatcroft, S.B. The Impact of Insulin Resistance on Endothelial Function, Progenitor Cells and Repair. Diab. Vasc. Dis. Res. 2007, 4, 103–111. [Google Scholar] [CrossRef]
- Wang, C.C.L.; Gurevich, I.; Draznin, B. Insulin Affects Vascular Smooth Muscle Cell Phenotype and Migration via Distinct Signaling Pathways. Diabetes 2003, 52, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Aljada, A.; Dhindsa, S.; Garg, R. Insulin as an Anti-Inflammatory and Antiatherosclerotic Hormone. Clin. Cornerstone 2003, 5 (Suppl. 4), S13–S20. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Dandona, P.; Fonseca, V. Cardiovascular Benefits of Exogenous Insulin. J. Clin. Endocrinol. Metab. 2012, 97, 3079–3091. [Google Scholar] [CrossRef]
- Guo, J.; Breen, D.M.; Pereira, T.J.; Dalvi, P.S.; Zhang, H.; Mori, Y.; Ghanim, H.; Tumiati, L.; Fantus, I.G.; Bendeck, M.P.; et al. The Effect of Insulin to Decrease Neointimal Growth After Arterial Injury Is Endothelial Nitric Oxide Synthase-Dependent. Atherosclerosis 2015, 241, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.M.; Chan, K.K.; Dhaliwall, J.K.; Ward, M.R.; Al Koudsi, N.; Lam, L.; De Souza, M.; Ghanim, H.; Dandona, P.; Stewart, D.J.; et al. Insulin Increases Reendothelialization and Inhibits Cell Migration and Neointimal Growth After Arterial Injury. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1060–1066. [Google Scholar] [CrossRef]
- Kim, T.; Chan, K.K.; Dhaliwall, J.K.; Huynh, N.; Suen, R.; Uchino, H.; Naigamwalla, D.; Bendeck, M.P.; Giacca, A. Anti-Atherogenic Effect of Insulin In Vivo. J. Vasc. Res. 2005, 42, 455–462. [Google Scholar] [CrossRef]
- Mori, Y.; Gonzalez Medina, M.; Liu, Z.; Guo, J.; Dingwell, L.S.; Chiang, S.; Kahn, C.R.; Husain, M.; Giacca, A. Roles of Vascular Endothelial and Smooth Muscle Cells in the Vasculoprotective Effect of Insulin in a Mouse Model of Restenosis. Diab. Vasc. Dis. Res. 2021, 18, 14791641211027324. [Google Scholar] [CrossRef] [PubMed]
- King, G.L.; Park, K.; Li, Q. Selective Insulin Resistance and the Development of Cardiovascular Diseases in Diabetes: The 2015 Edwin Bierman Award Lecture. Diabetes 2016, 65, 1462–1471. [Google Scholar] [CrossRef]
- Breen, D.M.; Dhaliwall, J.K.; Chan, K.K.; Guo, J.; Lam, L.; Bendeck, M.P.; Giacca, A. Insulin Inhibits and Oral Sucrose Increases Neointimal Growth after Arterial Injury in Rats. J. Vasc. Res. 2010, 47, 412–422. [Google Scholar] [CrossRef]
- Li, Q.; Fu, J.; Xia, Y.; Qi, W.; Ishikado, A.; Park, K.; Yokomizo, H.; Huang, Q.; Cai, W.; Rask-Madsen, C.; et al. Homozygous Receptors for Insulin and Not IGF-1 Accelerate Intimal Hyperplasia in Insulin Resistance and Diabetes. Nat. Commun. 2019, 10, 4427. [Google Scholar] [CrossRef]
- Sag, C.M.; Schnelle, M.; Zhang, J.; Murdoch, C.E.; Kossmann, S.; Protti, A.; Santos, C.X.C.; Sawyer, G.; Zhang, X.; Mongue-Din, H.; et al. Distinct Regulatory Effects of Myeloid Cell and Endothelial Cell NADPH Oxidase 2 on Blood Pressure. Circulation 2017, 135, 2163–2177. [Google Scholar] [CrossRef]
- Wirth, A.; Benyó, Z.; Lukasova, M.; Leutgeb, B.; Wettschureck, N.; Gorbey, S.; Orsy, P.; Horváth, B.; Maser-Gluth, C.; Greiner, E.; et al. G12-G13-LARG-Mediated Signaling in Vascular Smooth Muscle Is Required for Salt-Induced Hypertension. Nat. Med. 2008, 14, 64–68. [Google Scholar] [CrossRef]
- Groneberg, D.; König, P.; Wirth, A.; Offermanns, S.; Koesling, D.; Friebe, A. Smooth Muscle–Specific Deletion of Nitric Oxide–Sensitive Guanylyl Cyclase Is Sufficient to Induce Hypertension in Mice. Circulation 2010, 121, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; McCubbin, J.A.; Feinglos, M.N. Diet-Induced Type II Diabetes in C57BL/6J Mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef]
- Mori, Y.; Chiang, S.; Bendeck, M.P.; Giacca, A. Insulin Decreases Atherosclerotic Plaque Burden and Increases Plaque Stability via Nitric Oxide Synthase in Apolipoprotein E-Null Mice. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E335–E345. [Google Scholar] [CrossRef]
- Sata, M.; Maejima, Y.; Adachi, F.; Fukino, K.; Saiura, A.; Sugiura, S.; Aoyagi, T.; Imai, Y.; Kurihara, H.; Kimura, K.; et al. A Mouse Model of Vascular Injury That Induces Rapid Onset of Medial Cell Apoptosis Followed by Reproducible Neointimal Hyperplasia. J. Mol. Cell. Cardiol. 2000, 32, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Carpentier, A.; Adeli, K.; Giacca, A. Disordered Fat Storage and Mobilization in the Pathogenesis of Insulin Resistance and Type 2 Diabetes. Endocr. Rev. 2002, 23, 201–229. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; Li, Q.; Freund, B.; Feather, D.; Abramov, R.; Wu, I.-H.; Chen, K.; Yamamoto-Hiraoka, J.; Goldenbogen, J.; Sotiropoulos, K.B.; et al. Loss of Insulin Signaling in Vascular Endothelial Cells Accelerates Atherosclerosis in Apolipoprotein E Null Mice. Cell Metab. 2010, 11, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Xi, X.-P.; Graf, K.; Goetze, S.; Hsueh, W.A.; Law, R.E. Inhibition of MAP Kinase Blocks Insulin-Mediated DNA Synthesis and Transcriptional Activation of c-Fos by Elk-1 in Vascular Smooth Muscle Cells. FEBS Lett. 1997, 417, 283–286. [Google Scholar] [CrossRef]
- Li, Q.; Fu, J.; Park, K.; Wu, I.-H.; King, G.L. Insulin Stabilized Arterial Plaque by Regulating Vascular Smooth Muscle Cell Metabolism and Differentiation. Diabetes 2023, 72 (Suppl. 1), 40-OR. [Google Scholar] [CrossRef]
- Meln, I.; Wolff, G.; Gajek, T.; Koddebusch, J.; Lerch, S.; Harbrecht, L.; Hong, W.; Bayindir-Buchhalter, I.; Krunic, D.; Augustin, H.G.; et al. Dietary Calories and Lipids Synergistically Shape Adipose Tissue Cellularity during Postnatal Growth. Mol. Metab. 2019, 24, 139–148. [Google Scholar] [CrossRef]
- King, G.L.; Goodman, A.D.; Buzney, S.; Moses, A.; Kahn, C.R. Receptors and Growth-Promoting Effects of Insulin and Insulinlike Growth Factors on Cells from Bovine Retinal Capillaries and Aorta. J. Clin. Investig. 1985, 75, 1028–1036. [Google Scholar] [CrossRef]
- Bornfeldt, K.E.; Raines, E.W.; Nakano, T.; Graves, L.M.; Krebs, E.G.; Ross, R. Insulin-like Growth Factor-I and Platelet-Derived Growth Factor-BB Induce Directed Migration of Human Arterial Smooth Muscle Cells via Signaling Pathways That Are Distinct from Those of Proliferation. J. Clin. Investig. 1994, 93, 1266–1274. [Google Scholar] [CrossRef]
- Zhu, B.; Zhao, G.; Witte, D.P.; Hui, D.Y.; Fagin, J.A. Targeted Overexpression of IGF-I in Smooth Muscle Cells of Transgenic Mice Enhances Neointimal Formation through Increased Proliferation and Cell Migration after Intraarterial Injury. Endocrinology 2001, 142, 3598–3606. [Google Scholar] [CrossRef]
- Lim, H.-J.; Park, H.-Y.; Ko, Y.-G.; Lee, S.-H.; Cho, S.-Y.; Lee, E.J.; Jameson, J.L.; Jang, Y. Dominant Negative Insulin-like Growth Factor-1 Receptor Inhibits Neointimal Formation through Suppression of Vascular Smooth Muscle Cell Migration and Proliferation, and Induction of Apoptosis. Biochem. Biophys. Res. Commun. 2004, 325, 1106–1114. [Google Scholar] [CrossRef]
- Cittadini, A.; Monti, M.G.; Castiello, M.C.; D’arco, E.; Galasso, G.; Sorriento, D.; Saldamarco, L.; Paulis, A.D.; Napoli, R.; Iaccarino, G.; et al. Insulin-like Growth Factor-1 Protects from Vascular Stenosis and Accelerates Re-endothelialization in a Rat Model of Carotid Artery Injury. J. Thromb. Haemost. 2009, 7, 1920–1928. [Google Scholar] [CrossRef]
- Jacob, A.; Molkentin, J.D.; Smolenski, A.; Lohmann, S.M.; Begum, N. Insulin Inhibits PDGF-Directed VSMC Migration via NO/CGMP Increase of MKP-1 and Its Inactivation of MAPKs. Am. J. Physiol. Cell Physiol. 2002, 283, C704–C713. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Lin, Y.W.; Clemont, A.; Feener, E.P.; Hein, K.D.; Igarashi, M.; Yamauchi, T.; White, M.F.; King, G.L. Characterization of Selective Resistance to Insulin Signaling in the Vasculature of Obese Zucker (Fa/Fa) Rats. J. Clin. Investig. 1999, 104, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Symons, J.D.; McMillin, S.L.; Riehle, C.; Tanner, J.; Palionyte, M.; Hillas, E.; Jones, D.; Cooksey, R.C.; Birnbaum, M.J.; McClain, D.A.; et al. Contribution of Insulin and Akt1 Signaling to Endothelial Nitric Oxide Synthase in the Regulation of Endothelial Function and Blood Pressure. Circ. Res. 2009, 104, 1085–1094. [Google Scholar] [CrossRef]
- Banskota, N.K.; Taub, R.; Zellner, K.; King, G.L. Insulin, Insulin-Like Growth Factor I and Platelet-Derived Growth Factor Interact Additively in the Induction of the Protooncogene C-Myc and Cellular Proliferation in Cultured Bovine Aortic Smooth Muscle Cells. Mol. Endocrinol. 1989, 3, 1183–1190. [Google Scholar] [CrossRef]
- Hage Hassan, R.; Pacheco de Sousa, A.C.; Mahfouz, R.; Hainault, I.; Blachnio-Zabielska, A.; Bourron, O.; Koskas, F.; Górski, J.; Ferré, P.; Foufelle, F.; et al. Sustained Action of Ceramide on the Insulin Signaling Pathway in Muscle Cells. J. Biol. Chem. 2016, 291, 3019–3029. [Google Scholar] [CrossRef] [PubMed]
- Schubert, K.M.; Scheid, M.P.; Duronio, V. Ceramide Inhibits Protein Kinase B/Akt by Promoting Dephosphorylation of Serine 473. J. Biol. Chem. 2000, 275, 13330–13335. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LFD, Vehicle | LFD, Insulin | HFSD, Vehicle | HFSD, Insulin | |||||
|---|---|---|---|---|---|---|---|---|
| IRf/f Cre (−) | IRf/f Cre (+) | IRf/f Cre (−) | IRf/f Cre (+) | IRf/f Cre (−) | IRf/f Cre (+) | IRf/f Cre (−) | IRf/f Cre (+) | |
| Body Weight (g) | 25.7 ± 0.7 | 27.4 ± 0.7 | 26.1 ± 0.5 | 25.5 ± 0.6 | 31.4 ± 0.7 * | 33 ± 1.2 * | 30.2 ± 0.6 * | 29.3 ± 0.9 * |
| Fasted Plasma Glucose (mM) | 11.1 ± 0.6 | 10.0 ± 0.6 | 9.9 ± 0.5 | 9.9 ± 0.6 | 14.6 ± 0.6 * | 14 ± 0.8 * | 13.8 ± 0.3 * | 13 ± 0.4 * |
| Fed Plasma Glucose (mM) | 10.5 ± 0.3 | 10.1 ± 0.2 | 10.4 ± 0.2 | 9.5 ± 0.3 | 12.9 ± 0.2 * | 12.3 ± 0.4 * | 12.5 ± 0.4 * | 12.7 ± 0.7 * |
| Fed Insulin (pM) | 200 ± 85 | 200 ± 58 | 540 ± 120 $ | 469 ± 88 $ | 131± 23 | 88 ± 12 | 474± 110 $ | 507 ± 148$ |
| Fed IGF-1 (pg/mL) | 506 ± 81 | 404 ± 90 | 472 ± 45 | 402 ± 36 | 776 ± 64 * | 623 ± 79 * | 604 ± 60 * | 570 ± 46 * |
| Fed VEGF (pg/mL) | 21.7 ± 1.5 | 24.9 ± 1.43 | 23.4 ± 1.44 | 25.7 ± 1.86 | 25.5 ± 1.39 | 23.2 ± 1.42 | 27.7 ± 2.10 | 26.7 ± 1.70 |
| Fed Triglycerides (mM) | 1.4 ± 0.1 | 1.6 ± 0.1 | 1.1 ± 0.1 $ | 0.8 ± 0.2 $ | 1.7 ± 0.1 * | 1.5 ± 0.2 * | 1.3 ± 0.1 *$ | 1.3 ± 0.2 *$ |
| Fed Free Fatty Acids (mM) | 0.63 ± 0.04 | 0.58 ± 0.04 | 0.52 ± 0.03 $ | 0.49 ± 0.04 $ | 0.75 ± 0.05 * | 0.66 ± 0.04 * | 0.59 ± 0.02 *$ | 0.58 ± 0.02 *$ |
| Total cholesterol (mM) | 1.4 ± 0.18 | 1.3 ± 0.17 | 1.4 ± 0.31 | 1.4 ± 0.43 | 2.7 ± 0.31 * | 2.3 ± 0.38 * | 3.1 ± 1.0 * | 2.3 ± 0.44 * |
| LFD, Vehicle | LFD, Insulin | HFSD, Vehicle | HFSD, Insulin | |||||
|---|---|---|---|---|---|---|---|---|
| IRf/fCre (−) | IRf/f Cre (+) | IRf/f Cre (−) | IRf/f Cre (+) | IRf/f Cre (−) | IRf/f Cre (+) | IRf/f Cre (−) | IRf/f Cre (+) | |
| Body Weight (g) | 25.7 ± 0.7 | 26 ± 0.5 | 26.1 ± 0.5 | 25.8 ± 0.5 | 31.4 ± 0.7 * | 29.4 ± 0.4 * | 30.2 ± 0.6 * | 31 ± 0.4 * |
| Fasted Plasma Glucose (mM) | 11.1 ± 0.6 | 11.2 ± 0.3 | 9.9 ± 0.5 | 10.3 ± 0.7 | 14.6 ± 0.6 * | 14.6 ± 0.7 * | 13.8 ± 0.3 * | 14.5 ± 0.9 * |
| Fed Plasma Glucose (mM) | 10.5 ± 0.3 | 10.4 ± 0.3 | 10.4 ± 0.2 | 9.9 ± 0.6 | 12.8 ± 0.2 * | 12.5 ± 0.4 * | 12.8 ± 0.4 * | 12.2 ± 0.8 * |
| Fed Insulin (pM) | 200 ± 85 | 111 ± 26 | 540 ± 120 $ | 439 ± 103 $ | 131 ± 23 | 87 ± 12 | 474 ± 110 $ | 344 ± 55 $ |
| Fed IGF-1 (pg/mL) | 506 ± 81 | 562 ± 33 | 472 ± 45 | 376 ± 53 | 776 ± 64 * | 650 ± 14 * | 604 ± 60 * | 560 ± 44 * |
| Fed VEGF (pg/mL) | 21.7 ± 1.5 | 25.5 ± 1.39 | 23.4 ± 1.44 | 27.7 ± 2.10 | 25.5 ± 1.39 | 24.2 ± 1.44 | 27.7 ± 2.10 | 27.2 ± 1.09 |
| Fed Triglycerides (mM) | 1.4 ± 0.1 | 1.5 ± 0.2 | 1.1 ± 0.1 $ | 0.9 ± 0.1 $ | 1.7 ± 0.1 * | 1.5 ± 0.1 * | 1.3 ± 0.1 *$ | 1.5 ± 0.1 *$ |
| Fed Free Fatty Acids (mM) | 0.63 ± 0.04 | 0.54 ± 0.04 | 0.52 ± 0.03 $ | 0.40 ± 0.04 $ | 0.75 ± 0.05 * | 0.69 ± 0.02 * | 0.59 ± 0.02 *$ | 0.63 ± 0.02 *$ |
| Total cholesterol (mM) | 1.4 ± 0.18 | 1.7 ± 0.21 | 1.4 ± 0.31 | 1.1 ± 0.27 | 2.7 ± 0.31 * | 2.0 ± 0.16 * | 3.1 ± 1.0 * | 2.1 ± 0.19 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez Medina, M.; Liu, Z.; Wang, J.; Zhang, C.; Cash, S.B.; Cummins, C.L.; Giacca, A. Cell-Specific Effects of Insulin in a Murine Model of Restenosis Under Insulin-Sensitive and Insulin-Resistant Conditions. Cells 2024, 13, 1387. https://doi.org/10.3390/cells13161387
Gonzalez Medina M, Liu Z, Wang J, Zhang C, Cash SB, Cummins CL, Giacca A. Cell-Specific Effects of Insulin in a Murine Model of Restenosis Under Insulin-Sensitive and Insulin-Resistant Conditions. Cells. 2024; 13(16):1387. https://doi.org/10.3390/cells13161387
Chicago/Turabian StyleGonzalez Medina, Marel, Zhiwei Liu, Johny Wang, Cindy Zhang, Sarah B. Cash, Carolyn L. Cummins, and Adria Giacca. 2024. "Cell-Specific Effects of Insulin in a Murine Model of Restenosis Under Insulin-Sensitive and Insulin-Resistant Conditions" Cells 13, no. 16: 1387. https://doi.org/10.3390/cells13161387