
iPSC-Derived Cardiomyocytes as a Disease Model to Understand the Biology of Congenital Heart Defects

Abstract
:1. Introduction
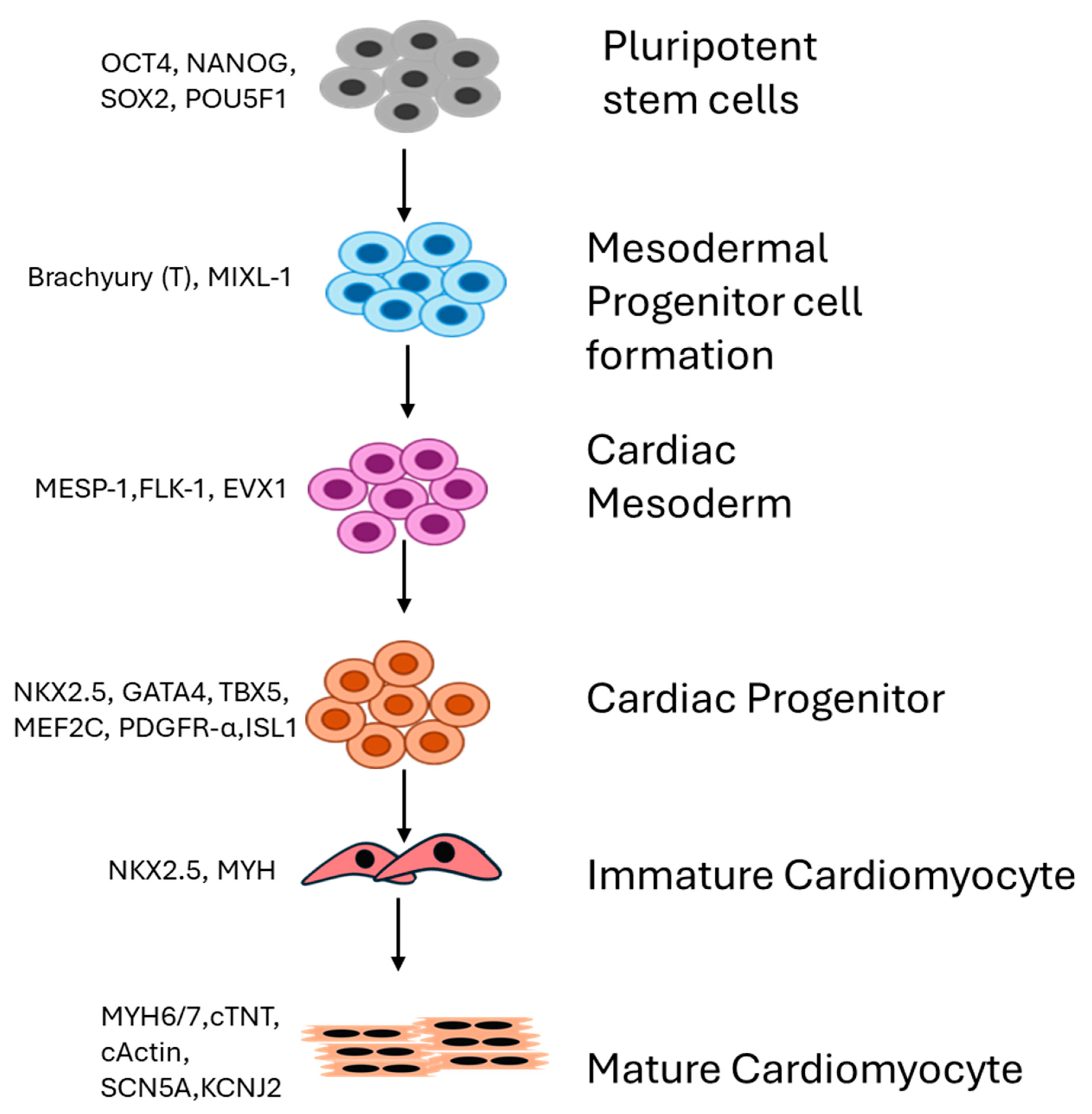
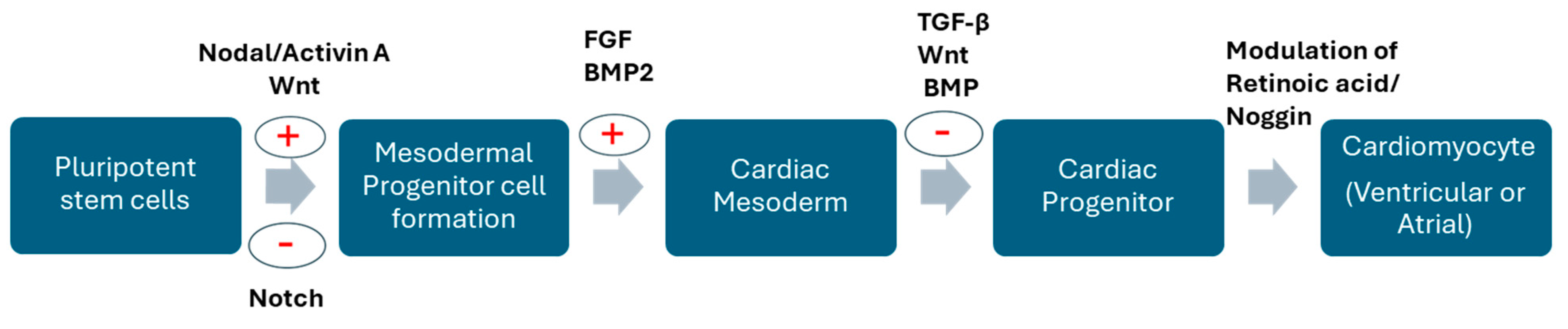
2. Differential Expression of Genes Drives Generation of Cardiomyocytes
3. iPSC-Derived Cardiomyocytes in Disease Modeling
3.1. hiPSCs in Tetrology of Fallot
3.2. hiPSCs in Hypoplastic Left Heart Syndrome
3.3. hiPSCs in Pulmonary Atresia with Intact Ventricular Septum
3.4. hiPSC in Drug Screening
4. Limitations of iPSCs as a Model System
5. Conclusions

{kind=link}
{kind=link}
| Condition | Disease Model | Experimental Methods | Observation | Reference |
|---|---|---|---|---|
| TOF | Patient-specific TOF-iPSC-derived CMs | Whole genome and transcriptome sequencing of iPSC-CMs from TOF patients and healthy individuals | (1) Somatic mutation in DNA-binding domain of P53 in one clone—May point to functional impact, (2) Increased DEG observed in control vs. TOF patient CMs, (3) Collagen-related genes are more affected, (4) BICC1 and MYH11 downregulated in CM d60 in TOF and mutation in FBLN2 and DAAM2 | Grunert et al., 2020 [40] |
| TOF and SVD | Patient-specific TOF and SVD-iPSC-derived CMs | RNA Sequencing Analysis of iPSC-CM from TOF, SVD, and healthy individuals | Significantly distinct transcriptomes in iPSC-CMs from SVD and TOF compared to non-CHD iPSC-CMs | Kitani et al., 2020 [41] |
| TOF and SVD | Patient-specific TOF and SVD-iPSC-derived CMs | RNA-seq datasets of Patient-specific TOF and SVD-iPSC-derived CMs from Gene Expression Omnibus database used to study genome-wide Alternative splicing (AS) events | Abnormal AS events observed in TOF and SVD iPSC-CMs compared to non-CHD iPSC-CMs | Xu et al., 2022 [42] |
| HLHS | Patient-specific HLHS-iPSC-derived CMs | Investigated cellular, structural, and functional properties of HLHS iPSC-CMs. Employed RNA-seq analysis and whole-exome sequencing analysis | Decreased beating rate, disorganised sarcomeres, dysfunctional sarcoplasmic reticulum, and deleterious variants in NOTCH receptor and genes involved in its signaling pathway | Yang et al., 2017 [72] |
| HLHS | Patient-specific HLHS-iPSC-derived CMs | iPSC-CMs from HLH patients, sarcomere immunostaining. Examined the impact of MYH6-R443P variant by inserting it into iPSCs from healthy individuals | Insertion of variant caused dysmorphic sarcomere phenotype and contractile dysfunction in healthy iPSC-CMs. Correcting the MYH6-R443P variant using CRISPR/Cas9 gene editing rescued the iPSC-CMs from reduced efficiency in CM differentiation, sacromere dysmorphism, and abnormal contraction rates | Kim et al., 2020 [44] |
| HLHS | Patient-specific HLHS-iPSC-derived CMs | Comprehensive analysis of HLHS-iPSC-CMs for testing the efficiency of cardiomyocyte differentiation, contractility and calcium handling, mPTP closure, mitochondrial dynamics, and respiration and corroborated the results with scRNA-seq analysis | Mitochondrial dysfunction and oxidative stress in HLHS; points to potential therapeutic interventions by targeting mPTP closure with sildenafil or suppression of UPR with TUDCA | Xu et al., 2022 [45] |
| HLHS | Patient-specific HLHS-iPSC-derived endocardial endothelial cells co-cultured with iPSC-CMs | Single-cell transcriptomic analysis of HLHS-iPSC endocardium and human fetal heart tissue | Defect in endocardial function and endocardium–myocardium cross-talk in HLHS | Miao et al., 2020 [73] |
| PA-IVS-1v | Patient-specific PA-IVS-1v-iPSC-CMs | Single-cell RNA sequencing and assessment of biological pathways and metabolic functions | Proportion of SHF progenitors reduced, FHF and epicardial progenitors increased. Seahorse assays show enhanced mitochondrial activity. Enhanced ATP and respiration in PA-IVS-1v compared to control and cardiac proliferation is compromised | Yu et al., 2024 [54] |
| PA-IVS | Patient-specific PA-IVS-iPSC-CMs | Single-cell transcriptomics and functional analysis of PA-IVS-iPSC-CMs and their engineered tissues models | Showed a downregulation of RV developmental pathways, contractility, and cardiac maturation | Lam et al., 2020 [57] |
Funding
Conflicts of Interest
References
- Jha, B.S.; Farnoodian, M.; Bharti, K. Regulatory considerations for developing a phase I investigational new drug application for autologous induced pluripotent stem cells-based therapy product. Stem Cells Transl. Med. 2020, 10, 198–208. [Google Scholar] [CrossRef]
- Balafkan, N.; Mostafavi, S.; Schubert, M.; Siller, R.; Liang, K.X.; Sullivan, G.; Bindoff, L.A. A method for differentiating human induced pluripotent stem cells toward functional cardiomyocytes in 96-well microplates. Sci. Rep. 2020, 10, 18498. [Google Scholar] [CrossRef] [PubMed]
- Ergir, E.; Oliver-De la Cruz, J.; Fernandes, S.; Cassani, M.; Niro, F.; Pereira-Sousa, D.; Vrbsky, J.; Vinarsky, V.; Perestrelo, A.; Debellis, D. Generation and Maturation of Human Ipsc-Derived 3d Organotypic Cardiac Microtissues in Long-Term Culture. Sci. Rep. 2022, 12, 17409. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Bao, X.; Wang, Y.; Lu, T.; Zhang, L. Human induced pluripotent stem cell (hiPSC)-derived cardiomyocyte modelling of cardiovascular diseases for natural compound discovery. Biomed. Pharmacother. 2023, 157, 113970. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, F.; Patel, P.; Kitsuka, T.; Arai, K. The exciting realities and possibilities of ips-derived cardiomyocytes. Bioengineering 2023, 10, 237. [Google Scholar] [CrossRef] [PubMed]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with als can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Hanna, J.; Markoulaki, S.; Schorderet, P.; Carey, B.W.; Beard, C.; Wernig, M.; Creyghton, M.P.; Steine, E.J.; Cassady, J.P.; Foreman, R.; et al. Direct reprogramming of terminally differentiated mature b lymphocytes to pluripotency. Cell 2008, 133, 250–264. [Google Scholar] [CrossRef]
- Moon, J.-H.; Heo, J.S.; Kim, J.S.; Jun, E.K.; Lee, J.H.; Kim, A.; Kim, J.; Whang, K.Y.; Kang, Y.-K.; Yeo, S.; et al. Reprogramming fibroblasts into induced pluripotent stem cells with Bmi1. Cell Res. 2011, 21, 1305–1315. [Google Scholar] [CrossRef]
- Huangfu, D.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 2008, 26, 1269–1275. [Google Scholar] [CrossRef]
- Mali, P.; Ye, Z.; Hommond, H.H.; Yu, X.; Lin, J.; Chen, G.; Zou, J.; Cheng, L. Improved efficiency and pace of generating induced pluripotent stem cells from human adult and fetal fibroblasts. Stem Cells 2008, 26, 1998–2005. [Google Scholar] [CrossRef]
- Shi, Y.; Desponts, C.; Do, J.T.; Hahm, H.S.; Schöler, H.R.; Ding, S. Induction of pluripotent stem cells from mouse embryonic fibroblasts by oct4 and klf4 with small-molecule Compounds. Cell Stem Cell 2008, 3, 568–574. [Google Scholar] [CrossRef]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef]
- González, F.; Boué, S.; Belmonte, J.C.I. Methods for making induced pluripotent stem cells: Reprogramming à la carte. Nat. Rev. Genet. 2011, 12, 231–242. [Google Scholar] [CrossRef]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef]
- Raad, F.S.; Khan, T.A.; Esser, T.U.; Hudson, J.E.; Seth, B.I.; Fujita, B.; Gandamala, R.; Tietze, L.F.; Zimmermann, W.-H. Chalcone-Supported Cardiac Mesoderm Induction in Human Pluripotent Stem Cells for Heart Muscle Engineering. ChemMedChem 2021, 16, 3300–3305. [Google Scholar] [CrossRef]
- Mishina, Y.; Suzuki, A.; Ueno, N.; Behringer, R.R. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev. 1995, 9, 3027–3037. [Google Scholar] [CrossRef]
- Winnier, G.; Blessing, M.; Labosky, P.A.; Hogan, B.L. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995, 9, 2105–2116. [Google Scholar] [CrossRef]
- Gu, S.; Feng, X.-H. TGF-β Signaling in Cancer. Acta Biochim. Biophys. Sin. 2018, 50, 941–949. [Google Scholar] [CrossRef]
- Yang, L.; Cai, C.-L.; Lin, L.; Qyang, Y.; Chung, C.; Monteiro, R.M.; Mummery, C.L.; Fishman, G.I.; Cogen, A.; Evans, S. Isl1Cre reveals a common Bmp pathway in heart and limb development. Development 2006, 133, 1575–1585. [Google Scholar] [CrossRef]
- Klaus, A.; Saga, Y.; Taketo, M.M.; Tzahor, E.; Birchmeier, W. Distinct roles of Wnt/β-catenin and Bmp signaling during early cardiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18531–18536. [Google Scholar] [CrossRef]
- Witman, N.; Zhou, C.; Beverborg, N.G.; Sahara, M.; Chien, K.R. Cardiac progenitors and paracrine mediators in cardiogenesis and heart regeneration. Semin. Cell Dev. Biol. 2019, 100, 29–51. [Google Scholar] [CrossRef]
- Lan, Y.; Pan, H.; Li, C.; Banks, K.M.; Sam, J.; Ding, B.; Elemento, O.; Goll, M.G.; Evans, T. TETs Regulate Proepicardial Cell Migration through Extracellular Matrix Organization during Zebrafish Cardiogenesis. Cell Rep. 2019, 26, 720–732.e4. [Google Scholar] [CrossRef]
- Evseenko, D.; Zhu, Y.; Schenke-Layland, K.; Kuo, J.; Latour, B.; Ge, S.; Scholes, J.; Dravid, G.; Li, X.; MacLellan, W.R.; et al. Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13742–13747. [Google Scholar] [CrossRef]
- Foley, A.C.; Mercola, M. Heart induction by Wnt antagonists depends on the homeodomain transcription factor Hex. Genes Dev. 2005, 19, 387–396. [Google Scholar] [CrossRef]
- Qyang, Y.; Martin-Puig, S.; Chiravuri, M.; Chen, S.; Xu, H.; Bu, L.; Jiang, X.; Lin, L.; Granger, A.; Moretti, A.; et al. The Renewal and Differentiation of Isl1+ Cardiovascular Progenitors Are Controlled by a Wnt/β-Catenin Pathway. Cell Stem Cell 2007, 1, 165–179. [Google Scholar] [CrossRef]
- Ai, D.; Fu, X.; Wang, J.; Lu, M.-F.; Chen, L.; Baldini, A.; Klein, W.H.; Martin, J.F. Canonical Wnt signaling functions in second heart field to promote right ventricular growth. Proc. Natl. Acad. Sci. USA 2007, 104, 9319–9324. [Google Scholar] [CrossRef]
- Mensah, I.K.; Emerson, M.L.; Tan, H.J.; Gowher, H. Cardiomyocyte Differentiation from mouse embryonic stem cells by wnt switch method. Cells 2024, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Varadkar, P.; Kraman, M.; Despres, D.; Ma, G.; Lozier, J.; McCright, B. Notch2 is required for the proliferation of cardiac neural crest-derived smooth muscle cells. Dev. Dyn. 2008, 237, 1144–1152. [Google Scholar] [CrossRef]
- MacGrogan, D.; Münch, J.; de la Pompa, J.L. Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 685–704. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, I.O.; Zhao, X.; Duester, G. Retinoic acid controls heart anteroposterior patterning by down-regulating Isl1 through the Fgf8 pathway. Dev. Dyn. 2008, 237, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, Y.-C.; Pratt, R.E.; Dzau, V.J.; Hodgkinson, C.P. Novel method of differentiating human induced pluripotent stem cells to mature cardiomyocytes via Sfrp2. Sci. Rep. 2023, 13, 3920. [Google Scholar] [CrossRef] [PubMed]
- Hirt, M.N.; Hansen, A.; Eschenhagen, T. Cardiac Tissue Engineering: State of the Art. Circ. Res. 2014, 114, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Choppe, J.; Kumar, A.; Engler, A.J.; McCulloch, A.D. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol. Biol. Cell 2017, 28, 1871–1882. [Google Scholar] [CrossRef]
- Ravenscroft, S.M.; Pointon, A.; Williams, A.W.; Cross, M.J.; Sidaway, J.E. Cardiac non-myocyte cells show enhanced pharmacological function suggestive of contractile maturity in stem cell derived cardiomyocyte microtissues. Toxicol. Sci. 2016, 152, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C., Jr.; Pabon, L.; et al. Fatty acids enhance the maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef]
- Chirico, N.; Kessler, E.L.; Maas, R.G.C.; Fang, J.; Qin, J.; Dokter, I.; Daniels, M.; Šarić, T.; Neef, K.; Buikema, J.-W.; et al. Small molecule-mediated rapid maturation of human induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res. Ther. 2022, 13, 531. [Google Scholar] [CrossRef] [PubMed]
- Parrotta, E.I.; Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Cuda, G. Modeling cardiac disease mechanisms using induced pluripotent stem cell-derived cardiomyocytes: Progress, promises and challenges. Int. J. Mol. Sci. 2020, 21, 4354. [Google Scholar] [CrossRef]
- Grunert, M.; Appelt, S.; Schönhals, S.; Mika, K.; Cui, H.; Cooper, A.; Cyganek, L.; Guan, K.; Sperling, S.R. Induced pluripotent stem cells of patients with Tetralogy of Fallot reveal transcriptional alterations in cardiomyocyte differentiation. Sci. Rep. 2020, 10, 10921. [Google Scholar] [CrossRef]
- Kitani, T.; Tian, L.; Zhang, T.; Itzhaki, I.; Zhang, J.Z.; Ma, N.; Liu, C.; Rhee, J.-W.; Romfh, A.W.; Lui, G.K.; et al. RNA sequencing analysis of induced pluripotent stem cell-derived cardiomyocytes from congenital heart disease patients. Circ. Res. 2020, 126, 923–925. [Google Scholar] [CrossRef]
- Xu, X.; Zou, R.; Liu, X.; Su, Q. Alternative splicing signatures of congenital heart disease and induced pluripotent stem cell-derived cardiomyocytes from congenital heart disease patients. Medicine 2022, 101, e30123. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Habibollah, S.; Tilgner, K.; Collin, J.; Barta, T.; Al-Aama, J.Y.; Tesarov, L.; Hussain, R.; Trafford, A.W.; Kirkwood, G.; et al. An Induced pluripotent stem cell model of hypoplastic left heart syndrome (hlhs) reveals multiple expression and functional differences in hlhs-derived cardiac myocytes. Stem Cells Transl. Med. 2014, 3, 416–423. [Google Scholar] [CrossRef]
- Kim, M.-S.; Fleres, B.; Lovett, J.; Anfinson, M.; Samudrala, S.S.K.; Kelly, L.J.; Teigen, L.E.; Cavanaugh, M.; Marquez, M.; Geurts, A.M.; et al. Contractility of induced pluripotent stem cell-cardiomyocytes with an myh6 head domain variant associated with hypoplastic left heart syndrome. Front. Cell Dev. Biol. 2020, 8, 440. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Jin, K.; Bais, A.S.; Zhu, W.; Yagi, H.; Feinstein, T.N.; Nguyen, P.K.; Criscione, J.D.; Liu, X.; Beutner, G. Uncompensated mitochondrial oxidative stress underlies heart failure in an ipsc-derived model of congenital heart disease. Cell Stem Cell 2022, 29, 840–855. [Google Scholar] [CrossRef]
- Diaz-Frias, J.; Guillaume, M. Tetralogy of Fallot; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Carli, D.; Moroni, A.; Eleonora, D.G.; Zonta, A.; Montin, D.; Licciardi, F.; Aidala, E.; Bordese, R.; Carlo, P.N.; Brusco, A. Atypical microdeletion 22q11. 2 in a patient with tetralogy of fallot. J. Genet. 2021, 100, 5. [Google Scholar] [CrossRef] [PubMed]
- Page, D.J.; Miossec, M.J.; Williams, S.G.; Monaghan, R.M.; Fotiou, E.; Cordell, H.J.; Sutcliffe, L.; Topf, A.; Bourgey, M.; Bourque, G.; et al. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of fallot. Circ. Res. 2019, 124, 553–563. [Google Scholar] [CrossRef]
- Cioffi, S.; Martucciello, S.; Fulcoli, F.G.; Bilio, M.; Ferrentino, R.; Nusco, E.; Illingworth, E. Tbx1 regulates brain vascularization. Hum. Mol. Genet. 2013, 23, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, R.M.; Paige, D.J.; Keavney, B.D. Identification of the major genetic contributors to tetralogy of fallot. Eur. J. Hum. Genet. 2020, 28, 286. [Google Scholar]
- Harvey, D.C.; Verma, R.; Sedaghat, B.; Hjelm, B.E.; Morton, S.U.; Seidman, J.G.; Kumar, S.R. Mutations in genes related to myocyte contraction and ventricular septum development in non-syndromic tetralogy of Fallot. Front. Cardiovasc. Med. 2023, 10, 1249605. [Google Scholar] [CrossRef]
- Liu, C.-F.; Wilcox, J.; Engelman, T.; Tang, W.H. Bicc1: A Potential biomarker for heart failure and cardio-renal syndrome. J. Card. Fail. 2024, 30, 137–138. [Google Scholar] [CrossRef]
- Kraus, M.R.-C.; Clauin, S.; Pfister, Y.; Di Maïo, M.; Ulinski, T.; Constam, D.; Bellanné-Chantelot, C.; Grapin-Botton, A. Two mutations in human BICC1 resulting in Wnt pathway hyperactivity associated with cystic renal dysplasia. Hum. Mutat. 2011, 33, 86–90. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, C.; Ye, S.; Xu, Z.; Lin, H.; Texter, K.; Shukla, V.; Ghadiali, S.; Ma, Q.; Garg, V.; et al. Abnormal progenitor cell differentiation and cardiomyocyte proliferation in hypoplastic right heart syndrome. Circulation 2024, 149, 888–891. [Google Scholar] [CrossRef] [PubMed]
- Paige, S.L.; Galdos, F.X.; Lee, S.; Chin, E.T.; Ranjbarvaziri, S.; Feyen, D.A.; Darsha, A.K.; Xu, S.; Ryan, J.A.; Beck, A.L.; et al. Patient-specific induced pluripotent stem cells implicate intrinsic impaired contractility in hypoplastic left heart syndrome. Circulation 2020, 142, 1605–1608. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Yoshida, M.; Tarui, S.; Hirata, M.; Nagai, Y.; Kasahara, S.; Naruse, K.; Ito, H.; Sano, S.; Oh, H. Directed differentiation of patient-specific induced pluripotent stem cells identifies the transcriptional repression and epigenetic modification of nkx2–5, hand1, and notch1 in hypoplastic left heart syndrome. PLoS ONE 2014, 9, e102796. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.; Keung, W.; Chan, C.; Geng, L.; Wong, N.; Brenière-Letuffe, D.; Li, R.A.; Cheung, Y. Single-cell transcriptomics of engineered cardiac tissues from patient-specific induced pluripotent stem cell-derived cardiomyocytes reveals abnormal developmental trajectory and intrinsic contractile defects in hypoplastic right heart syndrome. J. Am. Heart Assoc. 2020, 9, e016528. [Google Scholar] [CrossRef]
- Yu, Y.; Deschenes, I.; Zhao, M.-T. Precision medicine for long QT syndrome: Patient-specific iPSCs take the lead. Expert Rev. Mol. Med. 2023, 25, e5. [Google Scholar] [CrossRef]
- Schwartz, P.J. The Long QT Syndrome. Curr. Probl. Cardiol. 1997, 22, 297–351. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Amin, A.S.; Postema, P.G. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart 2021, 108, 332–338. [Google Scholar] [CrossRef]
- Berul, C.I. Congenital Long-QT Syndromes: Who’s at risk 26? Circulation 2008, 117, 2178–2180. [Google Scholar] [CrossRef]
- Modell, S.M.; Lehmann, M.H. The long QT syndrome family of cardiac ion channelopathies: A HuGE review. Anesth. Analg. 2006, 8, 143–155. [Google Scholar] [CrossRef]
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M. Spectrum of mutations in Long-QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE. Circulation 2000, 102, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Tester, D.J.; Ackerman, M.J. Genetics of long QT syndrome. Methodist DeBakey Cardiovasc. J. 2014, 10, 29–33. [Google Scholar] [CrossRef] [PubMed]
- van Mil, A.; Balk, G.M.; Neef, K.; Buikema, J.W.; Asselbergs, F.W.; Wu, S.M.; Doevendans, P.A.; Sluijter, J.P.G. Modelling inherited cardiac disease using human induced pluripotent stem cell-derived cardiomyocytes: Progress, pitfalls, and potential. Cardiovasc. Res. 2018, 114, 1828–1842. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-qt syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef]
- Wang, F.; Han, Y.; Sang, W.; Wang, L.; Liang, X.; Wang, L.; Xing, Q.; Guo, Y.; Zhang, J.; Zhang, L. In vitro drug screening using ipsc-derived cardiomyocytes of a long qt-syndrome patient carrying kcnq1 & trpm4 dual mutation: An experimental personalized treatment. Cells 2022, 11, 2495. [Google Scholar]
- Egashira, T.; Yuasa, S.; Suzuki, T.; Aizawa, Y.; Yamakawa, H.; Matsuhashi, T.; Ohno, Y.; Tohyama, S.; Okata, S.; Seki, T.; et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc. Res. 2012, 95, 419–429. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2016, 16, 115–130. [Google Scholar] [CrossRef]
- Casini, S.; Verkerk, A.O.; Remme, C.A. Human iPSC-derived cardiomyocytes for investigation of disease mechanisms and therapeutic strategies in inherited arrhythmia syndromes: Strengths and limitations. Cardiovasc. Drugs Ther. 2017, 31, 325–344. [Google Scholar] [CrossRef]
- Rao, K.S.; Kameswaran, V.; Bruneau, B.G. Modeling congenital heart disease: Lessons from mice, hPSC-based models, and organoids. Genes Dev. 2022, 36, 652–663. [Google Scholar] [CrossRef]
- Yang, C.; Xu, Y.; Yu, M.; Lee, D.; Alharti, S.; Hellen, N.; Shaik, N.A.; Banaganapalli, B.; Mohamoud, H.S.A.; Elango, R.; et al. Induced pluripotent stem cell modelling of HLHS underlines the contribution of dysfunctional NOTCH signalling to impaired cardiogenesis. Hum. Mol. Genet. 2017, 26, 3031–3045. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Tian, L.; Martin, M.; Paige, S.L.; Galdos, F.X.; Li, J.; Klein, A.; Zhang, H.; Ma, N.; Wei, Y.; et al. Intrinsic endocardial defects contribute to hypoplastic left heart syndrome. Cell Stem Cell 2020, 27, 574–589.e8. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pushpan, C.K.; Kumar, S.R. iPSC-Derived Cardiomyocytes as a Disease Model to Understand the Biology of Congenital Heart Defects. Cells 2024, 13, 1430. https://doi.org/10.3390/cells13171430
Pushpan CK, Kumar SR. iPSC-Derived Cardiomyocytes as a Disease Model to Understand the Biology of Congenital Heart Defects. Cells. 2024; 13(17):1430. https://doi.org/10.3390/cells13171430
Chicago/Turabian StylePushpan, Chithra K., and Subramanyan Ram Kumar. 2024. "iPSC-Derived Cardiomyocytes as a Disease Model to Understand the Biology of Congenital Heart Defects" Cells 13, no. 17: 1430. https://doi.org/10.3390/cells13171430