Human Multi-Lineage Liver Organoid Model Reveals Impairment of CYP3A4 Expression upon Repeated Exposure to Graphene Oxide

 , , ,
, , ,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Liver Organoid Formation and Culture
2.3. Cytotoxicity Assay
2.4. NMR-Based Exo-Metabolomics
2.5. LO 2D Projection Area Measurements
2.6. CYP3A4 Activity Assay
2.7. Albumin Quantification
2.8. Liver Organoid Treatment
2.9. Graphene Oxide Characterization
2.10. Cell Viability Assay
2.11. Gene Expression Analysis
2.12. DNA Quantification
2.13. Western Blot
2.14. Histology
2.15. SA-β-gal Activity Detection
2.16. Statistical Analysis
3. Results and Discussion
3.1. Structural and Functional Characterization of LOs
3.1.1. Self-Assembling of LOs and Characterization of Their Size Stability
3.1.2. Exo-Metabolome of LOs by NMR: Aerobic Glycolysis Supports LO Growth and Stabilization
3.1.3. LOs Do Not Exhibit Signs of Apoptosis or Cellular Senescence until Day 29
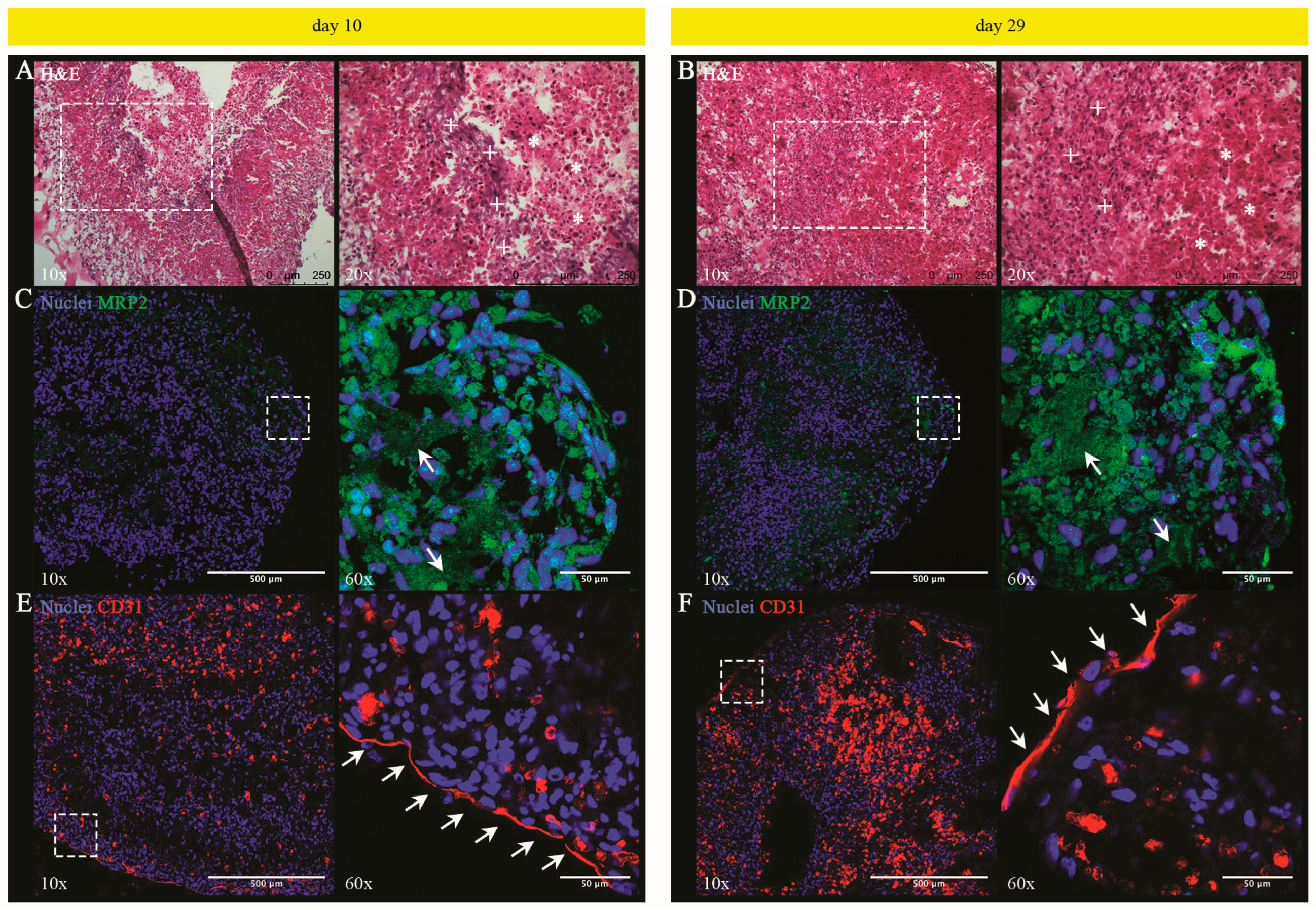
3.1.4. Histological and Phenotypic Analyses of LOs: Evidence of a Tissue Characterized by Hepatocyte-Derived Primitive Bile Canalicular Networks and LSEC-Formed Tube-like Structures
3.1.5. UHHs in LO Retain the Hepatocellular Functionality until Day 29
3.2. Repeated Exposure of LOs to GO and Toxicological Impact
3.2.1. Physical-Chemical Characterization of GO in LOM Medium and Evaluation of Penetration by Histological Analysis
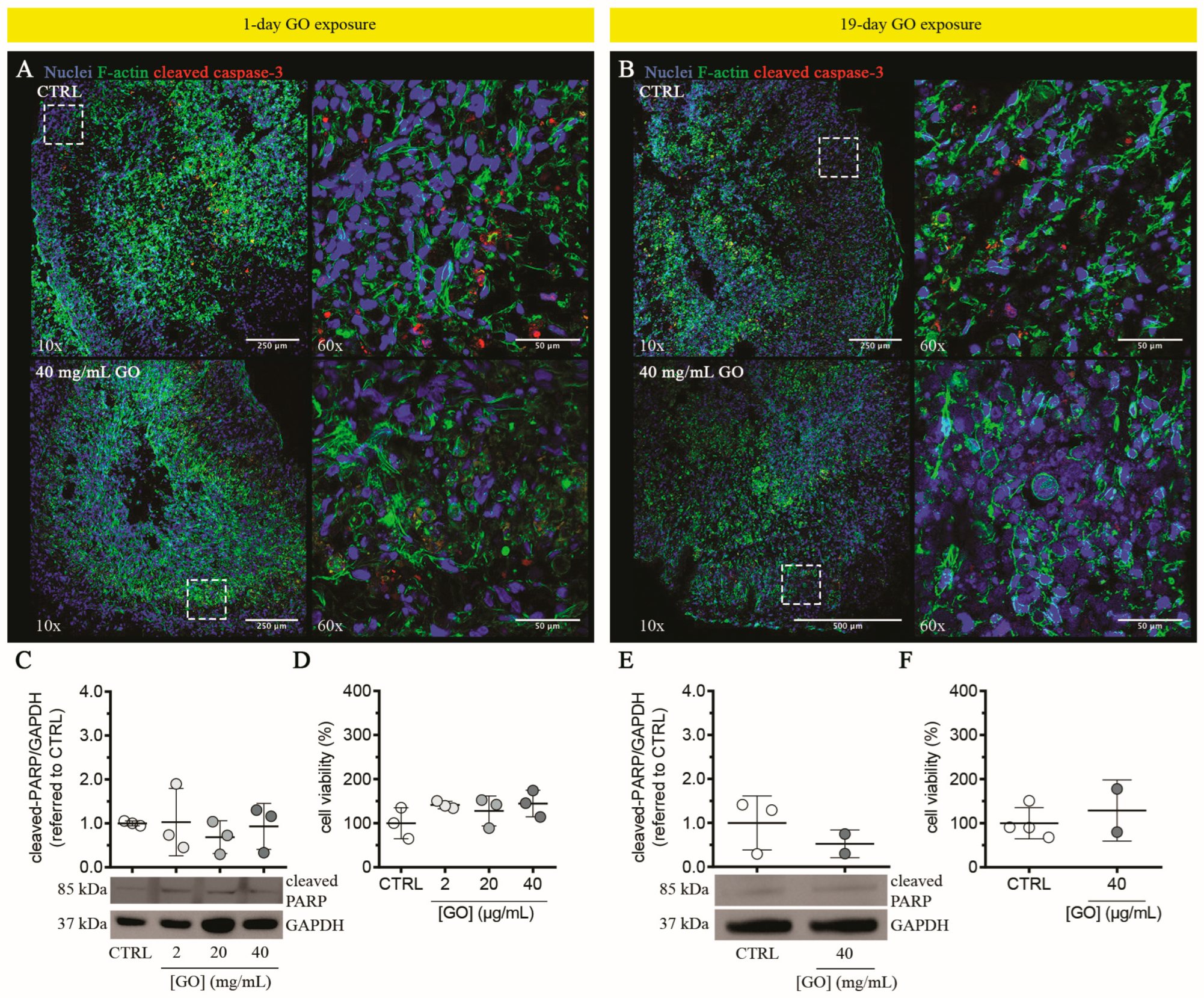
3.2.2. Graphene Oxide Does Not Induce Cytotoxicity in Liver Organoids upon Single or Repeated Exposure
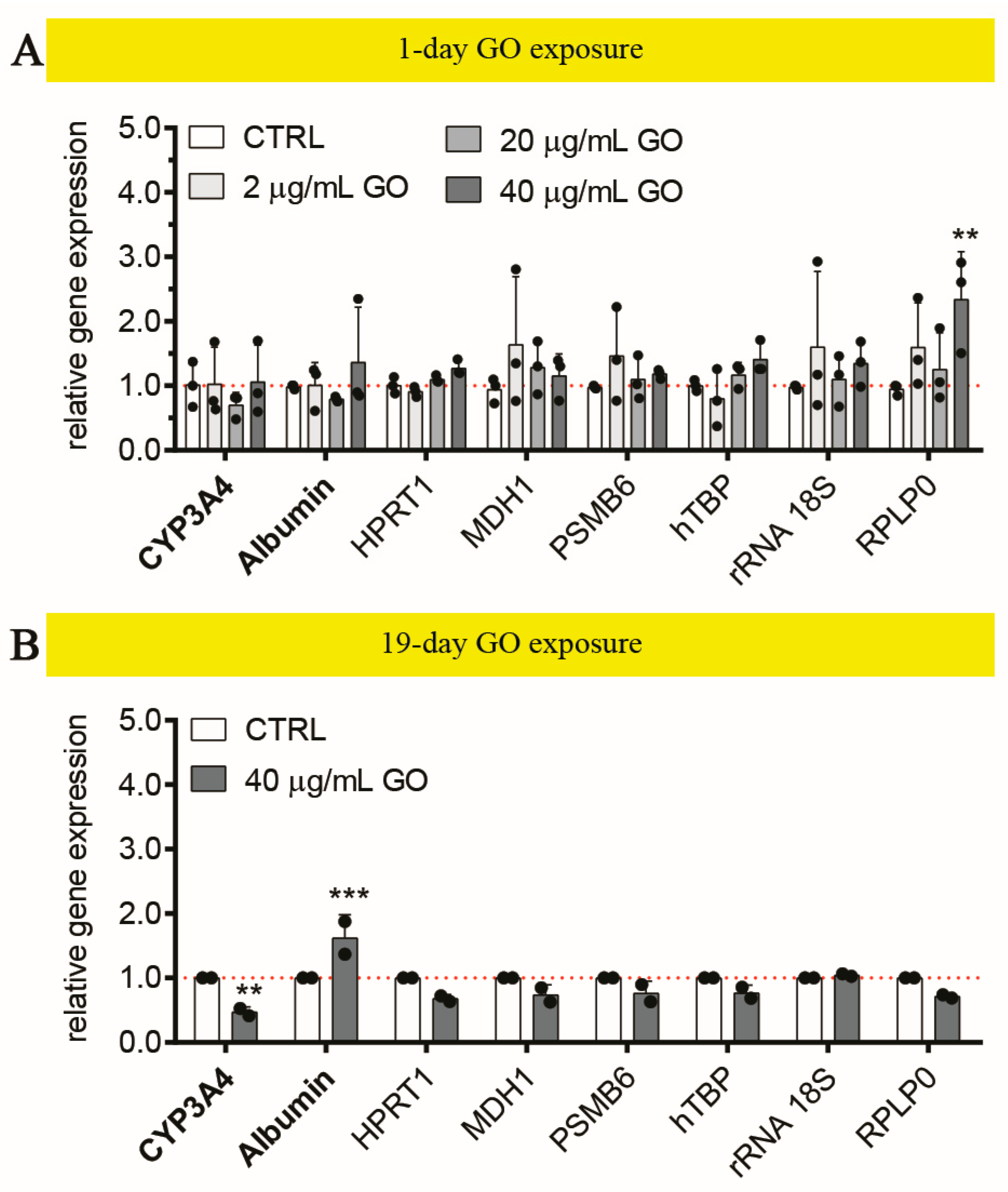
3.2.3. Graphene Oxide Down-Regulates CYP3A4 in Liver Organoids upon Repeated Exposure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2D | two-dimensional |
| 3D | three-dimensional |
| α-1-AT | α-1-antitrypsin |
| ABCG2 | ATP binding cassette subfamily G member 2 |
| ANOVA | analysis of variance |
| ATP | adenosine 5′-triphosphate |
| BCAA | branched-chain amino acid |
| BFC | 7-benzyloxy-4-trifluoromethylcoumarin |
| BSA | bovine serum albumin |
| CD31/PECAM-1 | cluster of differentiation 31/platelet endothelial cell adhesion molecule-1 |
| CDKN1A | cyclin dependent kinase inhibitor 1A |
| cDNA | complementary DNA |
| CoA | coenzyme A |
| CTRL | un-treated, control condition |
| CYP | cytochrome P450 (family…subfamily…member…) |
| D2O | deuterium oxide |
| DH | hydrodynamic diameter |
| DLS | dynamic light scattering |
| DMEM | Dulbecco’s modified Eagle medium |
| DNA | deoxyribonucleic acid |
| DPBS | Dulbecco’s phosphate buffered saline |
| ELISA | enzyme-linked immunosorbent assay |
| F-actin | filamentous actin |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GO | graphene oxide |
| H&E | hematoxylin and eosin |
| hbmMSC | human bone marrow-derived mesenchymal stromal cell |
| HFC | 7-hydroxy-4-trifluoromethylcoumarin |
| HHPM | hepatocyte high-performance medium |
| HPRT1 | hypoxanthine phosphoribosyltransferase 1 |
| KRT8/18 | keratin 8/18 |
| LDH | lactate dehydrogenase |
| LO | liver organoid |
| LOM | liver-organoid medium |
| LSEC | upcyte® liver sinusoidal endothelial cell |
| LSECm | LSEC culture medium |
| MDH1 | malate dehydrogenase 1 |
| MRP2/ABCC2 | multidrug resistance-associated protein 2/ATP binding cassette subfamily C member 2 |
| NASH | non-alcoholic steatohepatitis |
| NMR | nuclear magnetic resonance |
| ns | not significant |
| OECD | organisation for economic co-operation and development |
| OSM | oncostatin M |
| OXPHOS | oxidative phosphorylation |
| PARP | poly(ADP-ribose) polymerase |
| PC1 or 2 | principal component 1 or 2 |
| PCA | principal component analysis |
| PHH | primary human hepatocyte |
| PSMB6 | proteasome 20S subunit beta 6 |
| PUMA | p53 up-regulated modulator of apoptosis |
| qPCR | quantitative real time polymerase chain reaction |
| RNA | ribonucleic acid |
| ROI | region of interest |
| RPLP0 | ribosomal protein lateral stalk subunit P0 |
| rRNA 18S | ribosomal RNA 18S |
| SA-β-gal | senescence-associated β-galactosidase |
| SASP | senescent associated secretory phenotype |
| SD | standard deviation |
| TBP | TATA binding protein |
| TCA | tricarboxylic acid |
| TSP | trimethylsilylpropanoic acid |
| UHH | upcyte® human hepatocyte |
| VE-cadherin | vascular endothelial cadherin |
| XME | xenobiotic-metabolizing enzyme |
References
- Dirven, H.; Vist, G.E.; Bandhakavi, S.; Mehta, J.; Fitch, S.E.; Pound, P.; Ram, R.; Kincaid, B.; Leenaars, C.H.C.; Chen, M.; et al. Performance of preclinical models in predicting drug-induced liver injury in humans: A systematic review. Sci. Rep. 2021, 11, 6403. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.B. Drug Safety Sciences and the Bottleneck in Drug Development. Clin. Pharmacol. Ther. 2011, 89, 788–790. [Google Scholar] [CrossRef]
- Gerets, H.H.J.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.; Bomze, D.; Heinz, S.; Ramachandran, S.D.; Noerenberg, A.; Cohen, M.; Shibolet, O.; Sklan, E.; Braspenning, J.; Nahmias, Y. Long-term culture and expansion of primary human hepatocytes. Nat. Biotechnol. 2015, 33, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Zeilinger, K.; Freyer, N.; Damm, G.; Seehofer, D.; Knöspel, F. Cell sources for in vitro human liver cell culture models. Exp. Biol. Med. 2016, 241, 1684–1698. [Google Scholar] [CrossRef]
- Tolosa, L.; Jiménez, N.; Pelechá, M.; Castell, J.V.; Gómez-Lechón, M.J.; Donato, M.T. Long-term and mechanistic evaluation of drug-induced liver injury in Upcyte human hepatocytes. Arch. Toxicol. 2019, 93, 519–532. [Google Scholar] [CrossRef]
- Soldatow, V.Y.; LeCluyse, E.L.; Griffith, L.G.; Rusyn, I. In vitro models for liver toxicity testing. Toxicol. Res. 2013, 2, 23–39. [Google Scholar] [CrossRef]
- Tutty, M.A.; Movia, D.; Prina-Mello, A. Three-dimensional (3D) liver cell models—A tool for bridging the gap between animal studies and clinical trials when screening liver accumulation and toxicity of nanobiomaterials. Drug Deliv. Transl. Res. 2022, 12, 2048–2074. [Google Scholar] [CrossRef]
- Fraser, K.; Bruckner, D.M.; Dordick, J.S. Advancing Predictive Hepatotoxicity at the Intersection of Experimental, in Silico, and Artificial Intelligence Technologies. Chem. Res. Toxicol. 2018, 31, 412–430. [Google Scholar] [CrossRef]
- Baudy, A.R.; Otieno, M.A.; Hewitt, P.; Gan, J.; Roth, A.; Keller, D.; Sura, R.; Van Vleet, T.R.; Proctor, W.R. Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry. Lab Chip 2020, 20, 215–225. [Google Scholar] [CrossRef]
- Kermanizadeh, A.; Valli, J.; Sanchez, K.; Hutter, S.; Pawlowska, A.; Whyte, G.; Moritz, W.; Stone, V. Particulate and drug-induced toxicity assessed in novel quadruple cell human primary hepatic disease models of steatosis and pre-fibrotic NASH. Arch. Toxicol. 2022, 96, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.T.U.H.; Dan, Y.Y.; Chan, Y.-S.; Ng, H.-H. Emerging liver organoid platforms and technologies. Cell Regen. 2021, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Zhang, Y.; Lv, M.; Zang, G.; Ng, S.S.; Chen, X. Advances in 3D cell culture for liver preclinical studies. Acta Biochim. Biophys. Sin. 2021, 53, 643–651. [Google Scholar] [CrossRef]
- Xu, Q. Human Three-Dimensional Hepatic Models: Cell Type Variety and Corresponding Applications. Front. Bioeng. Biotechnol. 2021, 9, 730008. [Google Scholar] [CrossRef]
- Brooks, A.; Liang, X.; Zhang, Y.; Zhao, C.-X.; Roberts, M.S.; Wang, H.; Zhang, L.; Crawford, D.H.G. Liver organoid as a 3D in vitro model for drug validation and toxicity assessment. Pharmacol. Res. 2021, 169, 105608. [Google Scholar] [CrossRef]
- Kammerer, S. Three-Dimensional Liver Culture Systems to Maintain Primary Hepatic Properties for Toxicological Analysis In Vitro. Int. J. Mol. Sci. 2021, 22, 10214. [Google Scholar] [CrossRef]
- Yoshida, T.; Kobayashi, M.; Uomoto, S.; Ohshima, K.; Hara, E.; Katoh, Y.; Takahashi, N.; Harada, T.; Usui, T.; Elbadawy, M.; et al. The potential of organoids in toxicologic pathology: Role of toxicologic pathologists in in vitro chemical hepatotoxicity assessment. J. Toxicol. Pathol. 2022, 35, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Brown, P.C.; Chow, E.C.Y.; Ewart, L.; Ferguson, S.S.; Fitzpatrick, S.; Freedman, B.S.; Guo, G.L.; Hedrich, W.; Heyward, S.; et al. 3D cell culture models: Drug pharmacokinetics, safety assessment, and regulatory consideration. Clin. Transl. Sci. 2021, 14, 1659–1680. [Google Scholar] [CrossRef]
- Ramachandran, S.D.; Schirmer, K.; Münst, B.; Heinz, S.; Ghafoory, S.; Wölfl, S.; Simon-Keller, K.; Marx, A.; Oie, C.I.; Ebert, M.P.; et al. In vitro generation of functional liver organoid-like structures using adult human cells. PLoS ONE 2015, 10, e0139345. [Google Scholar] [CrossRef]
- OECD. Test No. 407: Repeated Dose 28-Day Oral Toxicity Study in Rodents; OECD Guidelines for the Testing of Chemicals, Section 4; OECD: Paris, France, 2008; ISBN 9789264070684. [Google Scholar] [CrossRef]
- Hoseini-Ghahfarokhi, M.; Mirkiani, S.; Mozaffari, N.; Abdolahi Sadatlu, M.A.; Ghasemi, A.; Abbaspour, S.; Akbarian, M.; Farjadain, F.; Karimi, M. Applications of Graphene and Graphene Oxide in Smart Drug/Gene Delivery: Is the World Still Flat? Int. J. Nanomed. 2020, 15, 9469–9496. [Google Scholar] [CrossRef]
- Biru, E.I.; Necolau, M.I.; Zainea, A.; Iovu, H. Graphene Oxide–Protein-Based Scaffolds for Tissue Engineering: Recent Advances and Applications. Polymers 2022, 14, 1032. [Google Scholar] [CrossRef] [PubMed]
- Peña-Bahamonde, J.; Nguyen, H.N.; Fanourakis, S.K.; Rodrigues, D.F. Recent advances in graphene-based biosensor technology with applications in life sciences. J. Nanobiotechnol. 2018, 16, 75. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Chen, X.; Huang, P. Graphene-based nanomaterials for bioimaging. Adv. Drug Deliv. Rev. 2016, 105, 242–254. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, W.; Ahmed, A.; Mahon, C.S.; Müllner, M.; Cao, B.; Xia, T. Engineering Protective Polymer Coatings for Liver Microtissues. Chem. Res. Toxicol. 2019, 32, 49–56. [Google Scholar] [CrossRef]
- Meiboom, S.; GillModified, D. Spin-Echo Method for Measuring Nuclear Relaxation Times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Romaldini, A.; Spanò, R.; Catalano, F.; Villa, F.; Poggi, A.; Sabella, S. Sub-Lethal Concentrations of Graphene Oxide Trigger Acute-Phase Response and Impairment of Phase-I Xenobiotic Metabolism in Upcyte® Hepatocytes. Front. Bioeng. Biotechnol. 2022, 10, 867728. [Google Scholar] [CrossRef]
- Bjornsson, T.D.; Callaghan, J.T.; Einolf, H.J.; Fischer, V.; Gan, L.; Grimm, S.; Kao, J.; King, S.P.; Miwa, G.; Ni, L.; et al. The conduct of in vitro and in vivo drug-drug interaction studies: A Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab. Dispos. 2003, 31, 815–832. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, D.; Sánchez-Moreno, P.; Del Rio Castillo, A.E.; Bonaccorso, F.; Gatto, F.; Bardi, G.; Martín, C.; Vázquez, E.; Catelani, T.; Sabella, S.; et al. Biotransformation and Biological Interaction of Graphene and Graphene Oxide during Simulated Oral Ingestion. Small 2018, 14, 1800227. [Google Scholar] [CrossRef]
- Di Cristo, L.; Grimaldi, B.; Catelani, T.; Vázquez, E.; Pompa, P.P.; Sabella, S. Repeated exposure to aerosolized graphene oxide mediates autophagy inhibition and inflammation in a three-dimensional human airway model. Mater. Today Bio 2020, 6, 100050. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 1993, 15, 532–537. [Google Scholar]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.-R.; Ueno, Y.; Zheng, Y.-W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- DeWane, G.; Salvi, A.M.; DeMali, K.A. Fueling the cytoskeleton—Links between cell metabolism and actin remodeling. J. Cell Sci. 2021, 134, jcs248385. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.R.; Warth, B.; Spilker, M.E.; Siuzdak, G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat. Biotechnol. 2018, 36, 316–320. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Allen, J.; Davey, H.M.; Broadhurst, D.; Heald, J.K.; Rowland, J.J.; Oliver, S.G.; Kell, D.B. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotechnol. 2003, 21, 692–696. [Google Scholar] [CrossRef]
- Behrends, V.; Williams, H.D.; Bundy, J.G. Metabolic footprinting: Extracellular metabolomic analysis. Methods Mol. Biol. 2014, 1149, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Kostidis, S.; Addie, R.D.; Morreau, H.; Mayboroda, O.A.; Giera, M. Quantitative NMR analysis of intra- and extracellular metabolism of mammalian cells: A tutorial. Anal. Chim. Acta 2017, 980, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Aranibar, N.; Borys, M.; Mackin, N.A.; Ly, V.; Abu-Absi, N.; Abu-Absi, S.; Niemitz, M.; Schilling, B.; Li, Z.J.; Brock, B.; et al. NMR-based metabolomics of mammalian cell and tissue cultures. J. Biomol. NMR 2011, 49, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Feng, Y.; Zhou, W.; Zhang, J.; Yu, Y.; Wang, X.; Zhang, P. 1H nuclear magnetic resonance-based extracellular metabolomic analysis of multidrug resistant Tca8113 oral squamous carcinoma cells. Oncol. Lett. 2015, 9, 2551–2559. [Google Scholar] [CrossRef]
- Cibecchini, G.; Veronesi, M.; Catelani, T.; Bandiera, T.; Guarnieri, D.; Pompa, P.P. Antiangiogenic Effect of Graphene Oxide in Primary Human Endothelial Cells. ACS Appl. Mater. Interfaces 2020, 12, 22507–22518. [Google Scholar] [CrossRef]
- Magrì, D.; Veronesi, M.; Sánchez-Moreno, P.; Tolardo, V.; Bandiera, T.; Pompa, P.P.; Athanassiou, A.; Fragouli, D. PET nanoplastics interactions with water contaminants and their impact on human cells. Environ. Pollut. 2021, 271, 116262. [Google Scholar] [CrossRef]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Posener, K.; Negelein, E. Über den Stoffwechsel der Carcinomzelle. Biochem. Z. 1924, 152, 309–344. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and Competition in the Evolution of ATP-Producing Pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Fitzgerald, G.; Soro-Arnaiz, I.; De Bock, K. The Warburg Effect in Endothelial Cells and its Potential as an Anti-angiogenic Target in Cancer. Front. Cell Dev. Biol. 2018, 6, 100. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Mann, G.; Mora, S.; Madu, G.; Adegoke, O.A.J. Branched-chain Amino Acids: Catabolism in Skeletal Muscle and Implications for Muscle and Whole-body Metabolism. Front. Physiol. 2021, 12, 702826. [Google Scholar] [CrossRef] [PubMed]
- Green, M.H. Are Fatty Acids Gluconeogenic Precursors? J. Nutr. 2020, 150, 2235–2238. [Google Scholar]
- Nakano, K.; Vousden, K.H. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Wang, J.; Thomas, H.R.; Li, Z.; Yeo, N.C.; Scott, H.E.; Dang, N.; Hossain, M.I.; Andrabi, S.A.; Parant, J.M. Puma, noxa, p53, and p63 differentially mediate stress pathway induced apoptosis. Cell Death Dis. 2021, 12, 659. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Müllauer, F.; Böck, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53- and Drug-Induced Apoptotic Responses Mediated by BH3-Only Proteins Puma and Noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef]
- Han, J.; Flemington, C.; Houghton, A.B.; Gu, Z.; Zambetti, G.P.; Lutz, R.J.; Zhu, L.; Chittenden, T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. USA 2001, 98, 11318–11323. [Google Scholar] [CrossRef]
- Chen, Q.; Ma, K.; Liu, X.; Chen, S.-H.; Li, P.; Yu, Y.; Leung, A.K.L.; Yu, X. Truncated PARP1 mediates ADP-ribosylation of RNA polymerase III for apoptosis. Cell Discov. 2022, 8, 3. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific Proteolytic Cleavage of Poly(ADP-ribose) Polymerase: An Early Marker of Chemotherapy-induced Apoptosis1. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Tewari, M.; Quan, L.T.; O’Rourke, K.; Desnoyers, S.; Zeng, Z.; Beidler, D.R.; Poirier, G.G.; Salvesen, G.S.; Dixit, V.M. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 1995, 81, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of Poly(ADP-ribose) Polymerase (PARP) Cleavage in Apoptosis. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Mattei, G.; Magliaro, C.; Giusti, S.; Ramachandran, S.D.; Heinz, S.; Braspenning, J.; Ahluwalia, A. On the adhesion-cohesion balance and oxygen consumption characteristics of liver organoids. PLoS ONE 2017, 12, e0173206. [Google Scholar] [CrossRef]
- Cui, Y.; König, J.; Buchholz, J.K.; Spring, H.; Leier, I.; Keppler, D. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol. Pharmacol. 1999, 55, 929–937. [Google Scholar]
- Kamisako, T.; Leier, I.; Cui, Y.; König, J.; Buchholz, U.; Hummel-Eisenbeiss, J.; Keppler, D. Transport of monoglucuronosyl and bisglucuronosyl bilirubin by recombinant human and rat multidrug resistance protein 2. Hepatology 1999, 30, 485–490. [Google Scholar] [CrossRef]
- Jedlitschky, G.; Hoffmann, U.; Kroemer, H.K. Structure and function of the MRP2 (ABCC2) protein and its role in drug disposition. Expert Opin. Drug Metab. Toxicol. 2006, 2, 351–366. [Google Scholar] [CrossRef]
- Schaefer, M.; Morinaga, G.; Matsui, A.; Schänzle, G.; Bischoff, D.; Süssmuth, R.D. Quantitative expression of hepatobiliary transporters and functional uptake of substrates in hepatic two-dimensional sandwich cultures: A comparative evaluation of upcyte and primary human hepatocytes. Drug Metab. Dispos. 2018, 46, 166–177. [Google Scholar] [CrossRef]
- Pandey, E.; Nour, A.S.; Harris, E.N. Prominent Receptors of Liver Sinusoidal Endothelial Cells in Liver Homeostasis and Disease. Front. Physiol. 2020, 11, 873. [Google Scholar] [CrossRef] [PubMed]
- Lalor, P. Human hepatic sinusoidal endothelial cells can be distinguished by expression of phenotypic markers related to their specialised functions in vivo. World J. Gastroenterol. 2006, 12, 5429. [Google Scholar] [CrossRef] [PubMed]
- Couvelard, A.; Scoazec, J.Y.; Feldmann, G. Expression of cell-cell and cell-matrix adhesion proteins by sinusoidal endothelial cells in the normal and cirrhotic human liver. Am. J. Pathol. 1993, 143, 738–752. [Google Scholar] [PubMed]
- DeLeve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef]
- Schaffner, F.; Popper, H. Capillarization of Hepatic Sinusoids in Man. Gastroenterology 1963, 44, 239–242. [Google Scholar] [CrossRef]
- DeLeve, L.D.; Wang, X.; Hu, L.; McCuskey, M.K.; McCuskey, R.S. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am. J. Physiol. Liver Physiol. 2004, 287, G757–G763. [Google Scholar] [CrossRef]
- Neubauer, K.; Wilfling, T.; Ritzel, A.; Ramadori, G. Platelet-endothelial cell adhesion molecule-1 gene expression in liver sinusoidal endothelial cells during liver injury and repair. J. Hepatol. 2000, 32, 921–932. [Google Scholar] [CrossRef]
- Baiocchini, A.; Del Nonno, F.; Taibi, C.; Visco-Comandini, U.; D’Offizi, G.; Piacentini, M.; Falasca, L. Liver sinusoidal endothelial cells (LSECs) modifications in patients with chronic hepatitis C. Sci. Rep. 2019, 9, 8760. [Google Scholar] [CrossRef]
- Kaden, T.; Noerenberg, A.; Boldt, J.; Sagawe, C.; Johannssen, T.; Rennert, K.; Raasch, M.; Evenburg, T. Generation & characterization of expandable human liver sinusoidal endothelial cells and their application to assess hepatotoxicity in an advanced in vitro liver model. Toxicology 2023, 483, 153374. [Google Scholar] [CrossRef]
- Vestweber, D. VE-Cadherin. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Malinova, T.S.; Angulo-Urarte, A.; Nüchel, J.; Tauber, M.; van der Stoel, M.M.; Janssen, V.; de Haan, A.; Groenen, A.G.; Tebbens, M.; Graupera, M.; et al. A junctional PACSIN2/EHD4/MICAL-L1 complex coordinates VE-cadherin trafficking for endothelial migration and angiogenesis. Nat. Commun. 2021, 12, 2610. [Google Scholar] [CrossRef] [PubMed]
- Dorland, Y.L.; Huveneers, S. Cell–cell junctional mechanotransduction in endothelial remodeling. Cell. Mol. Life Sci. 2017, 74, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Mariotti, M.; Thurston, G.; Smith, K.; Kunkel, R.; Brockhaus, M.; Lampugnani, M.G.; Martin-Padura, I.; Stoppacciaro, A.; Ruco, L.; et al. Vascular endothelial–cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 9815–9820. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.-S.; Nolan, D.J.; Butler, J.M.; James, D.; Babazadeh, A.O.; Rosenwaks, Z.; Mittal, V.; Kobayashi, H.; Shido, K.; Lyden, D.; et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 2010, 468, 310–315. [Google Scholar] [CrossRef]
- Géraud, C.; Evdokimov, K.; Straub, B.K.; Peitsch, W.K.; Demory, A.; Dörflinger, Y.; Schledzewski, K.; Schmieder, A.; Schemmer, P.; Augustin, H.G.; et al. Unique Cell Type-Specific Junctional Complexes in Vascular Endothelium of Human and Rat Liver Sinusoids. PLoS ONE 2012, 7, e34206. [Google Scholar] [CrossRef]
- Donato, M.T.; Jiménez, N.; Pelechá, M.; Tolosa, L. Oxidative-stress and long-term hepatotoxicity: Comparative study in Upcyte human hepatocytes and hepaRG cells. Arch. Toxicol. 2022, 96, 1021–1037. [Google Scholar] [CrossRef]
- Schaefer, M.; Schanzle, G.; Bischoff, D.; Sussmuth, R.D. Upcyte Human Hepatocytes: A Potent In Vitro Tool for the Prediction of Hepatic Clearance of Metabolically Stable Compounds. Drug Metab. Dispos. 2016, 44, 435–444. [Google Scholar] [CrossRef]
- Iwahori, T. CYP3A4 inducible model for in vitro analysis of human drug metabolism using a bioartificial liver. Hepatology 2003, 37, 665–673. [Google Scholar] [CrossRef]
- Achour, B.; Barber, J.; Rostami-Hodjegan, A. Expression of Hepatic Drug-Metabolizing Cytochrome P450 Enzymes and Their Intercorrelations: A Meta-Analysis. Drug Metab. Dispos. 2014, 42, 1349–1356. [Google Scholar] [CrossRef]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin-more than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, D.; Ursini, C.L.; Fresegna, A.M.; Ciervo, A.; Boccuni, F.; Ferrante, R.; Tombolini, F.; Maiello, R.; Chiarella, P.; Buresti, G.; et al. A follow-up study on workers involved in the graphene production process after the introduction of exposure mitigation measures: Evaluation of genotoxic and oxidative effects. Nanotoxicology 2022, 16, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Ursini, C.L.; Fresegna, A.M.; Ciervo, A.; Maiello, R.; Del Frate, V.; Folesani, G.; Galetti, M.; Poli, D.; Buresti, G.; Di Cristo, L.; et al. Occupational exposure to graphene and silica nanoparticles. Part II: Pilot study to identify a panel of sensitive biomarkers of genotoxic, oxidative and inflammatory effects on suitable biological matrices. Nanotoxicology 2021, 15, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Boccuni, F.; Ferrante, R.; Tombolini, F.; Natale, C.; Gordiani, A.; Sabella, S.; Iavicoli, S. Occupational exposure to graphene and silica nanoparticles. Part I: Workplace measurements and samplings. Nanotoxicology 2020, 14, 1280–1300. [Google Scholar] [CrossRef]
- Di Cristo, L.; Oomen, A.G.; Dekkers, S.; Moore, C.; Rocchia, W.; Murphy, F.; Johnston, H.J.; Janer, G.; Haase, A.; Stone, V.; et al. Grouping Hypotheses and an Integrated Approach to Testing and Assessment of Nanomaterials Following Oral Ingestion. Nanomaterials 2021, 11, 2623. [Google Scholar] [CrossRef]
- Tombolini, F.; Boccuni, F.; Ferrante, R.; Natale, C.; Marasco, L.; Mantero, E.; Del Rio Castillo, A.E.; Leoncino, L.; Pellegrini, V.; Sabella, S.; et al. An integrated and multi-technique approach to characterize airborne graphene flakes in the workplace during production phases. Nanoscale 2021, 13, 3841–3852. [Google Scholar] [CrossRef]
- Maiorano, G.; Sabella, S.; Sorce, B.; Brunetti, V.; Malvindi, M.A.; Cingolani, R.; Pompa, P.P. Effects of Cell Culture Media on the Dynamic Formation of Protein−Nanoparticle Complexes and Influence on the Cellular Response. ACS Nano 2010, 4, 7481–7491. [Google Scholar] [CrossRef]
- Li, J.; Diamante, G.; Ahn, I.S.; Wijaya, D.; Wang, X.; Chang, C.H.; Ha, S.; Immadisetty, K.; Meng, H.; Nel, A.; et al. Determination of the nanoparticle- and cell-specific toxicological mechanisms in 3D liver spheroids using scRNAseq analysis. Nano Today 2022, 47, 101652. [Google Scholar] [CrossRef]
- Cabral, H.; Matsumoto, Y.; Mizuno, K.; Chen, Q.; Murakami, M.; Kimura, M.; Terada, Y.; Kano, M.R.; Miyazono, K.; Uesaka, M.; et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotechnol. 2011, 6, 815–823. [Google Scholar] [CrossRef]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.-D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef]
- Niora, M.; Pedersbæk, D.; Münter, R.; Weywadt, M.F.D.V.; Farhangibarooji, Y.; Andresen, T.L.; Simonsen, J.B.; Jauffred, L. Head-to-Head Comparison of the Penetration Efficiency of Lipid-Based Nanoparticles into Tumor Spheroids. ACS Omega 2020, 5, 21162–21171. [Google Scholar] [CrossRef]
- Tang, L.; Gabrielson, N.P.; Uckun, F.M.; Fan, T.M.; Cheng, J. Size-Dependent Tumor Penetration and in Vivo Efficacy of Monodisperse Drug–Silica Nanoconjugates. Mol. Pharm. 2013, 10, 883–892. [Google Scholar] [CrossRef]
- Tchoryk, A.; Taresco, V.; Argent, R.H.; Ashford, M.; Gellert, P.R.; Stolnik, S.; Grabowska, A.; Garnett, M.C. Penetration and Uptake of Nanoparticles in 3D Tumor Spheroids. Bioconjug. Chem. 2019, 30, 1371–1384. [Google Scholar] [CrossRef]
- Wu, Y.; Feng, W.; Liu, R.; Xia, T.; Liu, S. Graphene Oxide Causes Disordered Zonation Due to Differential Intralobular Localization in the Liver. ACS Nano 2020, 14, 877–890. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, W.; Wu, B.; Yu, J.; Yu, H.; Zhang, X.-X.; Torres-Duarte, C.; Cherr, G.N. Low levels of graphene and graphene oxide inhibit cellular xenobiotic defense system mediated by efflux transporters. Nanotoxicology 2015, 10, 597–606. [Google Scholar] [CrossRef]
- Chatterjee, N.; Eom, H.-J.; Choi, J. A systems toxicology approach to the surface functionality control of graphene–cell interactions. Biomaterials 2014, 35, 1109–1127. [Google Scholar] [CrossRef]
- da Silva Frozza, C.O.; da Silva Ribeiro, T.; Gambato, G.; Menti, C.; Moura, S.; Pinto, P.M.; Staats, C.C.; Padilha, F.F.; Begnini, K.R.; de Leon, P.M.M.; et al. Proteomic analysis identifies differentially expressed proteins after red propolis treatment in Hep-2 cells. Food Chem. Toxicol. 2014, 63, 195–204. [Google Scholar] [CrossRef]
- Lantto, T.; Laakso, I.; Dorman, H.; Mauriala, T.; Hiltunen, R.; Kõks, S.; Raasmaja, A. Cellular Stress and p53-Associated Apoptosis by Juniperus communis L. Berry Extract Treatment in the Human SH-SY5Y Neuroblastoma Cells. Int. J. Mol. Sci. 2016, 17, 1113. [Google Scholar] [CrossRef]
- Strojny, B.; Sawosz, E.; Grodzik, M.; Jaworski, S.; Szczepaniak, J.; Sosnowska, M.; Wierzbicki, M.; Kutwin, M.; Orlińska, S.; Chwalibog, A. Nanostructures of diamond, graphene oxide and graphite inhibit CYP1A2, CYP2D6 and CYP3A4 enzymes and downregulate their genes in liver cells. Int. J. Nanomed. 2018, 13, 8561–8575. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romaldini, A.; Spanò, R.; Veronesi, M.; Grimaldi, B.; Bandiera, T.; Sabella, S. Human Multi-Lineage Liver Organoid Model Reveals Impairment of CYP3A4 Expression upon Repeated Exposure to Graphene Oxide. Cells 2024, 13, 1542. https://doi.org/10.3390/cells13181542
Romaldini A, Spanò R, Veronesi M, Grimaldi B, Bandiera T, Sabella S. Human Multi-Lineage Liver Organoid Model Reveals Impairment of CYP3A4 Expression upon Repeated Exposure to Graphene Oxide. Cells. 2024; 13(18):1542. https://doi.org/10.3390/cells13181542
Chicago/Turabian StyleRomaldini, Alessio, Raffaele Spanò, Marina Veronesi, Benedetto Grimaldi, Tiziano Bandiera, and Stefania Sabella. 2024. "Human Multi-Lineage Liver Organoid Model Reveals Impairment of CYP3A4 Expression upon Repeated Exposure to Graphene Oxide" Cells 13, no. 18: 1542. https://doi.org/10.3390/cells13181542
APA StyleRomaldini, A., Spanò, R., Veronesi, M., Grimaldi, B., Bandiera, T., & Sabella, S. (2024). Human Multi-Lineage Liver Organoid Model Reveals Impairment of CYP3A4 Expression upon Repeated Exposure to Graphene Oxide. Cells, 13(18), 1542. https://doi.org/10.3390/cells13181542