

Inflammatory Trajectory of Type 2 Diabetes: Novel Opportunities for Early and Late Treatment

 , , , , , ,
, , , , , , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
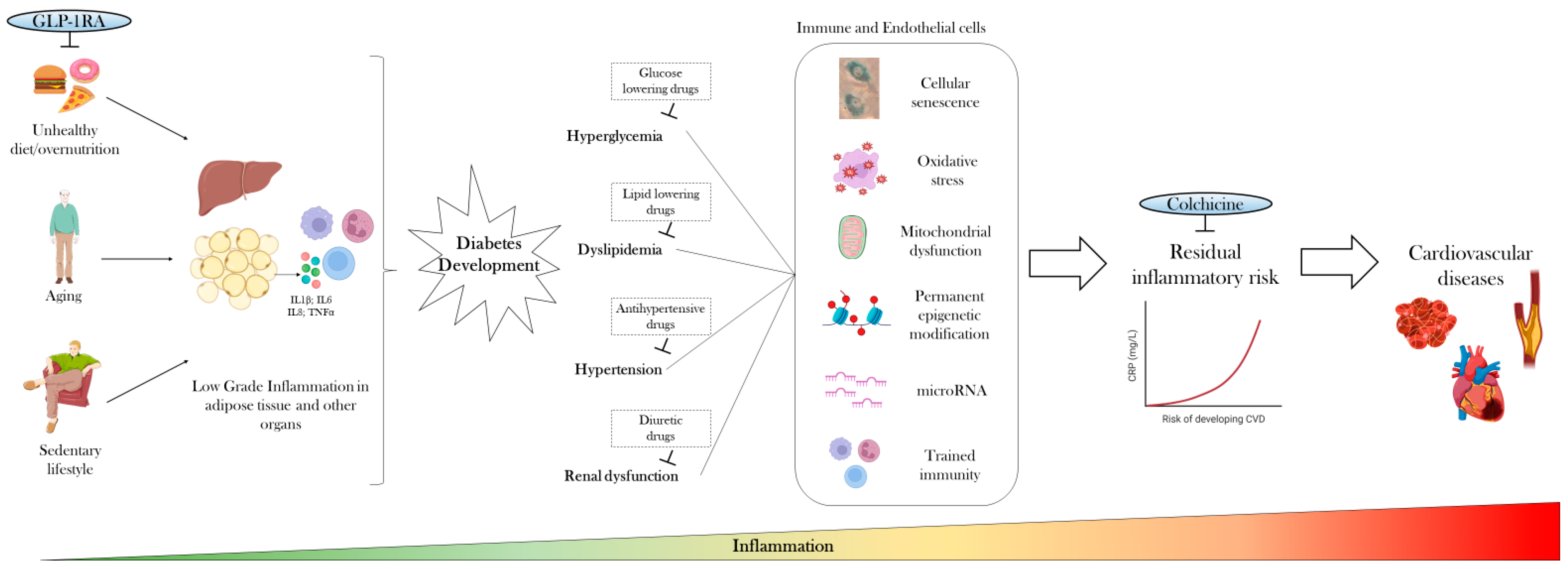
2. Low-Grade Inflammation Precedes Diabetes Development
3. Anti-Inflammatory Therapies Only Mildly Improve Glycemic Control
4. Targeting Cardiovascular Risk Factors and Common Therapies Only Partially Attenuate Inflammation
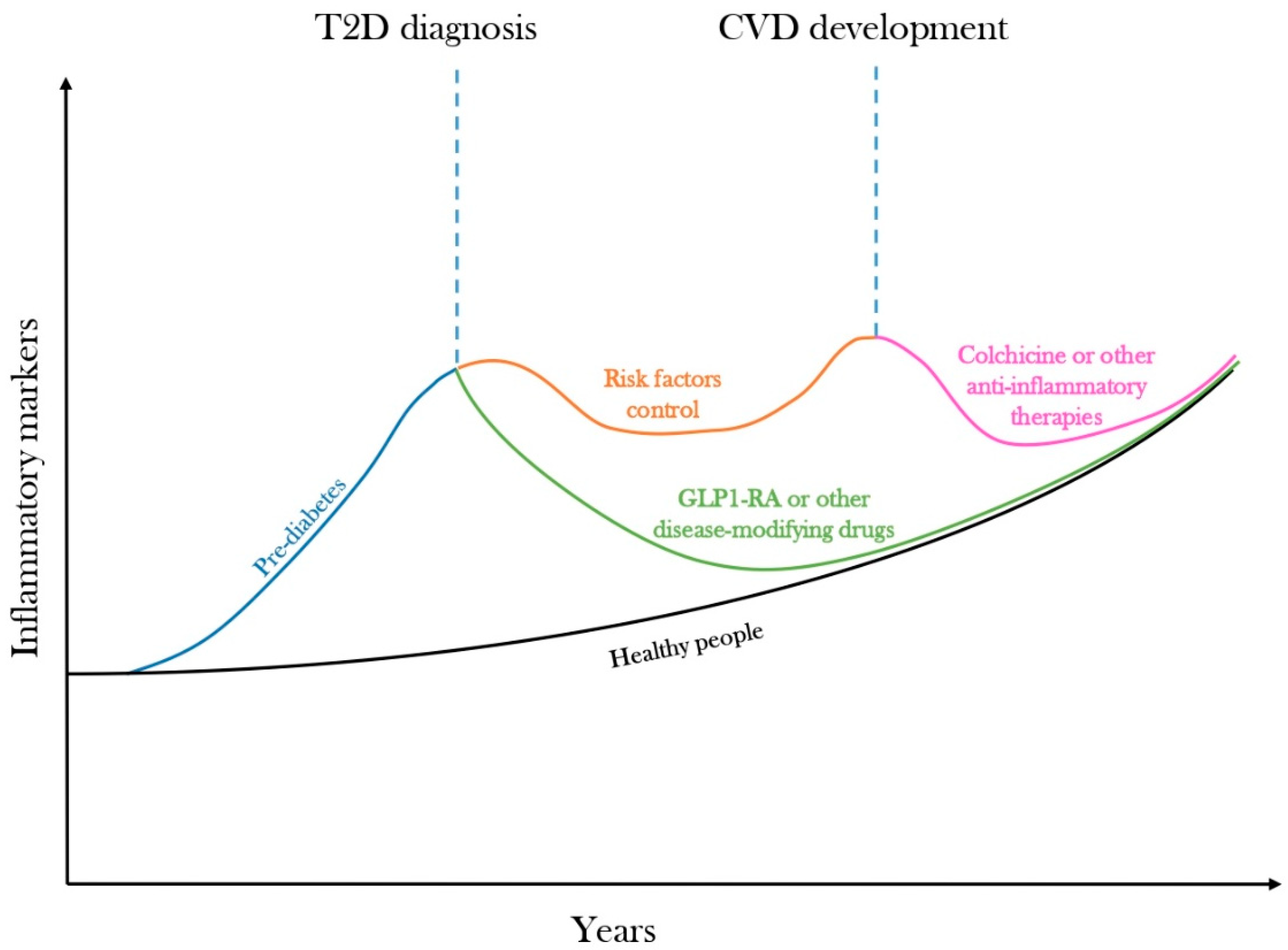
5. Residual Inflammatory Risk Predicts Cardiovascular Diseases
6. Disease-Modifying Drugs Could Curb the Inflammatory Trajectory of Diabetes
7. Targeting Low-Grade Inflammation with Colchicine for CVD Prevention in Diabetes
8. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. 2019, 14, 50–59. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Pollack, R.M.; Donath, M.Y.; LeRoith, D.; Leibowitz, G. Anti-inflammatory Agents in the Treatment of Diabetes and Its Vascular Complications. Diabetes Care 2016, 39 (Suppl. 2), S244–S252. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-dose colchicine for secondary prevention of cardiovascular disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- León-Pedroza, J.I.; González-Tapia, L.A.; del Olmo-Gil, E.; Castellanos-Rodríguez, D.; Escobedo, G.; González-Chávez, A. Low-grade systemic inflammation and the development of metabolic diseases: From the molecular evidence to the clinical practice. Cir. Cir. 2015, 83, 543–551. (In Spanish) [Google Scholar] [CrossRef] [PubMed]
- Banait, T.; Wanjari, A.; Danade, V.; Banait, S.; Jain, J. Role of High-Sensitivity C-reactive Protein (Hs-CRP) in Non-communicable Diseases: A Review. Cureus 2022, 14, e30225. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.; Bruunsgaard, H.; Weis, N.; Hendel, H.W.; Andreassen, B.U.; Eldrup, E.; Dela, F.; Pedersen, B.K. Circulating levels of TNF-alpha and IL-6-relation to truncal fat mass and muscle mass in healthy elderly individuals and in patients with type-2 diabetes. Mech. Ageing Dev. 2003, 124, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bao, W.; Liu, J.; Ouyang, Y.Y.; Wang, D.; Rong, S.; Xiao, X.; Shan, Z.L.; Zhang, Y.; Yao, P.; et al. Inflammatory markers and risk of type 2 diabetes: A systematic review and meta-analysis. Diabetes Care 2013, 36, 166–175. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Bradwin, G.; Hasan, A.A.; Rifai, N. Comparison of interleukin-6, C-reactive protein, and low-density lipoprotein cholesterol as biomarkers of residual risk in contemporary practice: Secondary analyses from the Cardiovascular Inflammation Reduction Trial. Eur. Heart J. 2020, 41, 2952–2961. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.J.; Norrie, J.; Caslake, M.J.; Gaw, A.; Ford, I.; Lowe, G.D.; O’Reilly, D.S.; Packard, C.J.; Sattar, N. West of Scotland Coronary Prevention Study. C-reactive protein is an independent predictor of risk for the development of diabetes in the West of Scotland Coronary Prevention Study. Diabetes 2002, 51, 1596–1600. [Google Scholar] [CrossRef] [PubMed]
- Duncan, B.B.; Schmidt, M.I.; Pankow, J.S.; Ballantyne, C.M.; Couper, D.; Vigo, A.; Hoogeveen, R.; Folsom, A.R.; Heiss, G. Atherosclerosis Risk in Communities Study. Low-grade systemic inflammation and the development of type 2 diabetes: The atherosclerosis risk in communities study. Diabetes 2003, 52, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S. Body mass index, diabetes, and C-reactive protein among, U.S. adults. Diabetes Care 1999, 22, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Kiyohara, Y.; Kubo, M.; Ninomiya, T.; Wakugawa, Y.; Yonemoto, K.; Iwase, M.; Iida, M. Elevated C-reactive protein is a predictor of the development of diabetes in a general Japanese population: The Hisayama Study. Diabetes Care 2005, 28, 2497–2500. [Google Scholar] [CrossRef] [PubMed]
- Dregan, A.; Charlton, J.; Chowienczyk, P.; Gulliford, M.C. Chronic inflammatory disorders and risk of type 2 diabetes mellitus, coronary heart disease, and stroke: A population-based cohort study. Circulation 2014, 130, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Gastaldelli, A.; Yki-Järvinen, H.; Scherer, P.E. Why does obesity cause diabetes? Cell Metab. 2022, 34, 11–20. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Gunton, J.E. Hypoxia-inducible factors and diabetes. J. Clin. Investig. 2020, 130, 5063–5073. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- de Candia, P.; Prattichizzo, F.; Garavelli, S.; Alviggi, C.; La Cava, A.; Matarese, G. The pleiotropic roles of leptin in metabolism, immunity, and cancer. J. Exp. Med. 2021, 218, e20191593. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Procaccini, C.; Garavelli, S.; Carbone, F.; Di Silvestre, D.; La Rocca, C.; Greco, D.; Colamatteo, A.; Lepore, M.T.; Russo, C.; De Rosa, G.; et al. Signals of pseudo-starvation unveil the amino acid transporter SLC7A11 as key determinant in the control of Treg cell proliferative potential. Immunity 2021, 54, 1543–1560.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Kusminski, C.M.; Scherer, P.E. Adiponectin, Leptin and Cardiovascular Disorders. Circ. Res. 2021, 128, 136–149. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rajkovic, N.; Zamaklar, M.; Lalic, K.; Jotic, A.; Lukic, L.; Milicic, T.; Singh, S.; Stosic, L.; Lalic, N.M. Relationship between obesity, adipocytokines and inflammatory markers in type 2 diabetes: Relevance for cardiovascular risk prevention. Int. J. Environ. Res. Public Health 2014, 11, 4049–4065. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zheng, C.; Yang, Q.; Cao, J.; Xie, N.; Liu, K.; Shou, P.; Qian, F.; Wang, Y.; Shi, Y. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis. 2016, 7, e2167. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Murano, I.; Barbatelli, G.; Parisani, V.; Latini, C.; Muzzonigro, G.; Castellucci, M.; Cinti, S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 2008, 49, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, S.; Ji, Y.; Kersten, S.; Qi, L. Mechanisms of inflammatory responses in obese adipose tissue. Annu. Rev. Nutr. 2012, 32, 261–286. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Caër, C.; Rouault, C.; Le Roy, T.; Poitou, C.; Aron-Wisnewsky, J.; Torcivia, A.; Bichet, J.C.; Clément, K.; Guerre-Millo, M.; André, S. Immune cell-derived cytokines contribute to obesity-related inflammation, fibrogenesis and metabolic deregulation in human adipose tissue. Sci. Rep. 2017, 7, 3000. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rehman, K.; Akash, M.S.H.; Liaqat, A.; Kamal, S.; Qadir, M.I.; Rasul, A. Role of Interleukin-6 in Development of Insulin Resistance and Type 2 Diabetes Mellitus. Crit. Rev. Eukaryot. Gene Expr. 2017, 27, 229–236. [Google Scholar] [CrossRef] [PubMed]
- da Silva Rosa, S.C.; Nayak, N.; Caymo, A.M.; Gordon, J.W. Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue. Physiol. Rep. 2020, 8, e14607. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tabula Muris Consortium. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Smith, U.; Li, Q.; Rydén, M.; Spalding, K.L. Cellular senescence and its role in white adipose tissue. Int. J. Obes. 2021, 45, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Espinosa De Ycaza, A.E.; Søndergaard, E.; Morgan-Bathke, M.; Carranza Leon, B.G.; Lytle, K.A.; Ramos, P.; Kirkland, J.L.; Tchkonia, T.; Jensen, M.D. Senescent cells in human adipose tissue: A cross-sectional study. Obesity 2021, 29, 1320–1327. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Matacchione, G.; Perugini, J.; Di Mercurio, E.; Sabbatinelli, J.; Prattichizzo, F.; Senzacqua, M.; Storci, G.; Dani, C.; Lezoche, G.; Guerrieri, M.; et al. Senescent macrophages in the human adipose tissue as a source of inflammaging. Geroscience 2022, 44, 1941–1960. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Villaret, A.; Galitzky, J.; Decaunes, P.; Estève, D.; Marques, M.A.; Sengenès, C.; Chiotasso, P.; Tchkonia, T.; Lafontan, M.; Kirkland, J.L.; et al. Adipose tissue endothelial cells from obese human subjects: Differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes 2010, 59, 2755–2763. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kondoh, H.; Hara, E. Targeting p21 for diabetes: Another choice of senotherapy. Cell Metab. 2022, 34, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; De Nigris, V.; Spiga, R.; Mancuso, E.; La Sala, L.; Antonicelli, R.; Testa, R.; Procopio, A.D.; Olivieri, F.; Ceriello, A. Inflammageing and metaflammation: The yin and yang of type 2 diabetes. Ageing Res. Rev. 2018, 41, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Mandrup-Poulsen, T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia 1996, 39, 1005–1029. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Vølund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Reduction in HbA1c with SGLT2 inhibitors vs. DPP-4 inhibitors as add-ons to metformin monotherapy according to baseline HbA1c: A systematic review of randomized controlled trials. Diabetes Metab. 2020, 46, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Brown, A.; Fischer, J.; Jain, A.; Littlejohn, T.; Nadeau, D.; Sussman, A.; Taylor, T.; Krol, A.; Magner, J. Efficacy and safety of acarbose in metformin-treated patients with type 2 diabetes. Diabetes Care 1998, 21, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Donath, M.Y.; Pradhan, A.D.; Thuren, T.; Pais, P.; Nicolau, J.C.; Glynn, R.J.; Libby, P.; Ridker, P.M. Anti-Inflammatory Therapy with Canakinumab for the Prevention and Management of Diabetes. J. Am. Coll. Cardiol. 2018, 71, 2392–2401. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Paquot, N.; Scheen, A.J. Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Expert. Opin. Investig. Drugs 2015, 24, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Goldfine, A.B.; Fonseca, V.; Jablonski, K.A.; Chen, Y.D.; Tipton, L.; Staten, M.A.; Shoelson, S.E. Targeting Inflammation Using Salsalate in Type 2 Diabetes Study Team. Salicylate (salsalate) in patients with type 2 diabetes: A randomized trial. Ann. Intern. Med. 2013, 159, 1–12. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Goldfine, A.B.; Fonseca, V.; Jablonski, K.A.; Pyle, L.; Staten, M.A.; Shoelson, S.E. TINSAL-T2D (Targeting Inflammation Using Salsalate in Type 2 Diabetes) Study Team. The effects of salsalate on glycemic control in patients with type 2 diabetes: A randomized trial. Ann. Intern. Med. 2010, 152, 346–357. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Faghihimani, E.; Aminorroaya, A.; Rezvanian, H.; Adibi, P.; Ismail-Beigi, F.; Amini, M. Salsalate improves glycemic control in patients with newly diagnosed type 2 diabetes. Acta Diabetol. 2013, 50, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Mohammadnia, N.; Los, J.; Opstal, T.S.J.; Fiolet, A.T.L.; Eikelboom, J.W.; Mosterd, A.; Nidorf, S.M.; Budgeon, C.A.; Tijssen, J.G.P.; Thompson, P.L.; et al. Colchicine and diabetes in patients with chronic coronary artery disease: Insights from the LoDoCo2 randomized controlled trial. Front. Cardiovasc. Med. 2023, 10, 1244529. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cocco, G.; Chu, D.C.; Pandolfi, S. Colchicine in clinical medicine. A guide for internists. Eur. J. Intern. Med. 2010, 21, 503–508. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice Committee. Summary of Revisions: Standards of Care in Diabetes—2024. Diabetes Care 2024, 47 (Suppl. 1), S5–S10. [Google Scholar] [CrossRef] [PubMed]
- Slieker, R.C.; van der Heijden, A.A.W.A.; van Leeuwen, N.; Mei, H.; Nijpels, G.; Beulens, J.W.J.; ‘t Hart, L.M. HbA1c is associated with altered expression in blood of cell cycle- and immune response-related genes. Diabetologia 2018, 61, 138–146. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thiem, K.; van Dierendonck, X.A.M.H.; Janssen, A.W.M.; Boogaard, J.P.; Riksen, N.P.; Tack, C.J.; Stienstra, R. A High Glycemic Burden Relates to Functional and Metabolic Alterations of Human Monocytes in Patients with Type 1 Diabetes. Diabetes 2020, 69, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Hulme, K.D.; Tong, Z.W.M.; Rowntree, L.C.; van de Sandt, C.E.; Ronacher, K.; Grant, E.J.; Dorey, E.S.; Gallo, L.A.; Gras, S.; Kedzierska, K.; et al. Increasing HbA1c is associated with reduced CD8+ T cell functionality in response to influenza virus in a TCR-dependent manner in individuals with diabetes mellitus. Cell Mol. Life Sci. 2024, 81, 35. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marfella, R.; Sardu, C.; D’Onofrio, N.; Prattichizzo, F.; Scisciola, L.; Messina, V.; La Grotta, R.; Balestrieri, M.L.; Maggi, P.; Napoli, C.; et al. Glycaemic control is associated with SARS-CoV-2 breakthrough infections in vaccinated patients with type 2 diabetes. Nat. Commun. 2022, 13, 2318. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Seo, Y.H.; Shin, H.Y. Relationship between hs-CRP and HbA1c in Diabetes Mellitus Patients: 2015–2017 Korean National Health and Nutrition Examination Survey. Chonnam. Med. J. 2021, 57, 62–67. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Seo, J.W.; Park, S.B. The Association of Hemoglobin A1c and Fasting Glucose Levels with hs-CRP in Adults Not Diagnosed with Diabetes from the KNHANES, 2017. J. Diabetes Res. 2021, 2021, 5585938. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, D.; Yu, Y.; Xu, Y.; Ge, J. Plasma Nesfatin-1: Potential Predictor and Diagnostic Biomarker for Cognitive Dysfunction in T2DM Patient. Diabetes Metab. Syndr. Obes. 2021, 14, 3555–3566. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Todingan, M.; Muhiddin, R.; Kurniawan, L.B. IL-6 Levels Analysis Controlled in Type 2 Diabetes Mellitus Patients and Uncontrolled. Indones. J. Clin. Pathol. Med. Lab. 2023, 29, 175–179. [Google Scholar] [CrossRef]
- Chen, W.; Liu, X.; Ye, S. Effects of metformin on blood and urine pro-inflammatory mediators in patients with type 2 diabetes. J. Inflamm. 2016, 13, 34. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mo, D.; Liu, S.; Ma, H.; Tian, H.; Yu, H.; Zhang, X.; Tong, N.; Liao, J.; Ren, Y. Effects of acarbose and metformin on the inflammatory state in newly diagnosed type 2 diabetes patients: A one-year randomized clinical study. Drug Des. Devel Ther. 2019, 13, 2769–2776. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hanefeld, M.; Marx, N.; Pfützner, A.; Baurecht, W.; Lübben, G.; Karagiannis, E.; Stier, U.; Forst, T. Anti-inflammatory effects of pioglitazone and/or simvastatin in high cardiovascular risk patients with elevated high sensitivity C-reactive protein: The PIOSTAT Study. J. Am. Coll. Cardiol. 2007, 49, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.; Savinko, T.; Wong, A.K.; et al. Anti-Inflammatory Effects of Metformin Irrespective of Diabetes Status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Novelle, M.G.; Ali, A.; Diéguez, C.; Bernier, M.; de Cabo, R. Metformin: A Hopeful Promise in Aging Research. Cold Spring Harb. Perspect. Med. 2016, 6, a025932. [Google Scholar] [CrossRef] [PubMed]
- Pfützner, A.; Marx, N.; Lübben, G.; Langenfeld, M.; Walcher, D.; Konrad, T.; Forst, T. Improvement of cardiovascular risk markers by pioglitazone is independent from glycemic control: Results from the pioneer study. J. Am. Coll. Cardiol. 2005, 45, 1925–1931. [Google Scholar] [CrossRef] [PubMed]
- Kothari, V.; Galdo, J.A.; Mathews, S.T. Hypoglycemic agents and potential anti-inflammatory activity. J. Inflamm. Res. 2016, 9, 27–38. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thévenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Nishiyama, M.; Taguchi, T.; Asai, M.; Yoshida, M.; Kambayashi, M.; Terada, Y.; Hashimoto, K. Insulin exhibits short-term anti-inflammatory but long-term proinflammatory effects in vitro. Mol. Cell Endocrinol. 2009, 298, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.siditalia.it/pdf/LG_379_diabete_ed2022_feb2023.pdf (accessed on 1 August 2024).
- Jukema, R.A.; Ahmed, T.A.N.; Tardif, J.C. Does low-density lipoprotein cholesterol induce inflammation? If so, does it matter? Current insights and future perspectives for novel therapies. BMC Med. 2019, 17, 197. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hulthe, J.; Fagerberg, B. Circulating oxidized LDL is associated with subclinical atherosclerosis development and inflammatory cytokines (AIR Study). Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1162–1167. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E. Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) Investigators. C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med. 2005, 352, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Park, C.S.; Yang, H.M.; Han, K.; Lee, H.S.; Kang, J.; Han, J.K.; Park, K.W.; Kang, H.J.; Koo, B.K.; Kim, H.S. J-shaped association between LDL cholesterol and cardiovascular events: A longitudinal primary prevention cohort of over 2.4 million people nationwide. J. Adv. Res. 2024, 58, 139–147. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ridker, P.M.; Rifai, N.; Rose, L.; Buring, J.E.; Cook, N.R. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N. Engl. J. Med. 2002, 347, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rane, M. Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circ. Res. 2021, 128, 1728–1746. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.; Ballantyne, C.; Sisk, C.; Shah, A.; Veltri, E.; Maccubbin, D. Comparison of effects of ezetimibe/simvastatin versus simvastatin versus atorvastatin in reducing C-reactive protein and low-density lipoprotein cholesterol levels. Am. J. Cardiol. 2007, 99, 1706–1713. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Galimberti, F.; Olmastroni, E.; Luscher, T.F.; Carugo, S.; Catapano, A.L.; Casula, M. META-LIPID Group. Effect of lipid-lowering therapies on C-reactive protein levels: A comprehensive meta-analysis of randomized controlled trials. Cardiovasc. Res. 2024, 120, 333–344. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Milajerdi, A.; Larijani, B.; Esmaillzadeh, A. Statins influence biomarkers of low grade inflammation in apparently healthy people or patients with chronic diseases: A systematic review and meta-analysis of randomized clinical trials. Cytokine 2019, 123, 154752. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Prattichizzo, F.; Sardu, C.; Paolisso, P.; D’Onofrio, N.; Scisciola, L.; La Grotta, R.; Frigé, C.; Ferraraccio, F.; Panarese, I.; et al. Evidence of an anti-inflammatory effect of PCSK9 inhibitors within the human atherosclerotic plaque. Atherosclerosis 2023, 378, 117180. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, N.; Prattichizzo, F.; Marfella, R.; Sardu, C.; Martino, E.; Scisciola, L.; Marfella, L.; Grotta, R.; Frigé, C.; Paolisso, G.; et al. SIRT3 mediates the effects of PCSK9 inhibitors on inflammation, autophagy, and oxidative stress in endothelial cells. Theranostics 2023, 13, 531–542. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bermudez, E.A.; Rifai, N.; Buring, J.; Manson, J.E.; Ridker, P.M. Interrelationships among circulating interleukin-6, C-reactive protein, and traditional cardiovascular risk factors in women. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1668–1673. [Google Scholar] [CrossRef] [PubMed]
- Rutter, M.K.; Meigs, J.B.; Sullivan, L.M.; D’Agostino, R.B., Sr.; Wilson, P.W. C-reactive protein, the metabolic syndrome, and prediction of cardiovascular events in the Framingham Offspring Study. Circulation 2004, 110, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Savoia, C.; Schiffrin, E.L. Reduction of C-reactive protein and the use of anti-hypertensives. Vasc. Health Risk Manag. 2007, 3, 975–983. [Google Scholar] [PubMed] [PubMed Central]
- Brull, D.J.; Sanders, J.; Rumley, A.; Lowe, G.D.; Humphries, S.E.; Montgomery, H.E. Impact of angiotensin converting enzyme inhibition on post-coronary artery bypass interleukin 6 release. Heart 2002, 87, 252–255. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ridker, P.M.; Danielson, E.; Rifai, N.; Glynn, R.J. Val-MARC Investigators. Valsartan, blood pressure reduction, and C-reactive protein: Primary report of the Val-MARC trial. Hypertension 2006, 48, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Dhindsa, S.; Ghanim, H.; Chaudhuri, A. Angiotensin II and inflammation: The effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J. Hum. Hypertens. 2007, 21, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Palmas, W.; Ma, S.; Psaty, B.; Goff, D.C., Jr.; Darwin, C.; Barr, R.G. Antihypertensive medications and C-reactive protein in the multi-ethnic study of atherosclerosis. Am. J. Hypertens. 2007, 20, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Schnell, O.; Amann-Zalan, I.; Jelsovsky, Z.; Moritz, A.; Bermejo, J.L.; Parkin, C.G.; Schweitzer, M.A.; Fisher, L.; Polonsky, W.H. Changes in A1C levels are significantly associated with changes in levels of the cardiovascular risk biomarker hs-CRP: Results from the SteP study. Diabetes Care 2013, 36, 2084–2089. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979, Erratum in N. Engl. J. Med. 1997, 337, 356. [Google Scholar] [CrossRef] [PubMed]
- Maseri, A. Inflammation, atherosclerosis, and ischemic events—Exploring the hidden side of the moon. N. Engl. J. Med. 1997, 336, 1014–1016. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Puri, R.; Anderson, T.; Ballantyne, C.M.; Cho, L.; Kastelein, J.J.; Koenig, W.; Somaratne, R.; Kassahun, H.; Yang, J.; et al. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA 2016, 316, 2373–2384. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Visseren, F.L.J.; Cater, N.B.; Salhi, N.; Soronen, J.; Ray, K.K.; Delgado, V.; Jukema, J.W.; Laufs, U.; Zamorano, J.-L.; et al. Addressing residual risk beyond statin therapy: New targets in the management of dyslipidaemias–A report from the European Society of Cardiology Cardiovascular Round Table. J. Clin. Lipidol. 2024. [Google Scholar] [CrossRef]
- Ridker, P.M. High-sensitivity C-reactive protein and cardiovascular risk: Rationale for screening and primary prevention. Am. J. Cardiol. 2003, 92, 17K–22K. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Emerging Risk Factors Collaboration; Kaptoge, S.; Di Angelantonio, E.; Lowe, G.; Pepys, M.B.; Thompson, S.G.; Collins, R.; Danesh, J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet 2010, 375, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Bohula, E.A.; Giugliano, R.P.; Leiter, L.A.; Verma, S.; Park, J.G.; Sever, P.S.; Lira Pineda, A.; Honarpour, N.; Wang, H.; Murphy, S.A.; et al. Inflammatory and Cholesterol Risk in the FOURIER Trial. Circulation 2018, 138, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Koenig, W.; Kastelein, J.J.; Mach, F.; Lüscher, T.F. Has the time finally come to measure hsCRP universally in primary and secondary cardiovascular prevention? Eur. Heart J. 2018, 39, 4109–4111. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; Giuliani, A.; Sabbatinelli, J.; Matacchione, G.; Ramini, D.; Bonfigli, A.R.; Rippo, M.R.; de Candia, P.; Procopio, A.D.; Olivieri, F.; et al. Prevalence of residual inflammatory risk and associated clinical variables in patients with type 2 diabetes. Diabetes Obes. Metab. 2020, 22, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Tian, M.; Wang, L.; Qian, H.; Zhang, S.; Pang, H.; Liu, Z.; Fang, L.; Shen, Z. C-reactive protein for predicting cardiovascular and all-cause mortality in type 2 diabetic patients: A meta-analysis. Cytokine 2019, 117, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Sharif, S.; Van der Graaf, Y.; Cramer, M.J.; Kapelle, L.J.; de Borst, G.J.; Visseren, F.L.J.; Westerink, J. SMART study group. Low-grade inflammation as a risk factor for cardiovascular events and all-cause mortality in patients with type 2 diabetes. Cardiovasc. Diabetol. 2021, 20, 220. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lawler, P.R.; Bhatt, D.L.; Godoy, L.C.; Lüscher, T.F.; Bonow, R.O.; Verma, S.; Ridker, P.M. Targeting cardiovascular inflammation: Next steps in clinical translation. Eur. Heart J. 2021, 42, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Jousilahti, P.; Tuomilehto, J.; Antikainen, R.; Sundvall, J.; Salomaa, V. Association of serum C-reactive protein level with sex-specific type 2 diabetes risk: A prospective finnish study. J. Clin. Endocrinol. Metab. 2009, 94, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Domazet, S.L.; Olesen, T.B.; Stidsen, J.V.; Svensson, C.K.; Nielsen, J.S.; Thomsen, R.W.; Jessen, N.; Vestergaard, P.; Andersen, M.K.; Hansen, T.; et al. Low-grade inflammation in persons with recently diagnosed type 2 diabetes: The role of abdominal adiposity and putative mediators. Diabetes Obes. Metab. 2024, 26, 2092–2101. [Google Scholar] [CrossRef] [PubMed]
- Blaum, C.; Brunner, F.J.; Kröger, F.; Braetz, J.; Lorenz, T.; Goßling, A.; Ojeda, F.; Koester, L.; Karakas, M.; Zeller, T.; et al. Modifiable lifestyle risk factors and C-reactive protein in patients with coronary artery disease: Implications for an anti-inflammatory treatment target population. Eur. J. Prev. Cardiol. 2021, 28, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Ripa, R.S.; Zobel, E.H.; von Scholten, B.J.; Jensen, J.K.; Binderup, T.; Diaz, L.J.; Curovic, V.R.; Hansen, T.W.; Rossing, P.; Kjaer, A. Effect of Liraglutide on Arterial Inflammation Assessed as [18F]FDG Uptake in Patients with Type 2 Diabetes: A Randomized, Double-Blind, Placebo-Controlled Trial. Circ. Cardiovasc. Imaging 2021, 14, e012174. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bray, J.J.H.; Foster-Davies, H.; Stephens, J.W. A systematic review examining the effects of sodium-glucose cotransporter-2 inhibitors (SGLT2is) on biomarkers of inflammation and oxidative stress. Diabetes Res. Clin. Pract. 2020, 168, 108368. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; De Nigris, V.; La Sala, L.; Procopio, A.D.; Olivieri, F.; Ceriello, A. “Inflammaging” as a Druggable Target: A Senescence-Associated Secretory Phenotype-Centered View of Type 2 Diabetes. Oxid. Med. Cell Longev. 2016, 2016, 1810327. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prattichizzo, F.; de Candia, P.; Ceriello, A. Diabetes and kidney disease: Emphasis on treatment with SGLT-2 inhibitors and GLP-1 receptor agonists. Metabolism 2021, 120, 154799. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; de Candia, P.; De Nigris, V.; Nicolucci, A.; Ceriello, A. Legacy effect of intensive glucose control on major adverse cardiovascular outcome: Systematic review and meta-analyses of trials according to different scenarios. Metabolism 2020, 110, 154308. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, N.; Prattichizzo, F.; Martino, E.; Anastasio, C.; Mele, L.; La Grotta, R.; Sardu, C.; Ceriello, A.; Marfella, R.; Paolisso, G.; et al. MiR-27b attenuates mitochondrial oxidative stress and inflammation in endothelial cells. Redox Biol. 2023, 62, 102681. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Giuliani, A.; Cirilli, I.; Prattichizzo, F.; Mensà, E.; Fulgenzi, G.; Sabbatinelli, J.; Graciotti, L.; Olivieri, F.; Procopio, A.D.; Tiano, L.; et al. The mitomiR/Bcl-2 axis affects mitochondrial function and autophagic vacuole formation in senescent endothelial cells. Aging 2018, 10, 2855–2873. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Giuliani, A.; Prattichizzo, F.; Micolucci, L.; Ceriello, A.; Procopio, A.D.; Rippo, M.R. Mitochondrial (Dys) Function in Inflammaging: Do MitomiRs Influence the Energetic, Oxidative, and Inflammatory Status of Senescent Cells? Mediat. Inflamm. 2017, 2017, 2309034, Erratum in Mediat. Inflamm. 2019, 2019, 8716351. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prattichizzo, F.; De Nigris, V.; Sabbatinelli, J.; Giuliani, A.; Castaño, C.; Párrizas, M.; Crespo, I.; Grimaldi, A.; Baranzini, N.; Spiga, R.; et al. CD31+ Extracellular Vesicles from Patients with Type 2 Diabetes Shuttle a miRNA Signature Associated with Cardiovascular Complications. Diabetes 2021, 70, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; Matacchione, G.; Giuliani, A.; Sabbatinelli, J.; Olivieri, F.; de Candia, P.; De Nigris, V.; Ceriello, A. Extracellular vesicle-shuttled miRNAs: A critical appraisal of their potential as nano-diagnostics and nano-therapeutics in type 2 diabetes mellitus and its cardiovascular complications. Theranostics 2021, 11, 1031–1045. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Murakami, T.; Inagaki, N.; Kondoh, H. Cellular Senescence in Diabetes Mellitus: Distinct Senotherapeutic Strategies for Adipose Tissue and Pancreatic β Cells. Front. Endocrinol. 2022, 13, 869414. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lau, E.Y.M.; Carroll, E.C.; Callender, L.A.; Hood, G.A.; Berryman, V.; Pattrick, M.; Finer, S.; Hitman, G.A.; Ackland, G.L.; Henson, S.M. Type 2 diabetes is associated with the accumulation of senescent T cells. Clin. Exp. Immunol. 2019, 197, 205–213. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Khosla, S.; Farr, J.N.; Tchkonia, T.; Kirkland, J.L. The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 2020, 16, 263–275. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Natarajan, R. Epigenetic Mechanisms in Diabetic Vascular Complications and Metabolic Memory: The 2020 Edwin Bierman Award Lecture. Diabetes 2021, 70, 328–337. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Edgar, L.; Akbar, N.; Braithwaite, A.T.; Krausgruber, T.; Gallart-Ayala, H.; Bailey, J.; Corbin, A.L.; Khoyratty, T.E.; Chai, J.T.; Alkhalil, M.; et al. Hyperglycemia Induces Trained Immunity in Macrophages and Their Precursors and Promotes Atherosclerosis. Circulation 2021, 144, 961–982. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thiem, K.; Keating, S.T.; Netea, M.G.; Riksen, N.P.; Tack, C.J.; van Diepen, J.; Stienstra, R. Hyperglycemic Memory of Innate Immune Cells Promotes In Vitro Proinflammatory Responses of Human Monocytes and Murine Macrophages. J. Immunol. 2021, 206, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Kangas, R.; Pöllänen, E.; Rippo, M.R.; Lanzarini, C.; Prattichizzo, F.; Niskala, P.; Jylhävä, J.; Sipilä, S.; Kaprio, J.; Procopio, A.D.; et al. Circulating miR-21, miR-146a and Fas ligand respond to postmenopausal estrogen-based hormone replacement therapy—A study with monozygotic twin pairs. Mech. Ageing Dev. 2014, 143–144, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.A.; Akbar, N.; Baidžajevas, K.; Choudhury, R.P. Trained immunity in diabetes and hyperlipidemia: Emerging opportunities to target cardiovascular complications and design new therapies. FASEB J. 2023, 37, e23231. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e14. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161.e12. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lacy, M.; Atzler, D.; Liu, R.; de Winther, M.; Weber, C.; Lutgens, E. Interactions between dyslipidemia and the immune system and their relevance as putative therapeutic targets in atherosclerosis. Pharmacol. Ther. 2019, 193, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Palmer, A.K.; Kirkland, J.L. Aging and adipose tissue: Potential interventions for diabetes and regenerative medicine. Exp. Gerontol. 2016, 86, 97–105. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, X.; Habiballa, L.; Aversa, Z.; Ng, Y.E.; Sakamoto, A.E.; Englund, D.A.; Pearsall, V.M.; White, T.A.; Robinson, M.M.; Rivas, D.A.; et al. Characterization of cellular senescence in aging skeletal muscle. Nat. Aging 2022, 2, 601–615. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B., Jr.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e4. [Google Scholar] [CrossRef] [PubMed]
- Baboota, R.K.; Spinelli, R.; Erlandsson, M.C.; Brandao, B.B.; Lino, M.; Yang, H.; Mardinoglu, A.; Bokarewa, M.I.; Boucher, J.; Kahn, C.R.; et al. Chronic hyperinsulinemia promotes human hepatocyte senescence. Mol. Metab. 2022, 64, 101558. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Tchkonia, T.; Kirkland, J.L. Senolytics: Potential for Alleviating Diabetes and Its Complications. Endocrinology 2021, 162, bqab058. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I. Vascular cell senescence: Contribution to atherosclerosis. Circ. Res. 2007, 100, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; De Nigris, V.; Mancuso, E.; Spiga, R.; Giuliani, A.; Matacchione, G.; Lazzarini, R.; Marcheselli, F.; Recchioni, R.; Testa, R.; et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. 2018, 15, 170–181. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Narasimhan, A.; Flores, R.R.; Robbins, P.D.; Niedernhofer, L.J. Role of Cellular Senescence in Type II Diabetes. Endocrinology 2021, 162, bqab136. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carroll, J.E.; Cole, S.W.; Seeman, T.E.; Breen, E.C.; Witarama, T.; Arevalo, J.M.G.; Ma, J.; Irwin, M.R. Partial sleep deprivation activates the DNA damage response (DDR) and the senescence-associated secretory phenotype (SASP) in aged adult humans. Brain Behav. Immun. 2016, 51, 223–229. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tylutka, A.; Morawin, B.; Gramacki, A.; Zembron-Lacny, A. Lifestyle exercise attenuates immunosenescence; flow cytometry analysis. BMC Geriatr. 2021, 21, 200. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yzydorczyk, C.; Li, N.; Chehade, H.; Mosig, D.; Bidho, M.; Keshavjee, B.; Armengaud, J.B.; Nardou, K.; Siddeek, B.; Benahmed, M.; et al. Transient postnatal overfeeding causes liver stress-induced premature senescence in adult mice. Sci. Rep. 2017, 7, 12911. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Torma, F.; Kerepesi, C.; Jókai, M.; Babszki, G.; Koltai, E.; Ligeti, B.; Kalcsevszki, R.; McGreevy, K.M.; Horvath, S.; Radák, Z. Alterations of the gut microbiome are associated with epigenetic age acceleration and physical fitness. Aging Cell 2024, 23, e14101. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Monickaraj, F.; Aravind, S.; Gokulakrishnan, K.; Sathishkumar, C.; Prabu, P.; Prabu, D.; Mohan, V.; Balasubramanyam, M. Accelerated aging as evidenced by increased telomere shortening and mitochondrial DNA depletion in patients with type 2 diabetes. Mol. Cell Biochem. 2012, 365, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Bonfigli, A.R.; Spazzafumo, L.; Prattichizzo, F.; Bonafè, M.; Mensà, E.; Micolucci, L.; Giuliani, A.; Fabbietti, P.; Testa, R.; Boemi, M.; et al. Leukocyte telomere length and mortality risk in patients with type 2 diabetes. Oncotarget 2016, 7, 50835–50844. [Google Scholar] [CrossRef]
- Prattichizzo, F.; Frigé, C.; Pellegrini, V.; Scisciola, L.; Santoro, A.; Monti, D.; Rippo, M.R.; Ivanchenko, M.; Olivieri, F.; Franceschi, C. Organ-specific biological clocks: Ageotyping for personalized anti-aging medicine. Ageing Res. Rev. 2024, 96, 102253. [Google Scholar] [CrossRef] [PubMed]
- Sabbatinelli, J.; Giuliani, A.; Kwiatkowska, K.M.; Matacchione, G.; Belloni, A.; Ramini, D.; Prattichizzo, F.; Pellegrini, V.; Piacenza, F.; Tortato, E.; et al. DNA Methylation-derived biological age and long-term mortality risk in subjects with type 2 diabetes. Cardiovasc. Diabetol. 2024, 23, 250. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Sattar, N.; Lee, M.M.Y.; Kristensen, S.L.; Branch, K.R.H.; Del Prato, S.; Khurmi, N.S.; Lam, C.S.P.; Lopes, R.D.; McMurray, J.J.V.; Pratley, R.E.; et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 2021, 9, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Tendal, B.; Mustafa, R.A.; Vandvik, P.O.; Li, S.; Hao, Q.; Tunnicliffe, D.; Ruospo, M.; Natale, P.; Saglimbene, V.; et al. Sodium-glucose cotransporter protein-2 (SGLT-2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists for type 2 diabetes: Systematic review and network meta-analysis of randomised controlled trials. BMJ 2021, 372, m4573, Erratum in BMJ 2022, 376, o109. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kosiborod, M.N.; Abildstrøm, S.Z.; Borlaug, B.A.; Butler, J.; Rasmussen, S.; Davies, M.; Hovingh, G.K.; Kitzman, D.W.; Lindegaard, M.L.; Møller, D.V.; et al. STEP-HFpEF Trial Committees and Investigators. Semaglutide in Patients with Heart Failure with Preserved Ejection Fraction and Obesity. N. Engl. J. Med. 2023, 389, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Kosiborod, M.N.; Petrie, M.C.; Borlaug, B.A.; Butler, J.; Davies, M.J.; Hovingh, G.K.; Kitzman, D.W.; Møller, D.V.; Treppendahl, M.B.; Verma, S.; et al. STEP-HFpEF DM Trial Committees and Investigators. Semaglutide in Patients with Obesity-Related Heart Failure and Type 2 Diabetes. N. Engl. J. Med. 2024, 390, 1394–1407. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.S.; Bhatt, D.L.; Hameed, I.; Anker, S.D.; Cheng, A.Y.Y.; Hernandez, A.F.; Jones, W.S.; Khan, M.S.; Petrie, M.C.; Udell, J.A.; et al. Effect of SGLT2 inhibitors on heart failure outcomes and cardiovascular death across the cardiometabolic disease spectrum: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2024, 12, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Sardu, C.; D’Onofrio, N.; Fumagalli, C.; Scisciola, L.; Sasso, F.C.; Siniscalchi, M.; Marfella, L.V.; D’Andrea, D.; Minicucci, F.; et al. SGLT-2 inhibitors and in-stent restenosis-related events after acute myocardial infarction: An observational study in patients with type 2 diabetes. BMC Med. 2023, 21, 71. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Simms-Williams, N.; Treves, N.; Yin, H.; Lu, S.; Yu, O.; Pradhan, R.; Renoux, C.; Suissa, S.; Azoulay, L. Effect of combination treatment with glucagon-like peptide-1 receptor agonists and sodium-glucose cotransporter-2 inhibitors on incidence of cardiovascular and serious renal events: Population based cohort study. BMJ 2024, 385, e078242, Erratum in BMJ 2024, 385, q1094. Erratum in BMJ 2024, 385, q1237. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marfella, R.; Prattichizzo, F.; Sardu, C.; Rambaldi, P.F.; Fumagalli, C.; Marfella, L.V.; La Grotta, R.; Frigé, C.; Pellegrini, V.; D’Andrea, D.; et al. GLP-1 receptor agonists-SGLT-2 inhibitors combination therapy and cardiovascular events after acute myocardial infarction: An observational study in patients with type 2 diabetes. Cardiovasc. Diabetol. 2024, 23, 10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prattichizzo, F.; La Sala, L.; Rydén, L.; Marx, N.; Ferrini, M.; Valensi, P.; Ceriello, A. Glucose-lowering therapies in patients with type 2 diabetes and cardiovascular diseases. Eur. J. Prev. Cardiol. 2019, 26 (Suppl. 2), 73–80. [Google Scholar] [CrossRef] [PubMed]
- Mosenzon, O.; Del Prato, S.; Schechter, M.; Leiter, L.A.; Ceriello, A.; DeFronzo, R.A.; Raz, I. From glucose lowering agents to disease/diabetes modifying drugs: A “SIMPLE” approach for the treatment of type 2 diabetes. Cardiovasc. Diabetol. 2021, 20, 92. [Google Scholar] [CrossRef]
- Hogan, A.E.; Gaoatswe, G.; Lynch, L.; Corrigan, M.A.; Woods, C.; O’Connell, J.; O’Shea, D. Glucagon-like peptide 1 analogue therapy directly modulates innate immune-mediated inflammation in individuals with type 2 diabetes mellitus. Diabetologia 2014, 57, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, M.; Safa, B.I.; Gonzalez, M.S.C.; Kim, S.F.; Echouffo-Tcheugui, J.B. Systemic and organ-specific anti-inflammatory effects of sodium-glucose cotransporter-2 inhibitors. Trends Endocrinol. Metab. 2024, 35, 425–438. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- La Grotta, R.; de Candia, P.; Olivieri, F.; Matacchione, G.; Giuliani, A.; Rippo, M.R.; Tagliabue, E.; Mancino, M.; Rispoli, F.; Ferroni, S.; et al. Anti-inflammatory effect of SGLT-2 inhibitors via uric acid and insulin. Cell Mol. Life Sci. 2022, 79, 273. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, W.; Xing, Y.; Kong, D.; Zhang, Z.; Ma, H.; Yang, L. Meta-analysis of the effect of sodium-dependent glucose transporter 2 inhibitors on C-reactive protein in type 2 diabetes. Medicine 2022, 101, e30553. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Drucker, D.J.; Rosen, C.F. Glucagon-like peptide-1 (GLP-1) receptor agonists, obesity and psoriasis: Diabetes meets dermatology. Diabetologia 2011, 54, 2741–2744. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. GLP-1 physiology informs the pharmacotherapy of obesity. Mol. Metab. 2022, 57, 101351. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.; Ruan, B.; Jiang, H.; Le, S.; Liu, Y.; Ao, X.; Huang, Y.; Shi, X.; Xue, R.; Fu, X.; et al. The Weight-loss Effect of GLP-1RAs Glucagon-Like Peptide-1 Receptor Agonists in Non-diabetic Individuals with Overweight or Obesity: A Systematic Review with Meta-Analysis and Trial Sequential Analysis of Randomized Controlled Trials. Am. J. Clin. Nutr. 2023, 118, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Riddle, M.C.; Cefalu, W.T.; Evans, P.H.; Gerstein, H.C.; Nauck, M.A.; Oh, W.K.; Rothberg, A.E.; le Roux, C.W.; Rubino, F.; Schauer, P.; et al. Consensus Report: Definition and Interpretation of Remission in Type 2 Diabetes. Diabetes Care 2021, 44, 2438–2444. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kahn, S.E.; Deanfield, J.E.; Jeppesen, O.K.; Emerson, S.S.; Boesgaard, T.W.; Colhoun, H.M.; Kushner, R.F.; Lingvay, I.; Burguera, B.; Gajos, G.; et al. Effect of Semaglutide on Regression and Progression of Glycemia in People with Overweight or Obesity but Without Diabetes in the SELECT Trial. Diabetes Care 2024, 47, 1350–1359. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Frías, J.P.; Davies, M.J.; Rosenstock, J.; Pérez Manghi, F.C.; Fernández Landó, L.; Bergman, B.K.; Liu, B.; Cui, X.; Brown, K. SURPASS-2 Investigators. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Del Prato, S.; Kahn, S.E.; Pavo, I.; Weerakkody, G.J.; Yang, Z.; Doupis, J.; Aizenberg, D.; Wynne, A.G.; Riesmeyer, J.S.; Heine, R.J.; et al. SURPASS-4 Investigators. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): A randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet 2021, 398, 1811–1824. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Vázquez, L.; Del Prato, S.; Franco, D.R.; Weerakkody, G.; Dai, B.; Landó, L.F.; Bergman, B.K.; Rodríguez, A. Achieving Normoglycemia with Tirzepatide: Analysis of SURPASS 1-4 Trials. Diabetes Care 2023, 46, 1986–1992. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Masoro, E.J. Caloric restriction and aging: An update. Exp. Gerontol. 2000, 35, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Kraus, W.E.; Bhapkar, M.; Huffman, K.M.; Pieper, C.F.; Krupa Das, S.; Redman, L.M.; Villareal, D.T.; Rochon, J.; Roberts, S.B.; Ravussin, E.; et al. CALERIE Investigators. 2 years of calorie restriction and cardiometabolic risk (CALERIE): Exploratory outcomes of a multicentre, phase 2, randomised controlled trial. Lancet Diabetes Endocrinol. 2019, 7, 673–683. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ferrannini, E.; Muscelli, E.; Frascerra, S.; Baldi, S.; Mari, A.; Heise, T.; Broedl, U.C.; Woerle, H.J. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014, 124, 499–508, Erratum in J. Clin. Investig. 2014, 124, 1868. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ferrannini, G.; Hach, T.; Crowe, S.; Sanghvi, A.; Hall, K.D.; Ferrannini, E. Energy Balance After Sodium-Glucose Cotransporter 2 Inhibition. Diabetes Care 2015, 38, 1730–1735. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- La Grotta, R.; Frigé, C.; Matacchione, G.; Olivieri, F.; de Candia, P.; Ceriello, A.; Prattichizzo, F. Repurposing SGLT-2 Inhibitors to Target Aging: Available Evidence and Molecular Mechanisms. Int. J. Mol. Sci. 2022, 23, 12325. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kogot-Levin, A.; Riahi, Y.; Abramovich, I.; Mosenzon, O.; Agranovich, B.; Kadosh, L.; Ben-Haroush Schyr, R.; Kleiman, D.; Hinden, L.; Cerasi, E.; et al. Mapping the metabolic reprogramming induced by sodium-glucose cotransporter 2 inhibition. JCI Insight 2023, 8, e164296. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- O’Keefe, J.H.; Weidling, R.; O’Keefe, E.L.; Franco, W.G. SGLT inhibitors for improving Healthspan and lifespan. Prog. Cardiovasc. Dis. 2023, 81, 2–9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rossing, P.; Inzucchi, S.E.; Vart, P.; Jongs, N.; Docherty, K.F.; Jhund, P.S.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; et al. DAPA-CKD and DAPA-HF Trial Committees and Investigators. Dapagliflozin and new-onset type 2 diabetes in patients with chronic kidney disease or heart failure: Pooled analysis of the DAPA-CKD and DAPA-HF trials. Lancet Diabetes Endocrinol. 2022, 10, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Lucisano, G.; Prattichizzo, F.; La Grotta, R.; Frigé, C.; De Cosmo, S.; Di Bartolo, P.; Di Cianni, G.; Fioretto, P.; Giorda, C.B.; et al. AMD Annals study group. The legacy effect of hyperglycemia and early use of SGLT-2 inhibitors: A cohort study with newly-diagnosed people with type 2 diabetes. Lancet Reg. Health Eur. 2023, 31, 100666. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Prattichizzo, F.; Ceriello, A. Positioning newer drugs in the management of type 2 diabetes. Lancet Diabetes Endocrinol. 2021, 9, 138–139. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; La Sala, L.; Ceriello, A. Two drugs are better than one to start T2DM therapy. Nat. Rev. Endocrinol. 2020, 16, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Roubille, F.; Bouabdallaoui, N.; Kouz, S.; Waters, D.D.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Grégoire, J.C.; Gamra, H.; Kiwan, G.S.; et al. Low-Dose Colchicine in Patients with Type 2 Diabetes and Recent Myocardial Infarction in the Colchicine Cardiovascular Outcomes Trial (COLCOT). Diabetes Care 2024, 47, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef]
- Available online: https://www.sec.gov/Archives/edgar/data/1114448/000137036818000045/a181018-99_1.htm (accessed on 1 August 2024).
- Nelson, K.; Fuster, V.; Ridker, P.M. Low-Dose Colchicine for Secondary Prevention of Coronary Artery Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2023, 82, 648–660. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrini, V.; La Grotta, R.; Carreras, F.; Giuliani, A.; Sabbatinelli, J.; Olivieri, F.; Berra, C.C.; Ceriello, A.; Prattichizzo, F. Inflammatory Trajectory of Type 2 Diabetes: Novel Opportunities for Early and Late Treatment. Cells 2024, 13, 1662. https://doi.org/10.3390/cells13191662
Pellegrini V, La Grotta R, Carreras F, Giuliani A, Sabbatinelli J, Olivieri F, Berra CC, Ceriello A, Prattichizzo F. Inflammatory Trajectory of Type 2 Diabetes: Novel Opportunities for Early and Late Treatment. Cells. 2024; 13(19):1662. https://doi.org/10.3390/cells13191662
Chicago/Turabian StylePellegrini, Valeria, Rosalba La Grotta, Francesca Carreras, Angelica Giuliani, Jacopo Sabbatinelli, Fabiola Olivieri, Cesare Celeste Berra, Antonio Ceriello, and Francesco Prattichizzo. 2024. "Inflammatory Trajectory of Type 2 Diabetes: Novel Opportunities for Early and Late Treatment" Cells 13, no. 19: 1662. https://doi.org/10.3390/cells13191662