
Employing Multi-Omics Analyses to Understand Changes during Kidney Development in Perinatal Interleukin-6 Animal Model

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals
2.3. miRNA and mRNA Isolation with Assessment of Quantity and Quality
2.4. Library Preparation and Sequencing for miRNA and mRNA
2.5. Methyl-Seq Library Preparation and Sequencing
2.6. Bioinformatics Analysis
2.6.1. Transcriptome and miRNA Expression Analysis
2.6.2. Whole Genome Bisulfite Sequencing Data Analysis for DNA Methylated Regions (DMRs)
2.6.3. Integrative Multi-Omics Data Analysis for Drawing Inferences
2.6.4. Principle Component Analysis, Gene Set Enrichment Analysis (GSEA), and Regulatory Network Analysis
2.7. Immunofluorescence Staining in Newborn Mouse Kidney
3. Results
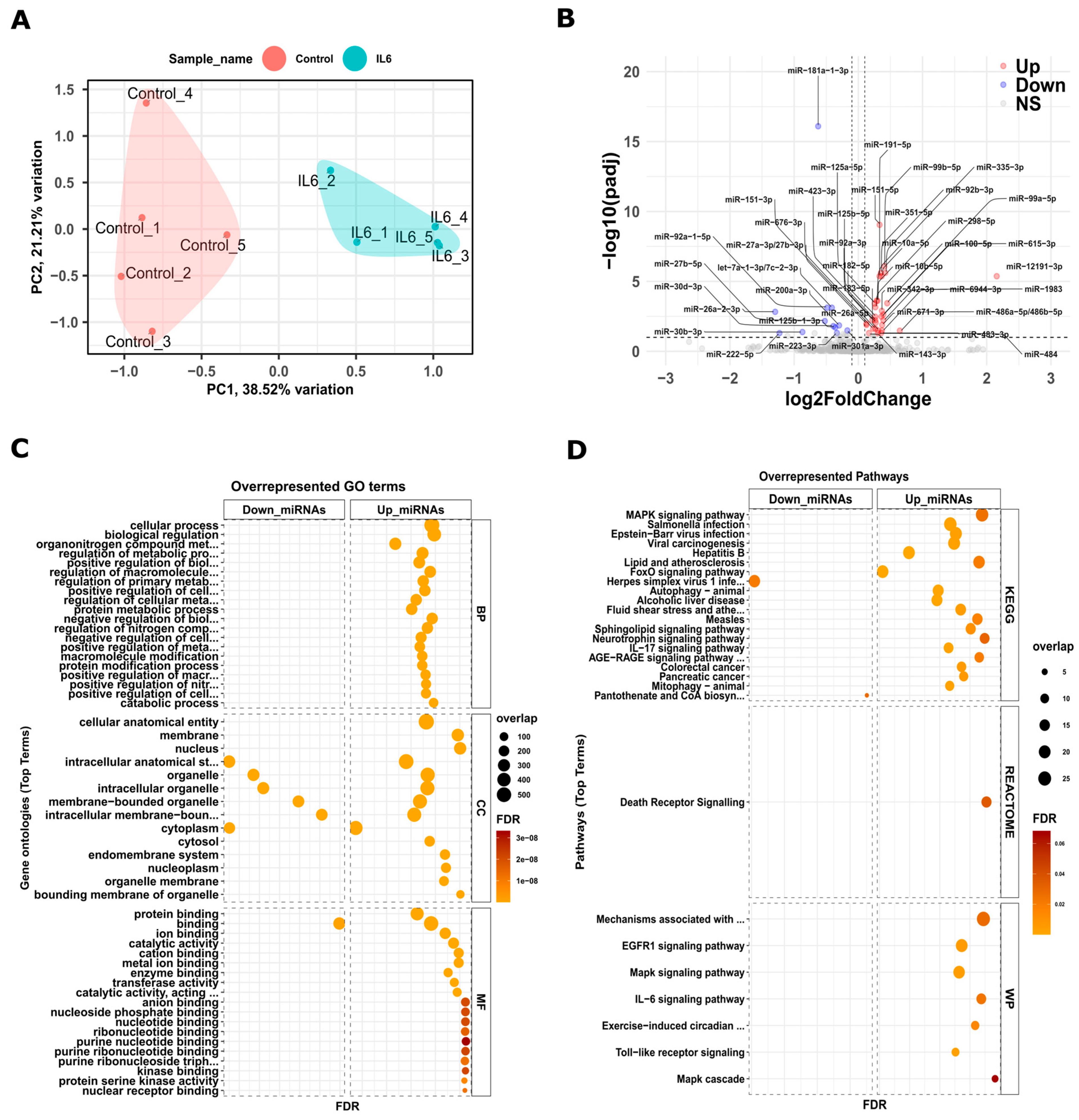
3.1. Analysis of Transcriptome of Newborn Kidneys from Pups Exposed to Intrauterine Interleukin-6
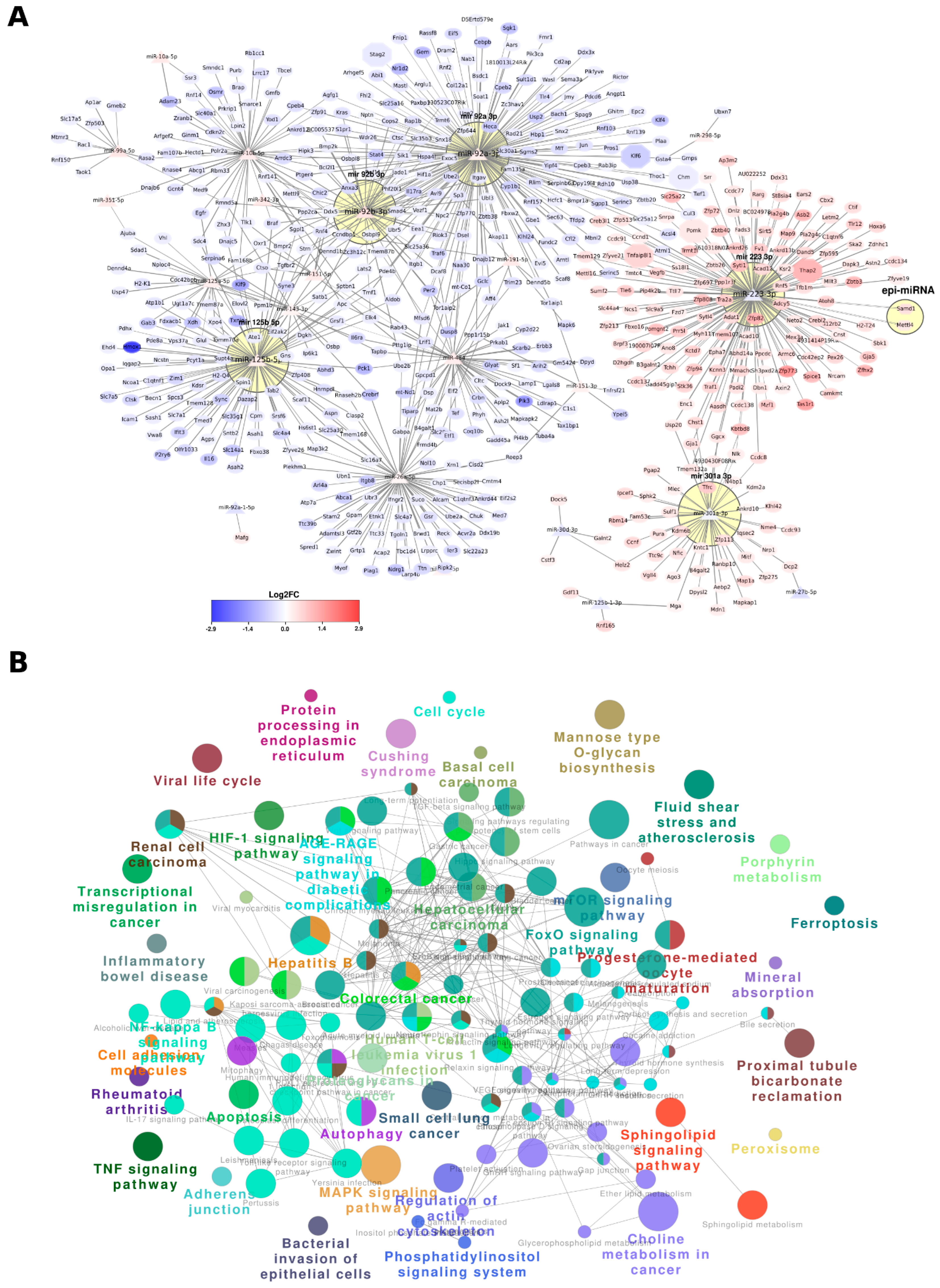
3.2. miRNAs and Their Interactions with Target Genes in Newborn Kidneys from Pups Exposed to Intrauterine Interleukin-6
3.3. Analysis of Whole Genome Bisulfite Sequencing of Newborn Kidneys from Pups Exposed to Intrauterine Interleukin-6
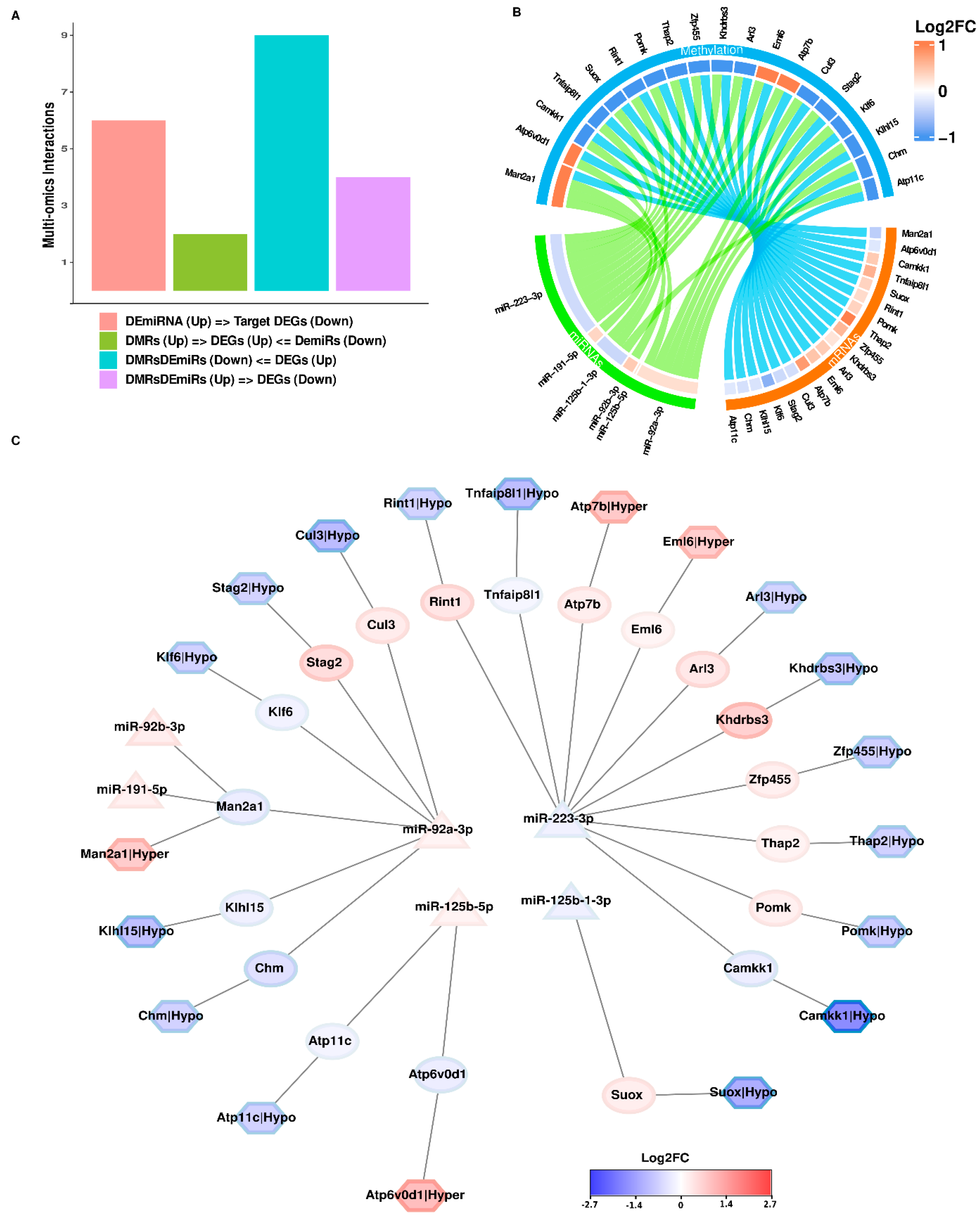
3.4. Novel Insights from Integrating 2 Multi-Omics Datasets from Newborn Kidneys of Pups Exposed to Intrauterine Interleukin-6
3.5. miRNA-Driven Regulatory Networks in Newborn Kidneys of Pups Exposed to Intrauterine Interleukin-6
3.6. Integrating 3 Multi-Omics Datasets from Newborn Kidneys of Pups Exposed to Intrauterine Interleukin-6
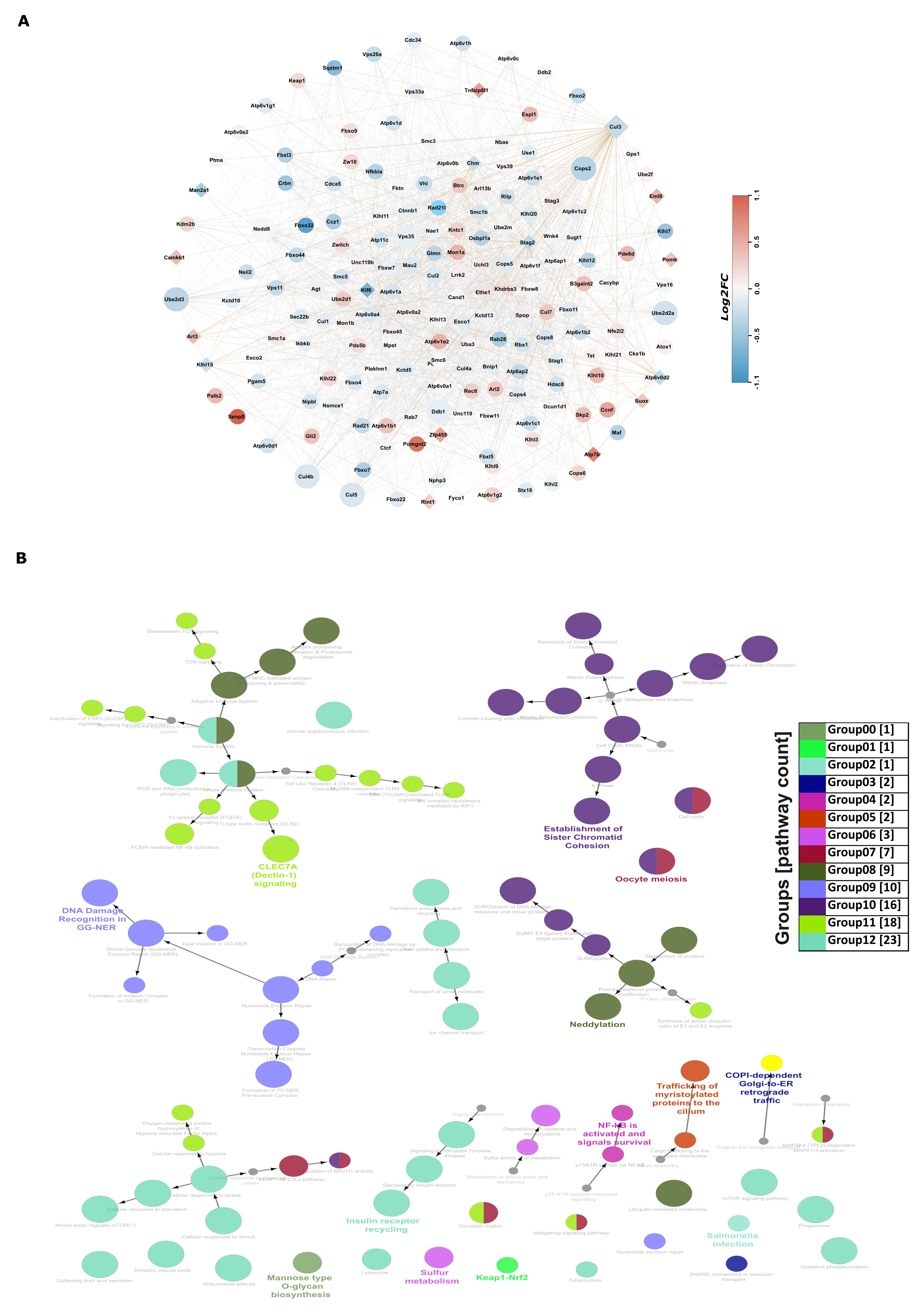
3.7. Novel Insights via Protein–Protein Interactions: Co-Expression-Based Functional Network Analysis of 19 Genes Identified in the Multi-Omics Datasets Analyses
3.8. Validation of Results Following Integration of Multi-Omics Datasets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iseki, K.; Ikemiya, Y.; Kinjo, K.; Inoue, T.; Iseki, C.; Takishita, S. Body Mass Index and the Risk of Development of End-Stage Renal Disease in a Screened Cohort. Kidney Int. 2004, 65, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Crook, E.D.; Jones, D.W.; Wofford, M.R.; Dubbert, P.M. Mechanisms of Obesity-Associated Cardiovascular and Renal Disease. Am. J. Med. Sci. 2002, 324, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Q.; Lumbers, E.R.; Oldmeadow, C.; Collins, C.E.; Johnson, V.; Keogh, L.; Sutherland, K.; Gordon, A.; Smith, R.; Rae, K.M.; et al. The Relationship between Maternal Adiposity during Pregnancy and Fetal Kidney Development and Kidney Function in Infants: The Gomeroi Gaaynggal Study. Physiol. Rep. 2019, 7, e14227. [Google Scholar] [CrossRef] [PubMed]
- Macumber, I.; Schwartz, S.; Leca, N. Maternal Obesity Is Associated with Congenital Anomalies of the Kidney and Urinary Tract in Offspring. Pediatr. Nephrol. 2017, 32, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Honein, M.A.; Moore, C.A.; Watkins, M.L. Subfertility and Prepregnancy Overweight/Obesity: Possible Interaction between These Risk Factors in the Etiology of Congenital Renal Anomalies. Birth Defects Res. A Clin. Mol. Teratol. 2003, 67, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Filler, G.; Rayar, M.S.; Da Silva, O.; Buffo, I.; Pepelassis, D.; Sharma, A.P. Should Prevention of Chronic Kidney Disease Start before Pregnancy? Int. Urol. Nephrol. 2008, 40, 483–488. [Google Scholar] [CrossRef]
- Blomberg, M.I.; Källén, B. Maternal Obesity, and Morbid Obesity: The Risk for Birth Defects in the Offspring. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 35–40. [Google Scholar] [CrossRef]
- Hsu, C.W.; Yamamoto, K.T.; Henry, R.K.; De Roos, A.J.; Flynn, J.T. Prenatal Risk Factors for Childhood CKD. J. Am. Soc. Nephrol. 2014, 25, 2105–2111. [Google Scholar] [CrossRef]
- Roytblat, L.; Rachinsky, M.; Fisher, A.; Greemberg, L.; Shapira, Y.; Douvdevani, A.; Gelman, S. Interleukin-6 Levels in Obese Patients. Obes. Res. 2000, 8, 673–675. [Google Scholar] [CrossRef]
- Eder, K.; Baffy, N.; Falus, A.; Fulop, A.K. The Major Inflammatory Mediator Interleukin-6 and Obesity. Inflamm. Res. 2009, 58, 727–736. [Google Scholar] [CrossRef]
- Ross, W.R.; McGill, J.B. Epidemiology of Obesity and Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2006, 13, 325–335. [Google Scholar] [CrossRef]
- Boubred, F.; Saint-Faust, M.; Buffat, C.; Ligi, I.; Grandvuillemin, I.; Simeoni, U. Developmental Origins of Chronic Renal Disease: An Integrative Hypothesis. Int. J. Nephrol. 2013, 2013, 346067. [Google Scholar] [CrossRef] [PubMed]
- Nenov, V.D.; Taal, M.W.; Sakharova, O.V.; Brenner, B.M. Multi-Hit Nature of Chronic Renal Disease. Curr. Opin. Nephrol. Hypertens. 2000, 9, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Schreuder, M.; Delemarre-van de Waal, H.; van Wijk, A. Consequences of Intrauterine Growth Restriction for the Kidney. Kidney Blood Press. Res. 2006, 29, 108–125. [Google Scholar] [CrossRef]
- Burton, G.J.; Fowden, A.L. The Placenta: A Multifaceted, Transient Organ. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140066. [Google Scholar] [CrossRef]
- Buhimschi, C.S.; Dulay, A.T.; Abdel-Razeq, S.; Zhao, G.; Lee, S.; Hodgson, E.J.; Bhandari, V.; Buhimschi, I.A. Fetal Inflammatory Response in Women with Proteomic Biomarkers Characteristic of Intra-Amniotic Inflammation and Preterm Birth. Br. J. Obs. Gyn. 2009, 16, 257–267. [Google Scholar] [CrossRef]
- Samuelsson, A.M.; Öhrn, I.; Dahlgren, J.; Eriksson, E.; Angelin, B.; Folkow, B.; Holmäng, A. Prenatal Exposure to Interleukin-6 Results in Hypertension and Increased Hypothalamic-Pituitary-Adrenal Axis Activity in Adult Rats. Endocrinology 2004, 145, 4897–4911. [Google Scholar] [CrossRef]
- Dahlgren, J.; Samuelsson, A.M.; Jansson, T.; Holmäng, A. Interleukin-6 in the Maternal Circulation Reaches the Rat Fetus in Mid-Gestation. Pediatr. Res. 2006, 60, 147–151. [Google Scholar] [CrossRef]
- Ramsay, J.E.; Ferrell, W.R.; Crawford, L.; Wallace, A.M.; Greer, I.A.; Sattar, N. Maternal Obesity Is Associated with Dysregulation of Metabolic, Vascular, and Inflammatory Pathways. J. Clin. Endocrinol. Metab. 2002, 87, 4231–4237. [Google Scholar] [CrossRef]
- Stewart, F.M.; Freeman, D.J.; Ramsay, J.E.; Greer, I.A.; Caslake, M.; Ferrell, W.R. Longitudinal Assessment of Maternal Endothelial Function and Markers of Inflammation and Placental Function throughout Pregnancy in Lean and Obese Mothers. J. Clin. Endocrinol. Metab. 2007, 92, 969–975. [Google Scholar] [CrossRef]
- Roberts, K.A.; Riley, S.C.; Reynolds, R.M.; Barr, S.; Evans, M.; Statham, A.; Hor, K.; Jabbour, H.N.; Norman, J.E.; Denison, F.C. Placental Structure and Inflammation in Pregnancies Associated with Obesity. Placenta 2011, 32, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal Immune Activation Alters Fetal Brain Development through Interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef]
- Srivastava, T.; Joshi, T.; Heruth, D.P.; Rezaiekhaligh, M.H.; Garola, R.E.; Zhou, J.; Boinpelly, V.C.; Ali, M.F.; Alon, U.S.; Sharma, M.; et al. A Mouse Model of Prenatal Exposure to Interleukin-6 to Study the Developmental Origin of Health and Disease. Sci. Rep. 2021, 11, 13260. [Google Scholar] [CrossRef]
- Suravajhala, P.; Kogelman, L.J.; Kadarmideen, H.N. Multi-Omic Data Integration and Analysis Using Systems Genomics Approaches: Methods and Applications in Animal Production, Health and Welfare. Genet. Sel. Evol. 2016, 48, 38. [Google Scholar] [CrossRef]
- Wörheide, M.A.; Krumsiek, J.; Kastenmüller, G.; Arnold, M. Multi-Omics Integration in Biomedical Research—A Metabolomics-Centric Review. Anal. Chim. Acta. 2021, 1141, 144–162. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-Omics Approaches to Disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Smith, G.D.; Ebrahim, S. ‘Mendelian Randomization’: Can Genetic Epidemiology Contribute to Understanding Environmental Determinants of Disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread Aligner: Fast, Accurate and Scalable Read Mapping by Seed-and-Vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality Control of RNA-Seq Experiments. Bioinform. Appl. Note 2012, 28, 2184–2185. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. Available online: https://journal.embnet.org/index.php/embnetjournal/article/view/200/479 (accessed on 7 October 2024). [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinform. Appl. Note 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, 917. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A Decade-Long Collection of Experimentally Supported miRNA–Gene Interactions. Nucleic Acids Res. 2017, 46, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Genome Analysis Circlize Implements and Enhances Circular Visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Blighe, K.; Lun, A. PCAtools: PCAtools: Everything Principal Components Analysis. Bioconductor. Available online: https://bioconductor.org/packages/PCAtools (accessed on 7 October 2024).
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H.; Willighagen, E.L. gprofiler2—An R package for Gene List Functional Enrichment Analysis and Namespace Conversion Toolset g:Profiler [version 2; peer review: 2 approved] version 1. F1000Research 2020, 9, ELIXIR-709. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Rasband, W.; Eliceiri, K. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein-Protein Association Networks and Functional Enrichment Analyses for any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Rajewsky, N. MicroRNA Target Predictions in Animals. Nat. Genet. 2006, 38, 8–13. [Google Scholar] [CrossRef]
- Sayed, D.; Abdellatif, M. MicroRNAs in Development and Disease. Physiol. Rev. 2011, 91, 827–887. [Google Scholar] [CrossRef]
- Marrone, A.K.; Ho, J. MicroRNAs: Potential Regulators of Renal Development Genes That Contribute to CAKUT. Pediatr. Nephrol. 2014, 29, 565–574. [Google Scholar] [CrossRef]
- Phua, Y.L.; Chu, J.Y.; Marrone, A.K.; Bodnar, A.J.; Sims-Lucas, S.; Ho, J. Renal Stromal miRNAs Are Required for Normal Nephrogenesis and Glomerular Mesangial Survival. Physiol. Rep. 2015, 3, e12537. [Google Scholar] [CrossRef]
- Li, Y.; Seah, M.K.; O’Neill, C. Mapping Global Changes in Nuclear Cytosine Base Modifications in the Early Mouse Embryo. Reproduction 2016, 151, 83–95. [Google Scholar] [CrossRef]
- Teh, A.L.; Pan, H.; Chen, L.; Ong, M.L.; Dogra, S.; Wong, J.; MacIsaac, J.L.; Mah, S.M.; McEwen, L.M.; Saw, S.M.; et al. The Effect of Genotype and in Utero Environment on Interindividual Variation in Neonate DNA Methylomes. Genome Res. 2014, 24, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Rakshit, G.; Komal; Dagur, P.; Jayaprakash, V. Multi-Omics Approaches in Drug Discovery. In CADD and Informatics in Drug Discovery. Interdisciplinary Biotechnological Advances; Rudrapal, M., Khan, J., Eds.; Springer: Singapore, 2023; pp. 79–98. ISBN 978-981-99-1315-2. [Google Scholar] [CrossRef]
- Tirado-Magallanes, R.; Rebbani, K.; Lim, R.; Pradhan, S.; Benoukraf, T. Whole Genome DNA Methylation: Beyond Genes Silencing. Oncotarget 2017, 8, 5629–5637. [Google Scholar] [CrossRef] [PubMed]
- Bahar Halpern, K.; Vana, T.; Walker, M.D. Paradoxical Role of DNA Methylation in Activation of FoxA2 Gene Expression during Endoderm Development. J. Biol. Chem. 2014, 289, 23882–23892. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-Promoted RNA Polymerase II Pausing Links DNA Methylation to Splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Chodavarapu, R.K.; Feng, S.; Bernatavichute, Y.V.; Chen, P.-Y.; Stroud, H.; Yu, Y.; Hetzel, J.A.; Kuo, F.; Kim, J.; Cokus, S.J.; et al. Relationship between Nucleosome Positioning and DNA Methylation. Nature 2010, 466, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.K.; Liu, Y.; Lay, F.D.; Liang, G.; Berman, B.P.; Jones, P.A. Genome-Wide Mapping of Nucleosome Positioning and DNA Methylation within Individual DNA Molecules. Genome Res. 2012, 22, 2497–2506. [Google Scholar] [CrossRef]
- Jimenez-Useche, I.; Ke, J.; Tian, Y.; Shim, D.; Howell, S.C.; Qiu, X.; Yuan, C. DNA Methylation Regulated Nucleosome Dynamics. Sci. Rep. 2013, 3, 2121. [Google Scholar] [CrossRef]
- Zhu, W.-G.; Srinivasan, K.; Dai, Z.; Duan, W.; Druhan, L.J.; Ding, H.; Yee, L.; Villalona-Calero, M.A.; Plass, C.; Otterson, G.A. Methylation of Adjacent CpG Sites Affects Sp1/Sp3 Binding and Activity in the p21(Cip1) Promoter. Mol. Cell. Biol. 2003, 23, 4056–4065. [Google Scholar] [CrossRef]
- Breiling, A.; Lyko, F. Epigenetic Regulatory Functions of DNA Modifications: 5-Methylcytosine and Beyond. Epigenetics Chromatin 2015, 8, 24. [Google Scholar] [CrossRef]
- Day, J.J.; Sweatt, J.D. DNA Methylation and Memory Formation. Nat. Neurosci. 2010, 13, 1319–1323. [Google Scholar] [CrossRef]
- Yoshida, K.; Maekawa, T.; Zhu, Y.; Renard-Guillet, C.; Chatton, B.; Inoue, K.; Uchiyama, T.; Ishibashi, K.-I.; Yamada, T.; Ohno, N. The Transcription Factor ATF7 Mediates Lipopolysaccharide-Induced Epigenetic Changes in Macrophages Involved in Innate Immunological Memory. Nat. Immunol. 2015, 16, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Hoy, W.E.; Douglas-Denton, R.N.; Hughson, M.D.; Cass, A.; Johnson, K.; Bertram, J.F. A Stereological Study of Glomerular Number and Volume: Preliminary Findings in a Multiracial Study of Kidneys at Autopsy. Kidney Int. Suppl. 2003, 83, S31–S317. [Google Scholar] [CrossRef] [PubMed]
- Hughson, M.; Farris, A.B., III; Douglas-Denton, R.; Hoy, W.E.; Bertram, J.F. Glomerular Number and Size in Autopsy Kidneys: The Relationship to Birth Weight. Kidney Int. 2003, 63, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Hughson, M.D.; Gobe, G.C.; Hoy, W.E.; Manning, R.D., Jr.; Douglas-Denton, R.; Bertram, J.F. Associations of Glomerular Number and Birth Weight with Clinicopathological Features of African Americans and Whites. Am. J. Kidney Dis. 2008, 52, 18–28. [Google Scholar] [CrossRef] [PubMed]
- White, S.L.; Perkovic, V.; Cass, A.; Chang, C.L.; Poulter, N.R.; Spector, T.; Haysom, L.; Craig, J.C.; Salmi, I.A.; Chadban, S.J.; et al. Is Low Birth Weight an Antecedent of CKD in Later Life? A Systematic Review of Observational Studies. Am. J. Kidney Dis. 2009, 54, 248–261. [Google Scholar] [CrossRef]
- Mu, M.; Wang, S.F.; Sheng, J.; Zhao, Y.; Li, H.Z.; Hu, C.L.; Tao, F.B. Birth Weight and Subsequent Blood Pressure: A Meta-Analysis. Arch. Cardiovasc. Dis. 2012, 105, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Maeoka, Y.; Ferdaus, M.Z.; Cornelius, R.J.; Sharma, A.; Su, X.T.; Miller, L.N.; Robertson, J.A.; Gurley, S.B.; Yang, C.L.; Ellison, D.H.; et al. Combined Kelch-Like 3 and Cullin 3 Degradation Is a Central Mechanism in Familial Hyperkalemic Hypertension in Mice. J. Am. Soc. Nephrol. 2022, 33, 584–600. [Google Scholar] [CrossRef]
- Kouranti, I.; Abdel Khalek, W.; Mazurkiewicz, S.; Loisel-Ferreira, I.; Gautreau, A.M.; Pintard, L.; Jeunemaitre, X.; Clauser, E. Cullin 3 Exon 9 Deletion in Familial Hyperkalemic Hypertension Impairs Cullin3-Ring-E3 Ligase (CRL3) Dynamic Regulation and Cycling. Int. J. Mol. Sci. 2022, 23, 5151. [Google Scholar] [CrossRef]
- Maeoka, Y.; Cornelius, R.J.; McCormick, J.A. Cullin 3 and Blood Pressure Regulation: Insights from Familial Hyperkalemic Hypertension. Hypertension 2023, 80, 912–923. [Google Scholar] [CrossRef]
- Cornelius, R.J.; Maeoka, Y.; McCormick, J.A. Renal Effects of Cullin 3 Mutations Causing Familial Hyperkalemic Hypertension. Curr. Opin. Nephrol. Hypertens. 2023, 32, 335–343. [Google Scholar] [CrossRef]
- Gomes, A.L.E.G.; Frappart, P.; Martins, R.A. RINT1 Loss Impairs Retinogenesis through TRP53-Mediated Apoptosis. Front. Cell Dev. Biol. 2020, 8, 559709. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | DMRs | DEGs | DEmiRs | |||||
|---|---|---|---|---|---|---|---|---|
| Gene_ID | Log2FC | p-Value | Log2FC | FDR | miRNA_id | Log2FC | FDR | |
| Man2a1 | 1.224 | 0.002 | −0.452 | 1.68933 × 10−8 | miR-92b-3p | 0.421 | 0.002 | |
| Man2a1 | 1.224 | 0.002 | −0.452 | 1.68933 × 10−8 | miR-191-5p | 0.33 | 0.002 | |
| Man2a1 | 1.224 | 0.002 | −0.452 | 1.68933 × 10−8 | miR-92a-3p | 0.265 | 0.014 | |
| Atp6v0d1 | 1.548 | 0.038 | −0.214 | 0.031 | miR-125b-5p | 0.266 | 0.015 | |
| Camkk1 | −2.714 | 2.23904 × 10−6 | 0.42 | 0.047 | miR-223-3p | −0.333 | 0.026 | |
| Tnfaip8l1 | −1.543 | 0.004 | 0.623 | 0.001 | miR-223-3p | −0.333 | 0.026 | |
| Suox | −1.929 | 0.004 | 0.353 | 0.048 | miR-125b-1-3p | −0.36 | 0.031 | |
| Rint1 | −1.046 | 0.008 | 0.29 | 0.01 | miR-223-3p | −0.333 | 0.026 | |
| Pomk | −1.217 | 0.016 | 0.355 | 0.036 | miR-223-3p | −0.333 | 0.026 | |
| Thap2 | −1.111 | 0.03 | 1.105 | 7.02311× 10−27 | miR-223-3p | −0.333 | 0.026 | |
| Zfp455 | −1.154 | 0.034 | 0.575 | 0.023 | miR-223-3p | −0.333 | 0.026 | |
| Khdrbs3 | −1.32 | 0.034 | 0.221 | 0.037 | miR-223-3p | −0.333 | 0.026 | |
| Arl3 | −1.018 | 0.036 | 0.409 | 0 | miR-223-3p | −0.333 | 0.026 | |
| Eml6 | 1.171 | 0.014 | 0.469 | 0.001 | miR-223-3p | −0.333 | 0.026 | |
| Atp7b | 1.294 | 0.017 | 0.806 | 0.005 | miR-223-3p | −0.333 | 0.026 | |
| Cul3 | −1.59 | 0.003 | −0.304 | 0.001 | miR-92a-3p | 0.265 | 0.014 | |
| Stag2 | −1.052 | 0.008 | −0.347 | 0.001 | miR-92a-3p | 0.265 | 0.014 | |
| Klf6 | −1.083 | 0.018 | −0.733 | 8.18305 × 10−5 | miR-92a-3p | 0.265 | 0.014 | |
| Klhl15 | −1.493 | 0.02 | −0.263 | 0.04 | miR-92a-3p | 0.265 | 0.014 | |
| Chm | −1.067 | 0.021 | −0.287 | 0.002 | miR-92a-3p | 0.265 | 0.014 | |
| Atp11c | −1.082 | 0.045 | −0.215 | 0.024 | miR-125b-5p | 0.266 | 0.015 | |
| Genes | Description | BP, CC, MF | PHENOTYPE or Associated Disease | Reference |
|---|---|---|---|---|
| Arl3 | ADP-ribosylation factor-like 3, | Mitotic cytokinesis, kidney development, cell cycle | Autosomal recessive polycystic kidney disease | PubMed ID: 16565502 |
| Atp11c | ATPase, class VI, type 11C | Pre-B cell differentiation, phospholipid translocation | Abnormal B cell differentiation | PubMed ID: 21423173; 21873635; |
| Atp6v0d1 | ATPase, H+ transporting, lysosomal V0 subunit D1 | Vacuolar transport | Abnormal kidney morphology | PubMed ID: 21873635 |
| Atp7b | ATPase, Cu++ transporting, beta polypeptide | Copper ion transport, intracellular copper ion homeostasis | Wilson disease, decreased body weight | PubMed ID: 11237756; 21873635; 6863890 |
| Camkk1 | Calcium/calmodulin-dependent protein kinase kinase 1, alpha | Intracellular signal transduction, positive regulation of protein kinase activity | Reproductive system phenotype | PubMed ID: 21873635; 17015467 |
| Chm | Choroidermia (RAB escort protein 1) | Vesicle-mediated transport, protein geranylgeranylation | Choroideremia, abnormal visceral yolk sac morphology | PubMed ID: 21873635; 15242790 |
| Cul3 | Cullin 3 | Negative regulation of transcription by RNA polymerase II, protein polyubiquitination, mitotic cell cycle, kidney development | Pseudo-hypoaldosteronism, abnormal cell cycle, abnormal gastrulation | PubMed ID: 17339333; 27708159; 25250572; 10500095 |
| Eml6 | Echinoderm microtubule-associated protein like 6 | Microtubule binding | Not Applicable | PubMed ID: 21873635 |
| Khdrbs3 | KH domain containing, RNA binding, signal transduction associated 3 | Regulation of mRNA splicing, via spliceosome, mRNA binding | Reproductive system phenotype | PubMed ID: 22196734; 21873635; 23637638 |
| Klf6 | Kruppel-like factor 6 | Cytokine-mediated signaling pathway, positive regulation of connective tissue replacement, DNA-binding transcription factor activity, RNA polymerase II-specific | Decreased body size and cell proliferation | PubMed ID: 10880228; 22820290; 21873635 |
| Klhl15 | Kelch-like 15 | Nuclear protein quality control by the ubiquitin–proteasome system | Not Applicable | PubMed ID: 21873635; |
| Man2a1 | Mannosidase 2, alpha 1 | In utero embryonic development, N-glycan processing, extracellular space | Abnormal renal glomerulus morphology, abnormal renal/urinary system physiology | PubMed ID: 21873635; 16754854; 11158608; 19710420 |
| Pomk | Protein-O-mannose kinase | Protein O-linked glycosylation, brain development, learning or memory, endoplasmic reticulum membrane | Hydrocephalus | PubMed ID: 21873635; 23929950 |
| Rint1 | RAD50 interactor 1 | Retrograde vesicle-mediated transport, Golgi to endoplasmic reticulum, cell cycle | Abnormal embryo development | PubMed ID: 21873635; 17470549 |
| Stag2 | Stromal antigen 2 | Sister chromatid cohesion, stem cell population maintenance, chromatin, chromatin binding | Abnormal mitosis, decreased B cell number, decreased body weight, decreased cell proliferation, decreased embryo size, decreased fibroblast proliferation | PubMed ID: 21873635; 20720539; 23920377;22780989; 32783938; 32249213 |
| Suox | Sulfite oxidase | Sulfur compound metabolic process, mitochondrion, molybdopterin cofactor binding | Not Applicable | PubMed ID: 21873635; 14651853; 18614015 |
| Thap2 | THAP domain containing apoptosis-associated protein 2 | Nucleus | Not Applicable | PubMed ID: 21873635 |
| Tnfaip8l1 | Tumor necrosis factor, alpha-induced protein 8-like 1 | Negative regulation of TOR signaling, regulation of apoptotic process, protein binding | Abnormal urinary bladder morphology, abnormal kidney morphology | PubMed ID: 21600655; 21873635; 21600655 |
| Zfp455 | Zinc finger protein 455 | RNA polymerase II cis-regulatory region sequence-specific DNA binding | Not Applicable | PubMed ID: 21873635 |
| Gene | N | Control Kidney | IL-6 Kidney | p-Value |
|---|---|---|---|---|
| Man2a1 | 5 | 3,207,680 (3,099,669, 3,810,075) | 2,415,094 (2,237,882, 2,553,436) | 0.032 |
| Klhl15 | 5 | 9,419,328 (8,376,423, 9,450,573) | 7,051,472 (6,556,358, 7,677,648) | 0.095 |
| Khdrbs3 | 5 | 12,632,619 (10,391,560, 4,424,368) | 9,188,871 (9,078,808, 9,727,293) | 0.032 |
| Atp11c | 5 | 4,517,416 (3,907,210, 5,916,229) | 3220225 (2574163, 3491679) | 0.008 |
| Arl3 | 5 | 62,159,247 (53,843,077, 64,752,901) | 54,465,211 (53,381,556, 62,712,896) | 0.548 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panzade, G.; Srivastava, T.; Heruth, D.P.; Rezaiekhaligh, M.H.; Zhou, J.; Lyu, Z.; Sharma, M.; Joshi, T. Employing Multi-Omics Analyses to Understand Changes during Kidney Development in Perinatal Interleukin-6 Animal Model. Cells 2024, 13, 1667. https://doi.org/10.3390/cells13191667
Panzade G, Srivastava T, Heruth DP, Rezaiekhaligh MH, Zhou J, Lyu Z, Sharma M, Joshi T. Employing Multi-Omics Analyses to Understand Changes during Kidney Development in Perinatal Interleukin-6 Animal Model. Cells. 2024; 13(19):1667. https://doi.org/10.3390/cells13191667
Chicago/Turabian StylePanzade, Ganesh, Tarak Srivastava, Daniel P. Heruth, Mohammad H. Rezaiekhaligh, Jianping Zhou, Zhen Lyu, Mukut Sharma, and Trupti Joshi. 2024. "Employing Multi-Omics Analyses to Understand Changes during Kidney Development in Perinatal Interleukin-6 Animal Model" Cells 13, no. 19: 1667. https://doi.org/10.3390/cells13191667