
Adipose Stromal Cell-Derived Secretome Attenuates Cisplatin-Induced Injury In Vitro Surpassing the Intricate Interplay between Proximal Tubular Epithelial Cells and Macrophages


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. MSC Culture and Production of Conditioned Medium (CM)
2.2. Ultra-Filtration of CM to Deplete Extracellular Vesicles
2.3. Thiol Measurement of CM
2.4. ciPTEC Culture and Cisplatin Treatment
2.5. ciPTEC Viability
2.6. ciPTEC Metabolic Activity
2.7. ciPTEC Migratory Capacity
2.8. ciPTEC Apoptosis
2.9. Nephrotoxicity PCR Array
2.10. Confirmatory RT-qPCR
2.11. Intracellular Reactive Oxygen Species Levels
2.12. Monocyte Isolation and Macrophage Culture
2.13. Macrophage Surface Marker Expression
2.14. Macrophage Phagocytosis
2.15. Indirect Co-Cultures of ciPTEC and Macrophages
2.16. Staining of ciPTECs on Transwell Membrane
2.17. Multiplex Assay to Quantify Secreted Factors in the Co-Culture System (Basolateral Side)
2.18. TNF-α Measurement in the Co-Culture System (Apical Side)
2.19. Statistical Analysis
3. Results
3.1. Cisplatin Decreases ciPTEC Viability, Metabolic Activity, and Migratory Capacity
3.1.1. ASC Secretome Attenuated Cisplatin-Induced Cell Toxicity and Promoted Migration of ciPTECs by Ameliorating Apoptosis, DNA Damage, and Oxidative Stress
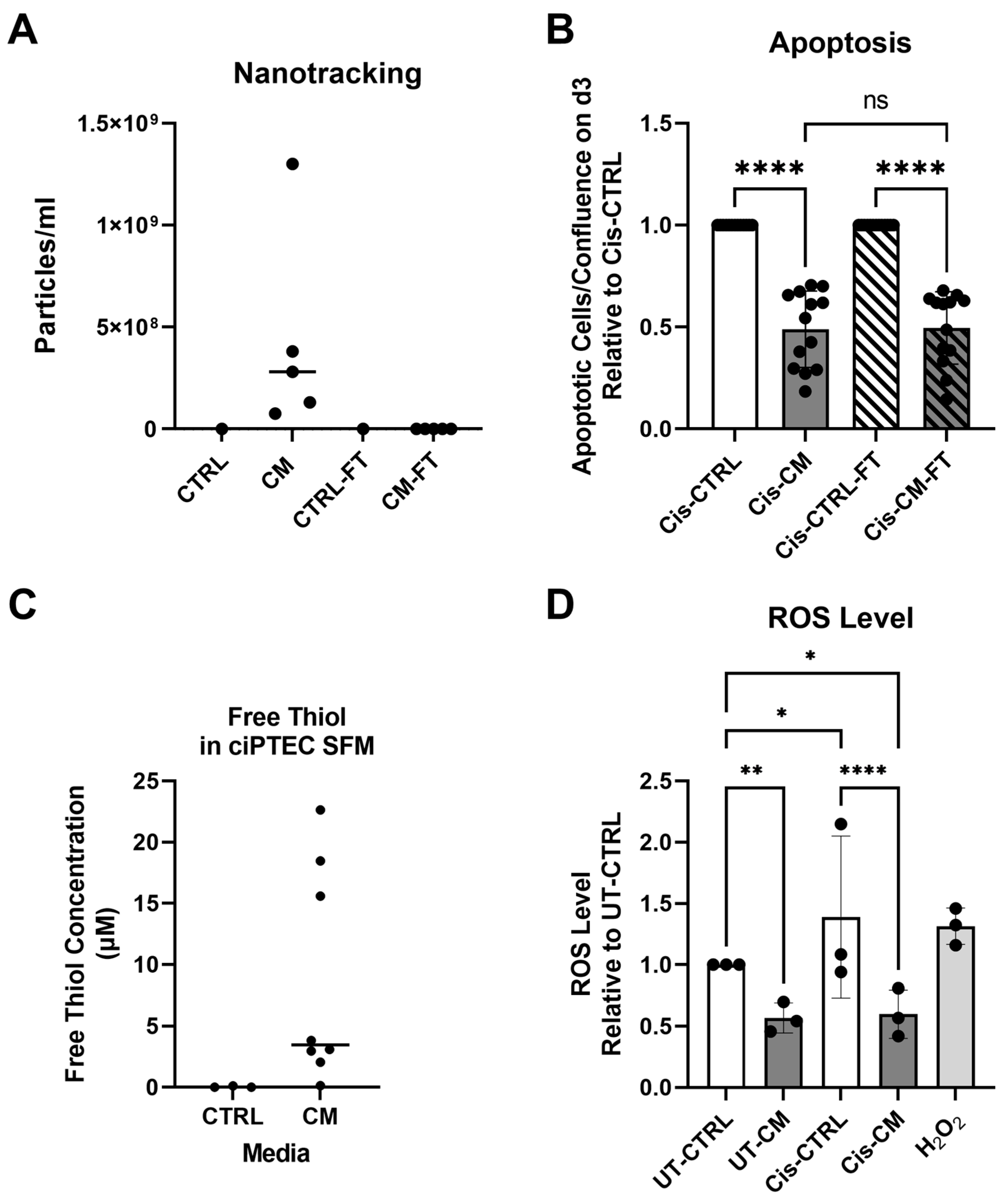
3.1.2. Anti-Apoptotic Effect of CM Was Not Affected by EV Depletion but Related to Its Free Thiol Content
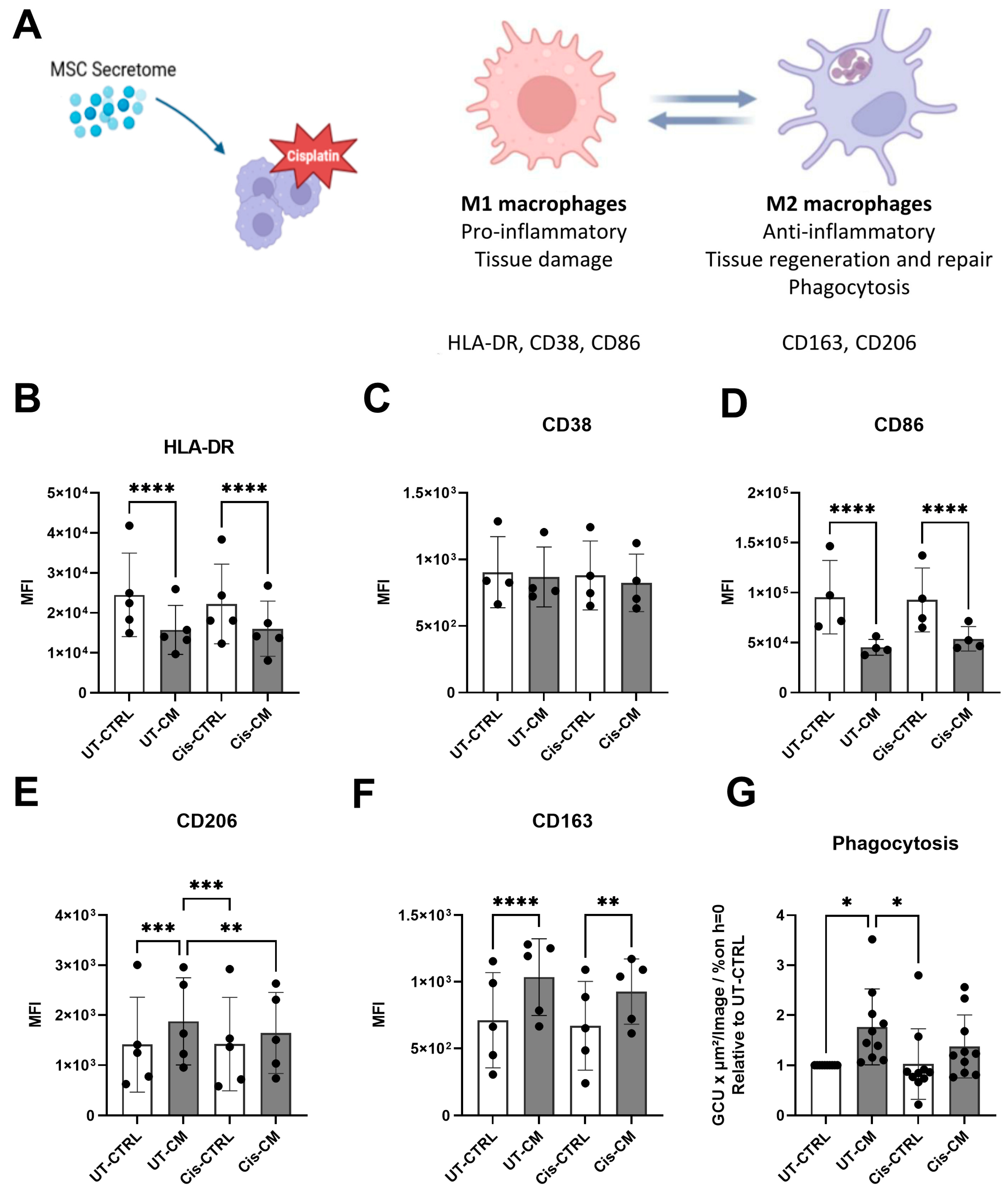
3.2. MSC-CM Promoted Macrophage Polarization towards the M2-like Phenotype
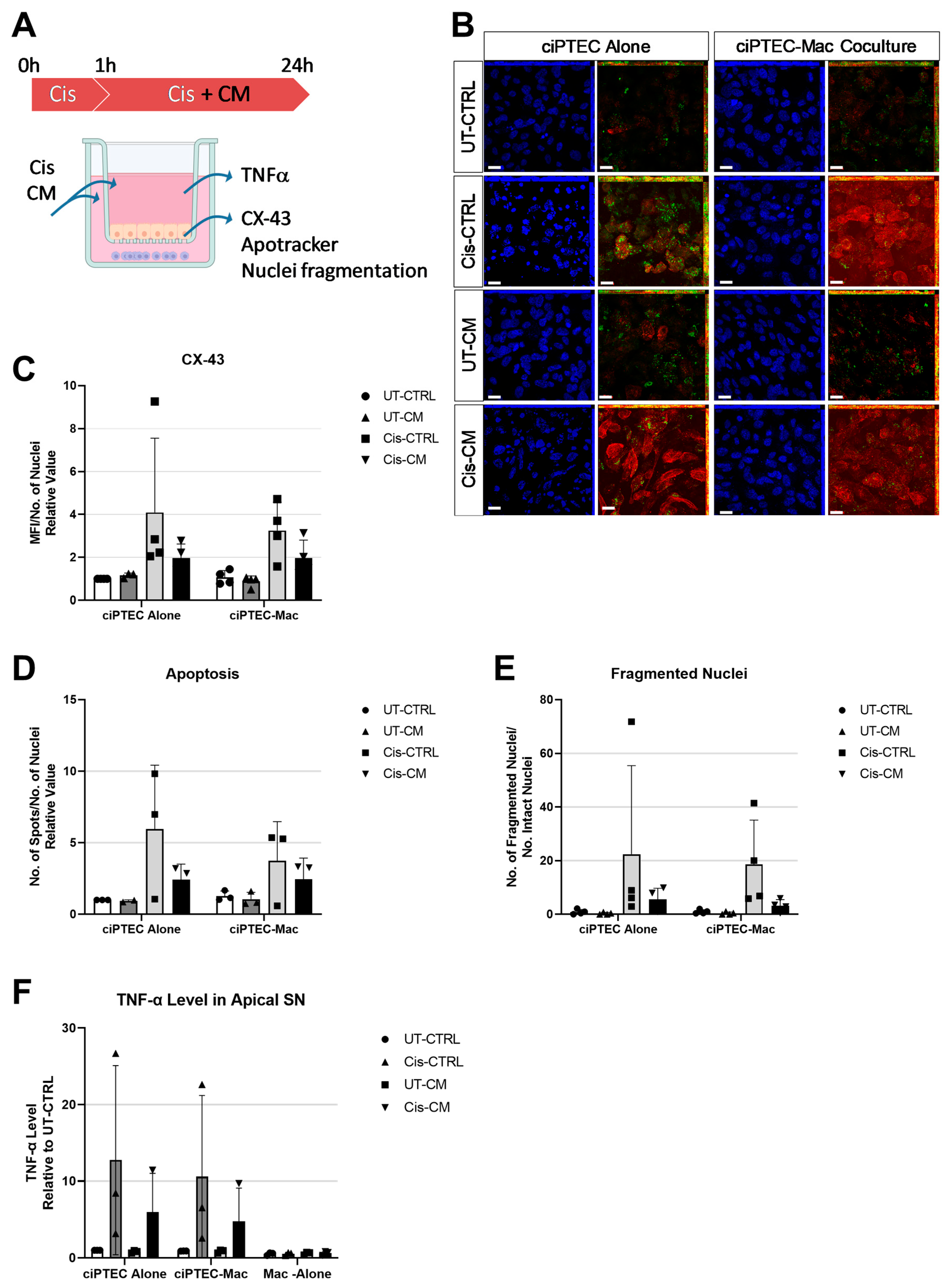
3.3. Macrophages Did Not Boost the Protective Effect of MSC-CM on ciPTECs
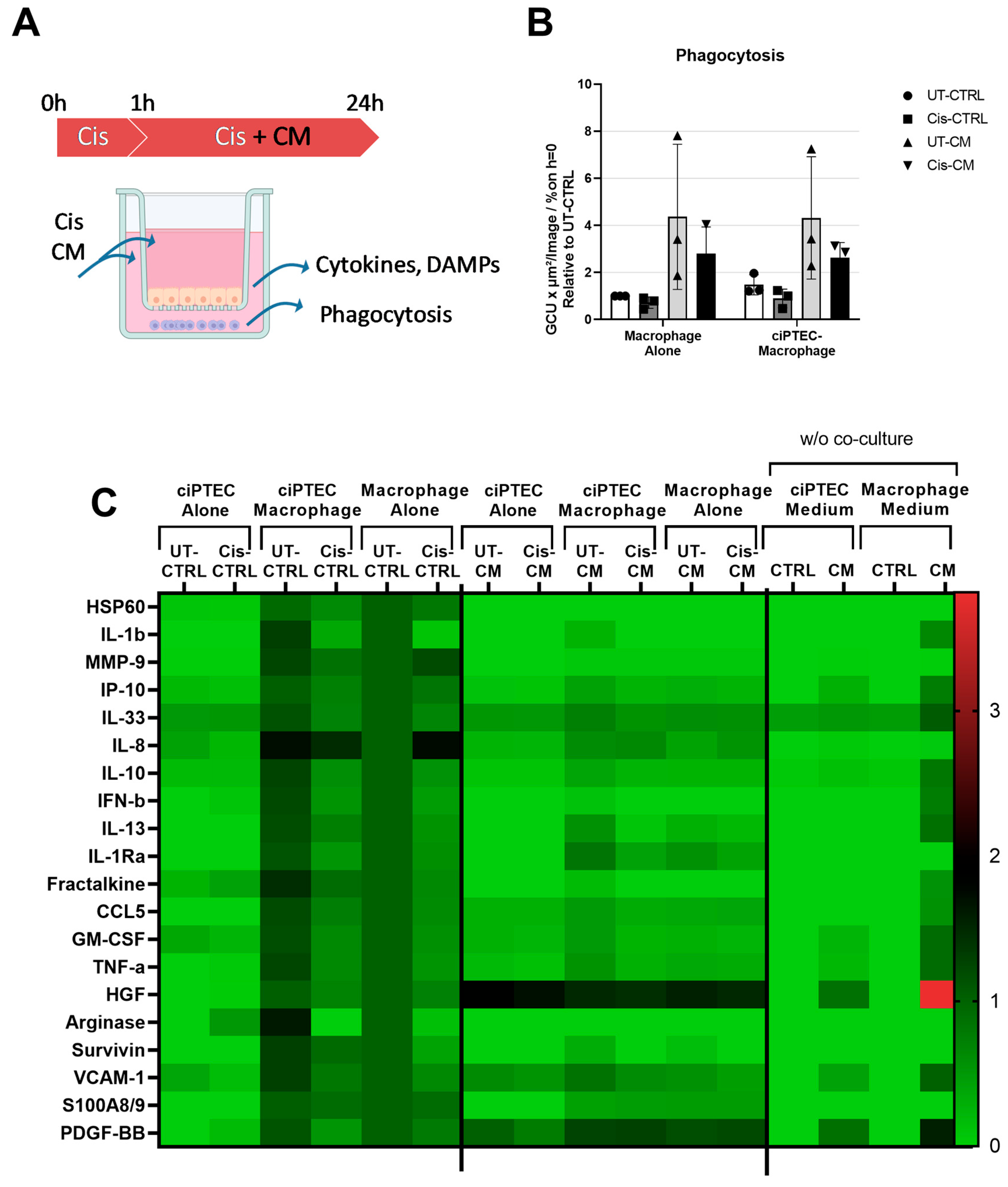
Despite Enhancing the Phagocytosis Capacity of Macrophages, CM Attenuated Macrophage Factor Secretion Triggered by ciPTECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torres Crigna, A.; Daniele, C.; Gamez, C.; Medina Balbuena, S.; Pastene, D.O.; Nardozi, D.; Brenna, C.; Yard, B.; Gretz, N.; Bieback, K. Stem/Stromal Cells for Treatment of Kidney Injuries With Focus on Preclinical Models. Front. Med. 2018, 5, 179. [Google Scholar] [CrossRef] [PubMed]
- Stavely, R.; Nurgali, K. The emerging antioxidant paradigm of mesenchymal stem cell therapy. Stem Cells Transl. Med. 2020, 9, 985–1006. [Google Scholar] [CrossRef]
- Rendra, E.; Scaccia, E.; Bieback, K. Recent advances in understanding mesenchymal stromal cells. F1000Research 2020, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Daneshmandi, L.; Shah, S.; Jafari, T.; Bhattacharjee, M.; Momah, D.; Saveh-Shemshaki, N.; Lo, K.W.; Laurencin, C.T. Emergence of the Stem Cell Secretome in Regenerative Engineering. Trends Biotechnol. 2020, 38, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Nonnekens, J.; Hoeijmakers, J.H. After surviving cancer, what about late life effects of the cure? EMBO Mol. Med. 2017, 9, 4–6. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, K.R.; Gadanec, L.K.; Qaradakhi, T.; Ali, B.A.; Zulli, A.; Apostolopoulos, V. Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers 2021, 13, 1572. [Google Scholar] [CrossRef]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef]
- Oh, G.S.; Kim, H.J.; Shen, A.; Lee, S.B.; Khadka, D.; Pandit, A.; So, H.S. Cisplatin-induced Kidney Dysfunction and Perspectives on Improving Treatment Strategies. Electrolyte Blood Press. 2014, 12, 55–65. [Google Scholar] [CrossRef]
- Yonezawa, A.; Inui, K. Organic cation transporter OCT/SLC22A and H(+)/organic cation antiporter MATE/SLC47A are key molecules for nephrotoxicity of platinum agents. Biochem. Pharmacol. 2011, 81, 563–568. [Google Scholar] [CrossRef]
- Motohashi, H.; Nakao, Y.; Masuda, S.; Katsura, T.; Kamba, T.; Ogawa, O.; Inui, K. Precise comparison of protein localization among OCT, OAT, and MATE in human kidney. J. Pharm. Sci. 2013, 102, 3302–3308. [Google Scholar] [CrossRef]
- Soni, H.; Kaminski, D.; Gangaraju, R.; Adebiyi, A. Cisplatin-induced oxidative stress stimulates renal Fas ligand shedding. Ren. Fail. 2018, 40, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Huen, S.C.; Cantley, L.G. Macrophages in Renal Injury and Repair. Annu. Rev. Physiol. 2017, 79, 449–469. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H. The Impact of Versatile Macrophage Functions on Acute Kidney Injury and Its Outcomes. Front. Physiol. 2019, 10, 1016. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.E.; Chade, A.R. Macrophage polarization in chronic kidney disease: A balancing act between renal recovery and decline? Am. J. Physiol. Ren. Physiol. 2019, 317, F1409–F1413. [Google Scholar] [CrossRef] [PubMed]
- Guiteras, R.; Flaquer, M.; Cruzado, J.M. Macrophage in chronic kidney disease. Clin. Kidney J. 2016, 9, 765–771. [Google Scholar] [CrossRef]
- Lech, M.; Grobmayr, R.; Ryu, M.; Lorenz, G.; Hartter, I.; Mulay, S.R.; Susanti, H.E.; Kobayashi, K.S.; Flavell, R.A.; Anders, H.J. Macrophage phenotype controls long-term AKI outcomes--kidney regeneration versus atrophy. J. Am. Soc. Nephrol. 2014, 25, 292–304. [Google Scholar] [CrossRef]
- Nash, W.T.; Yee, M.S.; Okusa, M.D. Myeloid Response to Acute Kidney Injury. Nephron 2023, 147, 39–43. [Google Scholar] [CrossRef]
- Lu, L.H.; Oh, D.J.; Dursun, B.; He, Z.; Hoke, T.S.; Faubel, S.; Edelstein, C.L. Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. J. Pharmacol. Exp. Ther. 2008, 324, 111–117. [Google Scholar] [CrossRef]
- Rui-Zhi, T.; Ke-Huan, X.; Yuan, L.; Xiao, L.; Bing-Wen, Z.; Tong-Tong, L.; Li, W. Renoprotective effect of isoliquiritigenin on cisplatin-induced acute kidney injury through inhibition of FPR2 in macrophage. J. Pharmacol. Sci. 2022, 148, 56–64. [Google Scholar] [CrossRef]
- Tan, R.-Z.; Wang, C.; Deng, C.; Zhong, X.; Yan, Y.; Luo, Y.; Lan, H.-Y.; He, T.; Wang, L. Quercetin protects against cisplatin-induced acute kidney injury by inhibiting Mincle/Syk/NF-κB signaling maintained macrophage inflammation. Phytother. Res. 2020, 34, 139–152. [Google Scholar] [CrossRef]
- Nakagawa, M.; Karim, M.R.; Izawa, T.; Kuwamura, M.; Yamate, J. Immunophenotypical Characterization of M1/M2 Macrophages and Lymphocytes in Cisplatin-Induced Rat Progressive Renal Fibrosis. Cells 2021, 10, 257. [Google Scholar] [CrossRef]
- Netsch, P.; Elvers-Hornung, S.; Uhlig, S.; Klüter, H.; Huck, V.; Kirschhöfer, F.; Brenner-Weiß, G.; Janetzko, K.; Solz, H.; Wuchter, P.; et al. Human mesenchymal stromal cells inhibit platelet activation and aggregation involving CD73-converted adenosine. Stem Cell Res. Ther. 2018, 9, 184. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, T.E.; Nagelkerke, A.; Nele, V.; Kauscher, U.; Stevens, M.M. Experimental artefacts can lead to misattribution of bioactivity from soluble mesenchymal stem cell paracrine factors to extracellular vesicles. J. Extracell. Vesicles 2020, 9, 1807674. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.P.; Bhatnagar, A. Role of thiols in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.; Schophuizen, C.M.S.; Wilmer, M.J.; Lahham, S.H.M.; Mutsaers, H.A.M.; Wetzels, J.F.M.; Bank, R.A.; van den Heuvel, L.P.; Hoenderop, J.G.; Masereeuw, R. A morphological and functional comparison of proximal tubule cell lines established from human urine and kidney tissue. Exp. Cell Res. 2014, 323, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Itoh, Y.; Matsuo, M.; Kawashiri, T.; Egashira, N.; Oishi, R. Involvement of both tumor necrosis factor-α-induced necrosis and p53-mediated caspase-dependent apoptosis in nephrotoxicity of cisplatin. Apoptosis 2007, 12, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Rendra, E.; Torres Criogna, A.; Daniele, C.; Sticht, C.; Kluth, M.A.; Cueppers, M.; Ganss, C.; Frank, M.H.; Gretz, N.; Bieback, K. Clinical-grade human skin-derived ABCB5+ mesenchymal stromal cells exert anti-apoptotic and anti-inflammatory effects in vitro and modulate mRNA expression in a cisplatin-induced kidney injury murine model. Front. Immunol. 2023, 14, 1228928. [Google Scholar] [CrossRef]
- Ershov, D.; Phan, M.-S.; Pylvänäinen, J.W.; Rigaud, S.U.; Le Blanc, L.; Charles-Orszag, A.; Conway, J.R.W.; Laine, R.F.; Roy, N.H.; Bonazzi, D.; et al. TrackMate 7: Integrating state-of-the-art segmentation algorithms into tracking pipelines. Nat. Methods 2022, 19, 829–832. [Google Scholar] [CrossRef]
- Bruno, S.; Grange, C.; Collino, F.; Deregibus, M.C.; Cantaluppi, V.; Biancone, L.; Tetta, C.; Camussi, G. Microvesicles derived from mesenchymal stem cells enhance survival in a lethal model of acute kidney injury. PLoS ONE 2012, 7, e33115. [Google Scholar] [CrossRef]
- Yuan, X.; Li, D.; Chen, X.; Han, C.; Xu, L.; Huang, T.; Dong, Z.; Zhang, M. Extracellular vesicles from human-induced pluripotent stem cell-derived mesenchymal stromal cells (hiPSC-MSCs) protect against renal ischemia/reperfusion injury via delivering specificity protein (SP1) and transcriptional activating of sphingosine kinase 1 and inhibiting necroptosis. Cell Death Dis. 2017, 8, 3200. [Google Scholar] [CrossRef]
- Abdulle, A.E.; Bourgonje, A.R.; Kieneker, L.M.; Koning, A.M.; la Bastide-van Gemert, S.; Bulthuis, M.L.C.; Dijkstra, G.; Faber, K.N.; Dullaart, R.P.F.; Bakker, S.J.L.; et al. Serum free thiols predict cardiovascular events and all-cause mortality in the general population: A prospective cohort study. BMC Med. 2020, 18, 130. [Google Scholar] [CrossRef] [PubMed]
- Roger, E.; Boutin, L.; Chadjichristos, C.E. The Role of Connexin 43 in Renal Disease: Insights from In Vivo Models of Experimental Nephropathy. Int. J. Mol. Sci. 2022, 23, 13090. [Google Scholar] [CrossRef] [PubMed]
- Miyaji, T.; Kato, A.; Yasuda, H.; Fujigaki, Y.; Hishida, A. Role of the increase in p21 in cisplatin-induced acute renal failure in rats. J. Am. Soc. Nephrol. 2001, 12, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Liebermann, D.A.; Hoffman, B. Gadd45 in stress signaling. J. Mol. Signal. 2008, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. The Role of HO-1 and Its Crosstalk with Oxidative Stress in Cancer Cell Survival. Cells 2021, 10, 2401. [Google Scholar] [CrossRef]
- Ku, H.C.; Cheng, C.F. Master Regulator Activating Transcription Factor 3 (ATF3) in Metabolic Homeostasis and Cancer. Front. Endocrinol. 2020, 11, 556. [Google Scholar] [CrossRef]
- Block, G.J.; Ohkouchi, S.; Fung, F.; Frenkel, J.; Gregory, C.; Pochampally, R.; DiMattia, G.; Sullivan, D.E.; Prockop, D.J. Multipotent stromal cells are activated to reduce apoptosis in part by upregulation and secretion of stanniocalcin-1. Stem Cells 2009, 27, 670–681. [Google Scholar] [CrossRef]
- Gowen, A.; Shahjin, F.; Chand, S.; Odegaard, K.E.; Yelamanchili, S.V. Mesenchymal Stem Cell-Derived Extracellular Vesicles: Challenges in Clinical Applications. Front. Cell Dev. Biol. 2020, 8, 149. [Google Scholar] [CrossRef]
- Skovronova, R.; Scaccia, E.; Calcat-i-Cervera, S.; Bussolati, B.; O’Brien, T.; Bieback, K. Adipose stromal cells bioproducts as cell-free therapies: Manufacturing and therapeutic dose determine in vitro functionality. J. Transl. Med. 2023, 21, 723. [Google Scholar] [CrossRef]
- Wolf, M.; Poupardin, R.W.; Ebner-Peking, P.; Andrade, A.C.; Blochl, C.; Obermayer, A.; Gomes, F.G.; Vari, B.; Maeding, N.; Eminger, E.; et al. A functional corona around extracellular vesicles enhances angiogenesis, skin regeneration and immunomodulation. J. Extracell. Vesicles 2022, 11, e12207. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Xu, Y.; Lu, Y.C.; Du, P.; Li, X.X.; Wang, C.C.; Guo, P.; Diao, L.; Lu, G.Z. Mesenchymal stromal cells pretreated with proinflammatory cytokines enhance skin wound healing via IL-6-dependent M2 polarization. Stem Cell Res. Ther. 2022, 13, 414. [Google Scholar] [CrossRef]
- Berg, T.; Hegelund-Myrbäck, T.; Öckinger, J.; Zhou, X.H.; Brännström, M.; Hagemann-Jensen, M.; Werkström, V.; Seidegård, J.; Grunewald, J.; Nord, M.; et al. Expression of MATE1, P-gp, OCTN1 and OCTN2, in epithelial and immune cells in the lung of COPD and healthy individuals. Respir. Res. 2018, 19, 68. [Google Scholar] [CrossRef] [PubMed]
- Schoeberl, A.; Gutmann, M.; Theiner, S.; Schaier, M.; Schweikert, A.; Berger, W.; Koellensperger, G. Cisplatin Uptake in Macrophage Subtypes at the Single-Cell Level by LA-ICP-TOFMS Imaging. Anal. Chem. 2021, 93, 16456–16465. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 01462. [Google Scholar] [CrossRef] [PubMed]
- Luque-Campos, N.; Bustamante-Barrientos, F.A.; Pradenas, C.; García, C.; Araya, M.J.; Bohaud, C.; Contreras-López, R.; Elizondo-Vega, R.; Djouad, F.; Luz-Crawford, P.; et al. The Macrophage Response Is Driven by Mesenchymal Stem Cell-Mediated Metabolic Reprogramming. Front. Immunol. 2021, 12, 624746. [Google Scholar] [CrossRef] [PubMed]
- Kleih, M.; Böpple, K.; Dong, M.; Gaissler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019, 10, 851. [Google Scholar] [CrossRef]
- Prakoura, N.; Kavvadas, P.; Chadjichristos, C.E. Connexin 43: A New Therapeutic Target Against Chronic Kidney Disease? Cell. Physiol. Biochem. 2018, 49, 998–1009. [Google Scholar] [CrossRef]
- Xu, H.Z.; Wang, M.; Li, Y.Z.; Shi, M.X.; Wang, Z.; Cao, C.J.; Hong, Y.; Hu, B.; Zhu, H.; Zhao, Z.; et al. Blocking connexin 43 and its promotion of ATP release from renal tubular epithelial cells ameliorates renal fibrosis. Cell Death Dis. 2022, 13, 511. [Google Scholar] [CrossRef]
- Kumar, R.; Herbert, P.E.; Warrens, A.N. An introduction to death receptors in apoptosis. Int. J. Surg. 2005, 3, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Atherton, S.S. Tumor necrosis factor-α in cisplatin nephrotoxicity: A homebred foe? Kidney Int. 2007, 72, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ramesh, G.; Norbury, C.C.; Reeves, W.B. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-α produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.J.; Rees, A.J.; Griffin, M.D.; Hughes, J.; Kurts, C.; Duffield, J. The Renal Mononuclear Phagocytic System. J. Am. Soc. Nephrol. 2012, 23, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Calle, P.; Jativa, S.; Torrico, S.; Muñoz, A.; García, M.; Sola, A.; Serra, D.; Mera, P.; Herrero, L.; Hotter, G. Infusion of Phagocytic Macrophages Overexpressing CPT1a Ameliorates Kidney Fibrosis in the UUO Model. Cells 2021, 10, 1650. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef]
- Sampangi, S.; Kassianos, A.J.; Wang, X.J.; Beagley, K.W.; Klein, T.; Afrin, S.; Healy, H.; Wilkinson, R. The Mechanisms of Human Renal Epithelial Cell Modulation of Autologous Dendritic Cell Phenotype and Function. PLoS ONE 2015, 10, 0134688. [Google Scholar] [CrossRef]
- Li, M.; Xie, H.B.; Liu, Y.K.; Xia, C.Y.; Cun, X.L.; Long, Y.; Chen, X.X.; Deng, M.; Guo, R.; Zhang, Z.R.; et al. Knockdown of hypoxia-inducible factor-1 alpha by tumor targeted delivery of CRISPR/Cas9 system suppressed the metastasis of pancreatic cancer. J. Control. Release 2019, 304, 204–215. [Google Scholar] [CrossRef]
- Lv, L.L.; Feng, Y.; Wu, M.; Wang, B.; Li, Z.L.; Zhong, X.; Wu, W.J.; Chen, J.; Ni, H.F.; Tang, T.T.; et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differ. 2020, 27, 210–226. [Google Scholar] [CrossRef]
- Jia, Y.; Chen, J.; Zheng, Z.; Tao, Y.; Zhang, S.; Zou, M.; Yang, Y.; Xue, M.; Hu, F.; Li, Y.; et al. Tubular epithelial cell-derived extracellular vesicles induce macrophage glycolysis by stabilizing HIF-1alpha in diabetic kidney disease. Mol. Med. 2022, 28, 95. [Google Scholar] [CrossRef]
- Lai, K.N.; Leung, J.C.K.; Chan, L.Y.Y.; Guo, H.; Tang, S.C.W. Interaction between proximal tubular epithelial cells and infiltrating monocytes/T cells in the proteinuric state. Kidney Int. 2007, 71, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.H.; Kim, H.K.; Na, J.Y.; Jin, C.; Seon, J.K. Anti-inflammatory effects of mesenchymal stem cell-conditioned media inhibited macrophages activation in vitro. Sci. Rep. 2022, 12, 4754. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Qiu, H.; Xue, M.; Zhang, S.; Zhang, X.; Xu, J.; Chen, J.; Yang, Y.; Xie, J. MSC-secreted TGF-beta regulates lipopolysaccharide-stimulated macrophage M2-like polarization via the Akt/FoxO1 pathway. Stem Cell Res. Ther. 2019, 10, 345. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Kim, H.J.; Jeong, H.J.; Lee, H.J.; Oh, J.Y. Mesenchymal Stem and Stromal Cells Harness Macrophage-Derived Amphiregulin to Maintain Tissue Homeostasis. Cell Rep. 2020, 30, 3806–3820.E6. [Google Scholar] [CrossRef] [PubMed]
- Stevens, H.Y.; Bowles, A.C.; Yeago, C.; Roy, K. Molecular Crosstalk Between Macrophages and Mesenchymal Stromal Cells. Front. Cell Dev. Biol. 2020, 8, 600160. [Google Scholar] [CrossRef]
- Giacomini, C.; Graneli, C.; Hicks, R.; Dazzi, F. The critical role of apoptosis in mesenchymal stromal cell therapeutics and implications in homeostasis and normal tissue repair. Cell. Mol. Immunol. 2023, 20, 570–582. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Liu, S.Y.; Zhao, M.; Wang, C.S.; Li, L.; Yuan, Y.J.; Li, L.; Liao, G.N.; Bresette, W.; Zhang, J.; et al. Injectable extracellular vesicle-released self-assembling peptide nanofiber hydrogel as an enhanced cell-free therapy for tissue regeneration. J. Control. Release 2019, 316, 93–104. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rendra, E.; Uhlig, S.; Moskal, I.; Thielemann, C.; Klüter, H.; Bieback, K. Adipose Stromal Cell-Derived Secretome Attenuates Cisplatin-Induced Injury In Vitro Surpassing the Intricate Interplay between Proximal Tubular Epithelial Cells and Macrophages. Cells 2024, 13, 121. https://doi.org/10.3390/cells13020121
Rendra E, Uhlig S, Moskal I, Thielemann C, Klüter H, Bieback K. Adipose Stromal Cell-Derived Secretome Attenuates Cisplatin-Induced Injury In Vitro Surpassing the Intricate Interplay between Proximal Tubular Epithelial Cells and Macrophages. Cells. 2024; 13(2):121. https://doi.org/10.3390/cells13020121
Chicago/Turabian StyleRendra, Erika, Stefanie Uhlig, Isabell Moskal, Corinna Thielemann, Harald Klüter, and Karen Bieback. 2024. "Adipose Stromal Cell-Derived Secretome Attenuates Cisplatin-Induced Injury In Vitro Surpassing the Intricate Interplay between Proximal Tubular Epithelial Cells and Macrophages" Cells 13, no. 2: 121. https://doi.org/10.3390/cells13020121
APA StyleRendra, E., Uhlig, S., Moskal, I., Thielemann, C., Klüter, H., & Bieback, K. (2024). Adipose Stromal Cell-Derived Secretome Attenuates Cisplatin-Induced Injury In Vitro Surpassing the Intricate Interplay between Proximal Tubular Epithelial Cells and Macrophages. Cells, 13(2), 121. https://doi.org/10.3390/cells13020121