Abstract
Protein homeostasis is essential for neuron longevity, requiring a balanced regulation between protein synthesis and degradation. The clearance of misfolded and aggregated proteins, mediated by autophagy and the ubiquitin–proteasome systems, maintains protein homeostasis in neurons, which are post-mitotic and thus cannot use cell division to diminish the burden of misfolded proteins. When protein clearance pathways are overwhelmed or otherwise disrupted, the accumulation of misfolded or aggregated proteins can lead to the activation of ER stress and the formation of stress granules, which predominantly attempt to restore the homeostasis by suppressing global protein translation. Alterations in these processes have been widely reported among studies investigating the toxic function of dipeptide repeats (DPRs) produced by G4C2 expansion in the C9orf72 gene of patients with amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). In this review, we outline the modalities of DPR-induced disruptions in protein homeostasis observed in a wide range of models of C9orf72-linked ALS/FTD. We also discuss the relative importance of each DPR for toxicity, possible synergies between DPRs, and discuss the possible functional relevance of DPR aggregation to disease pathogenesis. Finally, we highlight the interdependencies of the observed effects and reflect on the importance of feedback and feedforward mechanisms in their contribution to disease progression. A better understanding of DPR-associated disease pathogenesis discussed in this review might shed light on disease vulnerabilities that may be amenable with therapeutic interventions.
Keywords:
protein homeostasis; ALS; FTD; C9orf72; dipeptide repeats; autophagy; ER stress; proteasome; lysosome 1. Introduction
It is now well established that aging is associated with a progressive dysregulation in protein homeostasis [1,2]. Consistent with this, a key hallmark of many neurodegenerative diseases is the accumulation of toxic protein aggregates, ultimately leading to progressive loss of neuronal structure and function. This is the case for amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), two incurable diseases that share overlapping clinical manifestations, pathogenic mechanisms, and genetic risk factors [3,4]. In fact, many patients of both diseases have been found to carry a G4C2 hexanucleotide repeat expansion (HRE) in a non-coding region of the C9orf72 gene [5,6].
While, to date, most patients affected by C9orf72-linked ALS/FTD (C9-ALS/FTD) harbor between 100 and 1000 G4C2 repeats, the exact disease-causing threshold and how repeat length might alter disease progression, remains to be determined. Indeed, several studies have shown that expansions of around 30 repeats could be sufficient to be disease-causing [7,8,9]. In particular, the expansion is thought to cause disease via two mutually inclusive mechanisms: (1) a loss of function of the C9orf72 gene [10] and (2) a toxic gain of function driven by the HRE itself.
The relative importance of each of the two mechanisms represents a controversial, unanswered question at the core of the C9-ALS/FTD field. Indeed, since the discovery C9orf72-linked ALS/FTD, many conflicting results have been published regarding the relative importance of each of the two mechanisms. The lack of a clear correlation between repeat length, for example, appears to argue against the toxic gain-of-function as being a major driver of disease pathology. In line with this a recent phase I clinical trial (BIIB078) of an antisense oligonucleotide (ASO), targeting G4C2 repeat RNA and thus also DPR generation, failed to show any clinical benefits. It is unclear, however, whether the ASO administration indeed significantly lowered the levels of repeat RNAs and DPRs. Arguing instead against the loss-of-function hypothesis, patients homozygous for the C9orf72 repeat expansion, and thus expressing less C9orf72, do not show a more serve phenotype than those with C9orf72 haploinsufficiency [11]. In addition, C9orf72 knockout mice do not show any ALS/FTD-associated neurodegenerative phenotypes [12]. Although this remains an important unanswered question to be addressed in the field, the two mechanisms likely act in coordination, and it is therefore important to continue investigating both hypotheses.
Interestingly, the overexpression of a sufficient number of G4C2 repeats per se (i.e., without modulation of C9orf72 gene dosage) in vitro [13,14,15] as well as in mice [16,17], zebrafish [18,19], and Drosophila melanogaster [20,21] is sufficient to induce toxicity and neurodegeneration. These can be bidirectionally transcribed into repeat RNAs that subsequently aggregate, a general hallmark of non-coding repeat expansion diseases. Indeed, samples from patients with C9-ALS/FTD and patient-derived induced pluripotent stem cells (iPSCs) have been shown to contain nuclear, as well as some cytoplasmic, foci of aggregated RNA [18,20,22,23,24]. Further, these sense and anti-sense repeat RNAs can undergo non-canonical, repeat-associated non-AUG (RAN) translation to produce five distinct dipeptide repeats (DPRs): poly-PA, poly-PR, poly-GA, poly-GP, and poly-GR [25,26,27,28,29].
In this review, we focus on the role of DPRs in disrupting protein homeostasis pathways, and on recent evidence revealing how DPRs may contribute to multiple aspects of disease pathogenesis.
2. The Impact of C9-Associated Toxic Repeats on Protein Degradation Pathways
A key neuropathological hallmark in C9-ALS/FTD is the formation of star-shaped cytoplasmic DPR inclusions which are ubiquitin- and p62-positive [30,31], indicative of an involvement of the ubiquitin–proteasome system (UPS) and of macroautophagy (hereafter autophagy) in the targeted degradation of DPRs. Consistent with this, the pharmacological inhibition of either pathway leads to an increased accumulation and aggregation of distinct DPRs in cell culture [14,32]. However, several recent studies suggest that, while DPRs are targets of UPS- and autophagy-mediated clearance, they might also play a major role in disrupting both pathways (Figure 1), as discussed in detail in the next two chapters.
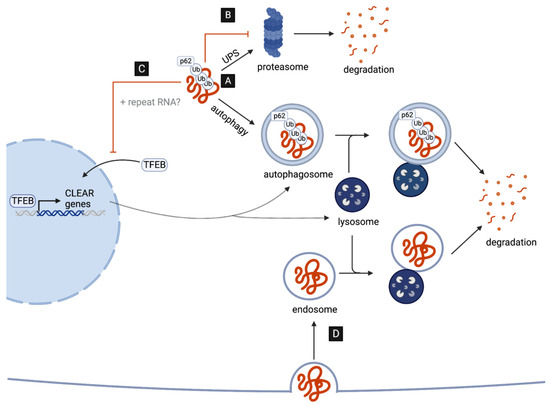
Figure 1.
DPRs as targets and disruptors of cellular clearance pathways. (A) C9-associated DPRs are targeted for degradation by the ubiquitin–proteasome system (UPS) and the autophagy pathway. (B) DPRs impair the UPS by directly interacting with and inhibiting the 26S proteasome. (C) DPRs (and repeat RNAs) disrupt the autophagy pathway reducing the nuclear translocation of TFEB, the master transcriptional regulator of autophagy genes (CLEAR network). (D) G4C2-induced impairment in lysosome function may aid the cell-to-cell transmission of DPRs via endocytosis.
2.1. Poly-GA Inhibits Protein Degradation via the UPS
The UPS is the major cellular pathway responsible for the degradation and recycling of short-lived, soluble proteins, and indeed appears to be important for DPR degradation [14,32]. In addition to being possible targets of UPS-mediated degradation, DPRs in fact appear to inhibit UPS activity [13,14,15,33,34,35,36,37]. Interestingly, several mutations in genes involved in the UPS have been associated with both ALS and FTD [4], highlighting the UPS as a common point of vulnerability in both neurodegenerative diseases. While all DPRs, with the notable exception of poly-PA, have been associated with disrupted UPS, the underlying mechanisms remain unclear.
Several studies have shown that UPS factors co-localize with cytoplasmic poly-GA inclusions in cell culture [13,14,15,33,34], in animal models, and in the brains of patients with C9-ALS/FTD [35]. Pointing to a more direct impairment of the UPS system by accumulation poly-GA, it has recently been shown that the 26S proteasome, which is ultimately required to degrade ubiquitin-tagged proteasomal substrates, is sequestered into poly-GA aggregates in cultured neurons [33]. In situ structural analysis of neuronal poly-GA aggregates suggests that these may force the sequestered proteasomes to become stuck in a highly transient intermediate state, which is usually associated with substrate translocation. This may lead to stalled degradation of ubiquitinated substrates, which could explain observed reductions in proteasome activity [33].
The specific consequences of Impaired proteasome function in the context of C9-linked ALS/FTD remains to be further investigated. However, independent of C9-ALS/FTD, the inhibition of the 26S proteasome has also been shown to lead to the cytoplasmic mislocalization and aggregation of TAR DNA-binding protein 43 (TDP43) [38,39], a second aggregation-prone protein associated with the vast majority of familiar and sporadic forms of ALS. Indeed, some TDP-43 pathology has been observed in patients with C9-ALS/FTD [28,40,41], although it appears to occur after DPR pathology [42,43]. Whether proteasomal inhibition by poly-GA may be a contributing factor to TDP-43 pathology therefore warrants further investigation. Indeed, primitive evidence suggests that poly-GA aggregates are able to induce TDP-43 mislocalization in cell culture, and that this mislocalization is dependent on GA-induced inhibition of the proteasome [34]. Interestingly, proteasome inhibition was shown to occur in a cell-autonomous and non-cell-autonomous manner. Specifically, cytoplasmic TDP-43 was observed in rat primary hippocampal neurons cultured with cell supernatant from GA-transduced cells. Such TDP-43 mislocalization was eliminated when depleting the culture media of poly-GA with anti-GA antibodies. While is remains to be determined whether this is through cell-to-cell transmission, this mechanism may help to explain why DPR inclusions and TDP-43 pathology predominantly occur in distinct neurons within the brains of patients with C9-ALS/FTD [41].
2.2. C9-Associated Toxic Repeats Disrupt Autophagosome and Lysosome Biogenesis
In addition to the UPS, the autophagy pathway is another essential contributor to intracellular protein clearance, which predominantly targets insoluble, aggregated, and long-lived proteins. The autophagy pathway involves the recognition of ubiquitin-tagged proteins destined to clearance by adaptor proteins, such as p62/SQSTM1, promoting engulfment by the forming autophagosome, a specialized double-membrane organelle [44]. In neurons, autophagosomes are predominantly formed in distal regions of the axon and, as they undergo retrograde transport toward the soma, they mature and ultimately fuse with lysosomes, a process critical for neuronal longevity [45]. Importantly, lysosomes contain digestive enzymes, activated by a low pH, which eventually break down the autophagic cargo, allowing the degraded products to be recycled back to the cell [46].
Emerging evidence suggests that both repeat RNAs and DPRs may disrupt multiple steps of the autophagy pathway. The overexpression of G4C2 repeats in Drosophila motor neurons, for example, leads to an accumulation of Drosophila p62, accompanied by a reduction in the number of autophagosomal vesicles in both the soma and distal axons in vivo [47,48]. These data suggest that autophagy initiation, and specifically autophagosome formation, may be impaired. In line with this, G4C2 repeat expression results in reductions in the levels of mature autophagosomes in cell culture [49].
How could autophagy induction be affected by the expression of G4C2 repeats? One possibility is that reductions in autophagosome formation may be, at least in part, a result of disruptions in TFEB, the master transcriptional regulator of autophagy and lysosomal biogenesis (Figure 1C). Specifically, G4C2 toxicity appears to inhibit the nuclear import of TFEB in Drosophila as well as in cell culture [47,49]. Remarkably, dysfunction in nucleo-cytoplasmic transport and the nuclear pore complex (NPC) is emerging as a key contributor to disease pathogenesis [50]. This includes disruptions induced by repeat RNAs in Drosophila neurons and patient-derived iPSCs [51,52], as well as by DPRs. In fact, using Xenopus laevis oocytes as a model system, poly-PR has specifically been shown to bind to the central channel nuclear pore to induce a block in the transport of macromolecules between the nucleus and cytoplasm [53]. Additionally, both cytoplasmic poly-GA and poly-GR aggregates appear to sequester components of the NPC in the brains of DPR-expressing mice and patients with C9-ALS/FTD [35,54]. This includes the nucleoporin POM121, which when overexpressed leads to a rescue of TFEB nuclear localization and autophagy initiation in cell culture [49].
In addition to promoting autophagosome formation, TFEB is essential in mammals and in flies for the expression of genes required for lysosome biogenesis and function [55,56,57]. Thus, it is also possible that lysosomes might be defective in the presence of repeat RNAs and DPRs. Consistent with this possibility, G4C2 overexpression results in reduced cleavage and activation of a Drosophila cathepsin and might result in reduced lysosome acidification. Indeed, the overexpression of vacuolar ATPase (V-ATPase) genes, encoding the main proton pump required for lysosome acidification, appear to suppress G4C2-induced neurodegeneration in Drosophila [47]. Interestingly, the specific overexpression of only poly-GA in human cells has been shown to lead to the accumulation of mature autophagosomes, as well as p62 and ubiquitin, which is indicative of lysosomal impairment but not necessarily disruptions in autophagosome formation [58]. This discrepancy may be a result of the different system used, as well as the differences arising from the expression of a single DPR versus all DPRs simultaneously (e.g., G4C2 repeat overexpression).
Despite the accumulating evidence of the G4C2-induced impairment of autophagy and lysosomal biogenesis, it is important to note that C9orf72-deficient cells also display reduced levels of autophagy initiation and impaired lysosome biogenesis, leading to enhanced DPR accumulation and increased neurotoxicity, indicating that the product of C9orf72 might play a role in physiologic activation of autophagy [59,60,61,62,63,64,65]. This is not surprising as C9orf72 is likely to act as an GDP/GTP exchange factor (GEF) for a number of RabGTPases regulating the early steps of autophagy and endocytic trafficking [66].
While reduced autophagy may thus represent an important contributor to C9-ALS/FTD pathogenesis, the consequences of reduced autophagy might extend beyond the impaired degradation of the DPRs themselves. Intriguingly, Marchi et al. recently proposed that reduced lysosome acidification may represent a mechanism by which endocytosed poly-GA aggregates can circumvent lysosomal degradation, thus enabling the retention and spread of poly-GA aggregates via the endocytic–exosomal pathway [67]. Although the mechanisms of cell-to-cell transmission may vary, all DPRs have indeed been observed to spread between cells in vitro [34,68,69,70]. Thus, it will be interesting to study (in the future) whether the endocytic–exosomal pathway represents a predominant mechanism of cell-to-cell transmission in C9-ALS/FTD (Figure 1D).
3. DPRs as Modulators of Stress Responses
In addition to activating the UPS, autophagy, endolysosomal, and secretory pathways, one additional way that neurons are able to respond to proteotoxic stress elicited by DPRs is by triggering intracellular stress response mechanisms that eventually repress translation and rebalance protein homeostasis. As it is the case of protein clearance pathways, an increasing body of evidence indicates that C9-associated DPRs both activate as well as disrupt stress pathways in multiple ways (Figure 2). The modulation of stress further reinforces disease pathology as described below.
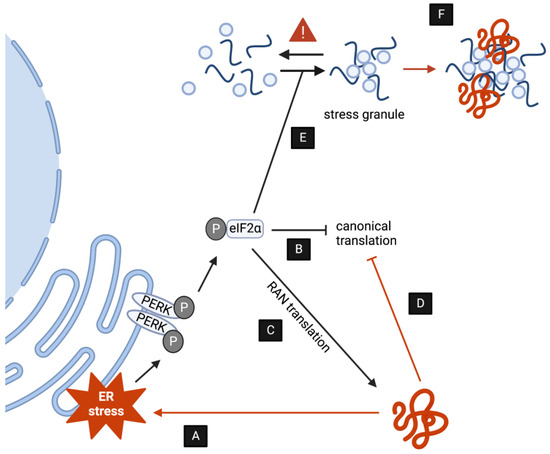
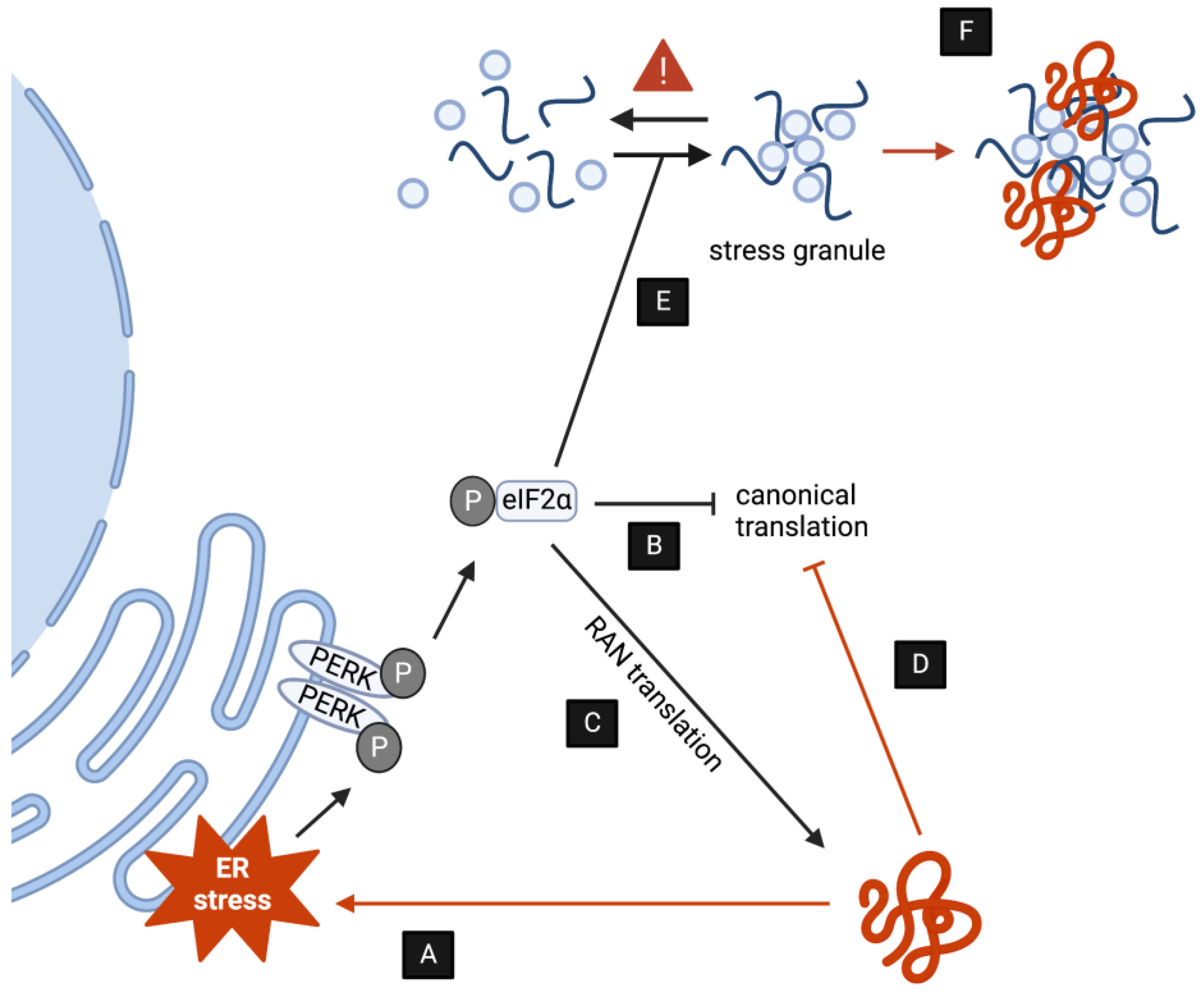
Figure 2.
DPRs induce toxicity by upsetting stress responses. (A,B) DPR accumulation induces ER stress via PERK and other kinases, ultimately decreasing translation as a compensatory mechanism. (C) The induction of ER stress by DPRs promotes RAN translation. (D) DPRs interact directly with the ribosome blocking polypeptide formation. (E) DPRs indirectly stimulate the formation of SGs. (F) (some) DPRs are subjected to LLPS transitions and might directly alter SG dynamics.
3.1. DPRs Induce Chronic ER stress
A major part of cell stress responses occurs at the endoplasmic reticulum (ER) and results in the activation of the unfolded protein response (UPR), which eventually suppresses global translation and induces catabolic pathways, such as autophagy and ER associated protein degradation (Figure 2A). While acute ER stress can thus help to maintain protein homeostasis, sustained ER stress is well established to be cytotoxic and is in fact a hallmark of different forms of ALS [71]. Indeed, UPR markers are upregulated in distinct brain regions of patients with C9 ALS/FTD [72,73]. In the cerebellum in particular, the upregulation of phosphorylation of PERK, eIF2α, a regulatory subunit of the eukaryotic initiation factor 2 (eIF2), and IRE1α, all of which are major regulators of ER stress, is associated with the presence of poly-GA aggregates (Figure 2B) [73]. In line with this, the overexpression of poly-GA has been demonstrated to induce ER stress in cell culture [13,74]. Recent studies have further demonstrated that poly-PR can also promote ER stress in cell culture [75,76]. Importantly, pharmacological inhibition of ER stress suppresses DPR-induced neurotoxicity, suggesting that ER stress might represent a major aspect of pathogenesis [13,77,78,79].
Interestingly, while phosphorylation of eIF2α is required to inhibit canonical translation upon the activation of ER stress, a number of studies have demonstrated that p-eIF2α enhances the RAN translation of G4C2 transcripts [80,81,82,83,84] as well as other repeat RNAs [85]. Indeed, the RAN translation of myotonic dystrophy type 2-associated repeat transcripts was shown to be reduced in PERK knockout cells [85]. While the exact mechanisms underlying RAN translation remain unclear, these studies point toward a pathogenic feed-forward loop, whereby DPRs enhance their own production via the ER stress response (Figure 2C). Interestingly, C9orf72 has been demonstrated to enhance the interaction between eIF2α and eIF2B5, an eIF2-specific GEF. As a result, C9orf72 knockout per se leads to global translation inhibition in vivo [86]. Future work will be required to assess whether disease-associated C9orf72 haploinsufficiency could also contribute to an enhanced RAN translation of G4C2 transcripts. A number of studies have demonstrated that DPR toxicity can also suppress global translation by interacting directly with ribosomal components (Figure 2D) [87,88,89]. In particular, poly-GR/PR has been shown to bind to and block the polypeptide exit site of the ribosome, leading to a block in translation [89].
3.2. DPRs Disrupt Stress Granule Homeostasis
Another cellular response to proteotoxic stress is the formation of cytoplasmic stress granules (SGs). This subgroup of ribonucleoprotein granules are membrane-less organelles that form in the cytoplasm concomitant to global translational suppression to sequester away non-translating mRNA, translation initiation complexes, and RNA binding proteins (RBPs) [90,91]. Consistent with this, ER stress is an established inducer of SG formation [92]. In line with the role of DPRs in inducing ER stress, it is therefore not surprising that the overexpression of DPRs per se appears to increase spontaneous formation of SGs [14,78,80,83,93,94,95,96,97]. Indeed, DPR-induced SG formation has been shown to be dependent on the phosphorylation of eIF2α [95], as well as by the activation of the c-Jun N-terminal kinase (JNK) [79]. Taken together, DPRs thus appear to indirectly increase SG formation through ER stress signaling cascades (Figure 2E).
Beyond the indirect involvement in SG formation, emerging evidence suggests that DPRs can directly interact with SGs to change their properties and dynamics [78]. Physiological SG formation relies on liquid–liquid phase separation (LLPS), a type of phase transition whereby macromolecules can reversibly and spontaneously separate from the soluble phase (e.g., the cytoplasm) into a concentrated, condensed state [98]. This is driven in part by proteins with low-complexity domains (LCDs), composed of a high number of uncharged polar amino acids, which are found in many RBPs. Intriguingly, the involvement of LCDs in driving LLPS also points toward an involvement of the arginine- and glycine-rich DPRs, poly-PR, and poly-GR in SG formation. Indeed, poly-PR has been shown to undergo LLPS in vitro [95], and the interactomes of both DPRs are enriched in common SG components, including the RBPs hnRNPA1, TIA1, and FUS [78,87,93,94,99]. Importantly, the arginine-containing DPRs appear to reduce the liquid-like properties of hnRNPA1, TIA1, and FUS liquid droplets in vitro [93,95]. This may, at least in part, be due to increased beta sheet content of the liquid droplets, making them less dynamic [95]. Taken together, poly-GR/PR may thus contribute to disease pathogenesis by promoting the transition of SGs into a more solid-like state (Figure 2F). Indeed, a number of ALS associated mutations in the LCDs of RBPs have been linked to perturbed SG dynamics [100], suggesting that this may be a common mechanism underlying ALS disease pathogenesis.
Finally, emerging evidence suggests that DPRs may hijack SG formation to promote their own aggregation. In particular, knocking out G3BP1/2, a core driver of SG formation [101,102,103], leads to a virtual abolishment of cytoplasmic poly-GR inclusions [104]. Consistent with this, G3BP1/2 knockdown suppresses G4C2 toxicity in vivo [93]. The converse has also been shown by the overexpression of SG genes [99]. Further work will need to be carried out to decipher the relationship between the LLPS-mediated formation of SGs and protein aggregates, although several studies have shown that a liquid phase can precede the formation of solid protein aggregates of several ALS- and FTD-associated proteins [105,106,107,108,109].
4. Open Questions
Just over 10 years since the discovery of the G4C2 expansion in a non-coding region of the C9orf72 gene, a myriad of disease mechanisms have been proposed to explain the effects of C9orf72’s loss of function as well as of the toxicity associated with repeat RNA and DPR production. However, a clear understanding of the relative importance of each disease mechanism, necessary to identify vulnerabilities that could be amenable to pharmacologic and medical interventions, is dramatically lacking. ALS/FTD, associated with C9orf72 G4C2 expansion, remains an incurable disease and progress is marred in no small part by the pleiotropism of DPRs effects.
In this review, we have presented accumulating evidence revealing that multifaceted disruptions of protein homeostasis could be a major trait associated with DPR toxicity. Specifically, DPR toxicity appears to cause the inhibition of protein clearance pathways as well as the induction of chronic ER stress and aberrant dynamics of SG. While correlative studies were obtained from a wealth of in vitro, cellular, and in vivo model systems capable of investigating multiple aspects of ALS/FTD, the relevance of DPR-induced disruptions in protein homeostasis to pathogenesis remains poorly understood.
In the following, closing paragraphs, we discuss what we believe to be the key aspects that may help to untangle the intricacies of these pleiotropic DPR effects, moving the field toward the establishment of cause–effect relationships with specific relevance to disease pathology.
4.1. Which DPR Is the Most Toxic in Patients?
Several discrepancies regarding the contribution of DPR toxicity to disease progression become apparent when comparing model systems and tissues from patients suffering from ALS/FTD with the C9orf72 G4C2 expansion (Table 1). In particular, the overexpression of the arginine-containing DPRs, but not poly-GA or poly-PA, appears to induce degeneration in the Drosophila eye [20,110]. In contrast, the overexpression of either poly-GA [111] or poly-GR [54] in mice causes neurodegenerative phenotypes, while poly-PR overexpression leads to notably milder and low penetrance phenotypes, with 60% of mice displaying no neurogenerative phenotypes by the age of 14 months [112]. Importantly, despite extensive post-mortem analysis of patient tissue, most studies have failed to demonstrate a clear spatial correlation between the presence of DPR inclusions and neurodegeneration [30,31,40,113,114]. In fact, DPR-positive inclusions appear to be quite rare, at least in comparison to the number of TDP43-positive inclusions found within the brains of patients with C9 ALS/FTD [41]. Intriguingly, one recent study has demonstrated a moderate correlation between the density of poly-GR aggregates and the extent of neurodegeneration in the frontal cortex [115], where the cytoplasmic poly-GR aggregates appear to be relatively abundant [116]. While this appears to be in line with the observation that poly-GR is toxic in Drosophila and mice, it remains inherently difficult to interpret the functional relevance from correlative observations across model systems.

Table 1.
Evidence of DPR toxicity from the tissues of patients with C9 ALS/FTD and in vivo model systems. Relative abundance of each DPR in patient tissue is indicated by “+” (++++ > +). DPR = dipeptide repeat, ND = neurodegeneration, UPS = ubiquitin–proteasome system, SGs = stress granules.
4.2. How Do DPRs Induce Toxicity in Physiologically Relevant Conditions?
The level of DPR expression used in cell culture and within in vivo model systems, which largely rely on G4C2 overexpression or the treatment with synthetic DPRs, are likely much higher than in patients. Indeed, DPRs are expressed at relatively low levels in patient-derived iPSCs. This highlights the need to validate the reported effects of DPR toxicity in patient tissues and patient-derived cells, in which the toxicity of endogenous levels of DPRs can be specifically investigated. Along these lines, it may also be important to consider a recent study showing that common protein tags can affect the toxicity of DPRs in vivo [117], again emphasizing the importance of studying endogenous DPRs.
In recent years, a lot of effort has been made to investigate the specific toxicity of each individual DPR. While this has revealed both some overlapping and some distinct pathways that appear to be targeted by each DPR (Table 1), these studies leave an important question unanswered: how might the interaction between DPRs contribute to toxicity? Initial evidence suggests, for example, that the co-expression of poly-PA and GA in the chick embryonic spinal cord can reduce poly-GA aggregation and toxicity [118]. Conversely, in another co-expression study, poly-GA was found to promote the aggregation and thereby partially reduce the toxicity of poly-GR [119]. Again, whether such functional interactions also occur between endogenous DPRs under physiological conditions will need to be explored. Along these lines, it may be important to consider that DPRs generated from the sense strand (poly-GA, GP, and GR) appear to be relatively more abundant in patients with C9 ALS/FTD than those formed only by the antisense strand (poly-PA and poly-PR) (Table 1) [113]. Again, how this ratio may affect the toxicity of DPRs will need to be investigated thoroughly.
4.3. Why Is Each DPR Toxic?
The conformation of each DPR might widely affect toxicity as implied by the fact that the aggregation status can alter DPR toxicity. Considering their differing biochemical properties, it is likely that the toxic form will be different for each DPR. The hydrophobic and uncharged poly-GA, for example, is highly aggregation-prone and indeed appears to induce toxicity by sequestering components, such as the UPS, into their aggregates. Strikingly, disruption of the aggregation propensity via the insertion of proline residues completely abolished GA toxicity in primary neuronal cell culture [35], suggesting that poly-GA is most toxic in its aggregated form. Whether the positively charged poly-GR, for example, is more toxic in its soluble form could provide important improvements to understanding the mechanisms underlying DPR toxicity. These considerations have particular importance when evaluating how DPRs are cleared by protein degradation and how their disruptions of protein homeostasis may contribute to disease progression. The possible DPR-induced reduction in autophagic clearance, for example, may not affect the toxicity of soluble DPRs but may be a critical enhancer of the aggregation-prone poly-GA.
5. Conclusions
While the correlation between DPR toxicity and disruptions in protein homeostasis are evident, cause–effect relationships are currently hard to derive from the large body of data obtained in different systems displaying DPR accumulation. For example, while ER stress appears to be a hallmark of DPR toxicity, it is unclear how DPR toxicity may induce ER stress and whether this also contributes to DPR-induced disease progression.
Some insight may be gained from studying the interdependencies of protein homeostasis pathways (Figure 3). Specifically, while these pathways normally act in close concert to respond to proteotoxic stress, it is important to consider that crosstalk may instead perpetuate C9-ALS/FTD disease progression. Proteasome inhibition, for example, is a well-established inducer of ER stress [120,121], which leads to the suppression of global translation to reduce the burden of protein accumulation. In the context of DPR toxicity, however, another outcome emerges, as ER stress promotes the RAN translation of G4C2 transcripts and therefore paradoxically enhances the burden of toxic DPR accumulation [80,81,82,83,84]. It is therefore conceivable that the inhibition of the UPS may not only reduce the clearance of DPRs but may also indirectly increase their translation (Figure 3A).
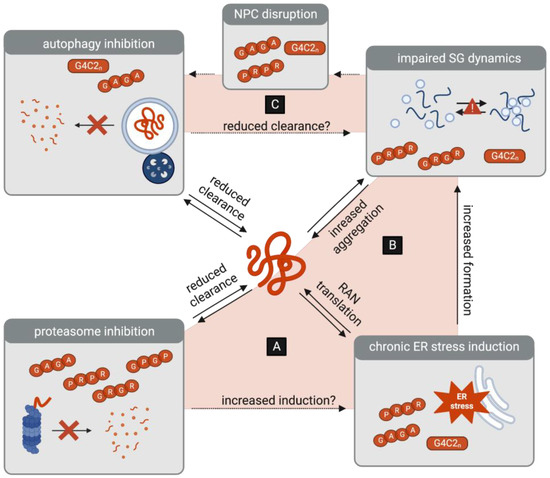
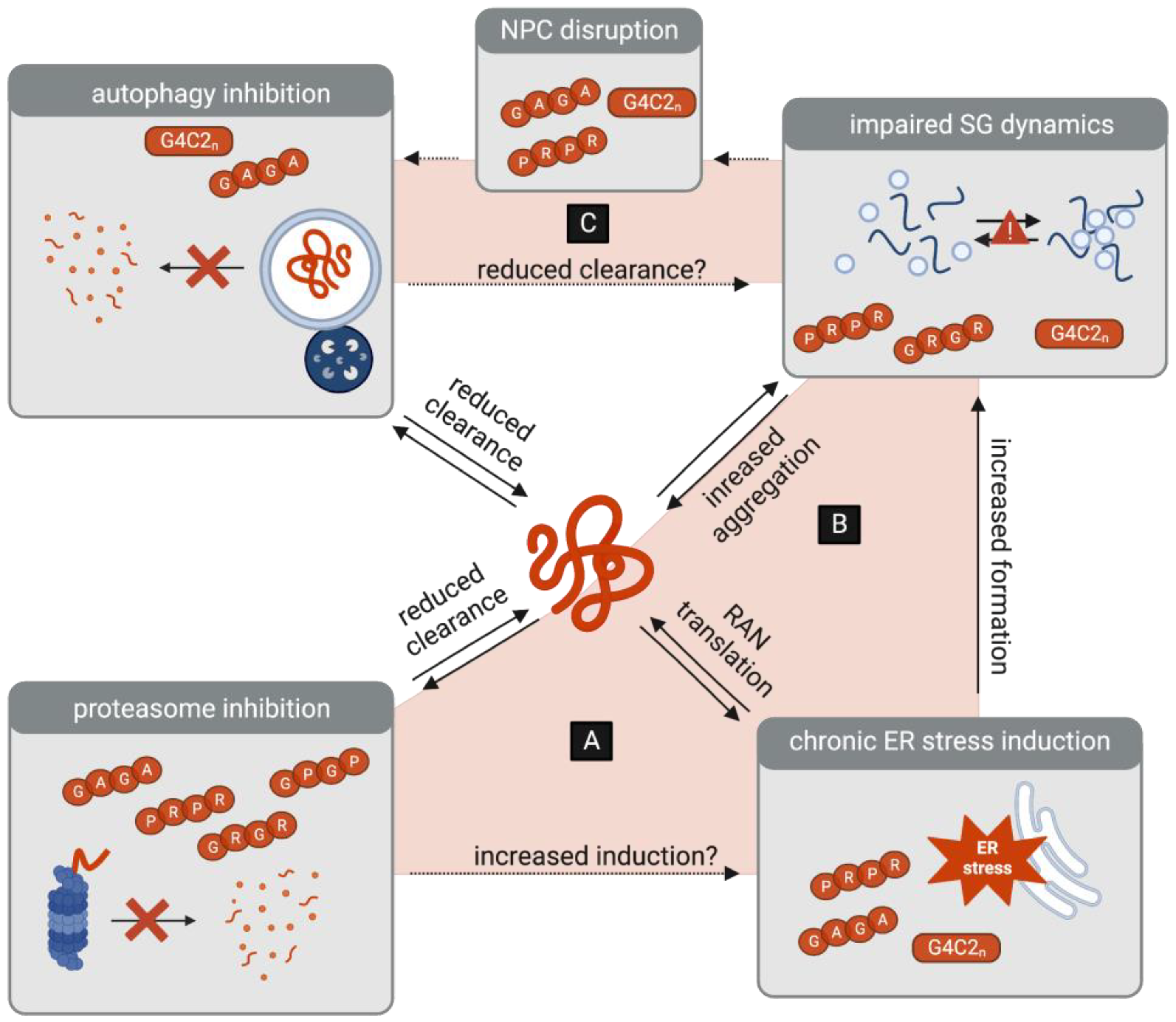
Figure 3.
Interplay between protein homeostasis pathways as a potential amplifier of DPR toxicity. (A) DPR-associated inhibition of the proteasome may be a trigger of ER stress, which in turn leads to the increased RAN translation of G4C2 transcripts. (B) DPR-induced ER stress leads to the formation of stress granules (SGs), which in turn may lead to the increased aggregation of DPRs. (C) DPR-associated inhibition of autophagy may lead to the reduced clearance of SGs. Conversely, DPR-associated SGs contribute to the disruptions in the nuclear pore complex (NPC), which may further inhibit the initiation of autophagy.
ER stress induction and subsequent translational suppression also leads to the formation of SGs, which sequester non-translating mRNA, translation initiation complexes, and RBPs. Indeed, proteasome inhibition has been linked to increased SG formation [108,122,123]. DPRs, however, appear to trigger the persistence of SGs by reducing their liquid-like properties and may even use SGs to mediate their own aggregation. Again, the burden of toxic DPR accumulation is thereby perpetuated. With increased DPR translation and aggregation, it is conceivable that proteasome activity may be further inhibited, ER stress may be chronically induced, and aberrant SG formation may be further enhanced. Taken together, this reveals a self-sustaining feedback loop that perpetuates DPR toxicity (Figure 3B).
Along these lines, it is also interesting to consider the crosstalk between autophagy, stress granules, and the NPC. Importantly, autophagy is emerging as a mediator of SG clearance [108,124,125]. Could the DPR-induced impairments in autophagy therefore also contribute to the persistence of aberrant SGs? Moreover, the persistence of SGs may conversely reduce the TFEB-mediated transcriptional activation of the autophagy pathway, via NPC disruptions. Specifically, DPR-induced SGs have been shown to sequester components of the NPC, including the nucleoporin POM121 [54,126], which is a known modulator of TFEB-mediated autophagy [49]. As a result, DPRs and SGs further accumulate in a vicious cycle (Figure 3C).
Ultimately, while the crosstalk between catabolic and anabolic pathways is essential for maintaining protein homeostasis, their interplay appears to create self-sustaining pathogenic loops, potentially amplifying the pathogenic effects of the DPRs. Deciphering the contributions of feedback and feedforward interdependencies will be crucial to identifying key molecular targets to develop future therapies.
Author Contributions
Conceptualization, P.H.S. and T.V. Writing—original draft preparation P.H.S., G.C. and T. V. Writing—review and editing P.H.S. and T.V. All authors have read and agreed to the published version of the manuscript.
Funding
Work in the Vaccari lab is supported by Telethon Italia (investigator grant GMR22T1078) and by the Italian Ministry for Universities and Research (MUR PRIN2020CL55XW and PRIN2022JKEBB8). PHS is recipient of a PhD fellowship from the European Union’s Horizon 2020 Research and Innovation ITN-ETN Programme SAND (Secretion and Autophagy and their roles in Neurodegeneration) under the Marie Skłodowska-Curie grant agreement 860035.
Acknowledgments
Figures were prepared with the help of BioRender.
Conflicts of Interest
The authors declare no conflict of interest.
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in als and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.C.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Byrne, S.; Heverin, M.; Elamin, M.; Walsh, C.; Hardiman, O. Intermediate repeat expansion length in C9orf72 may be pathological in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 148–150. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, Z.; Chen, X.; Cao, B.; Wei, Q.; Ou, R.; Zhao, B.; Song, W.; Wu, Y.; Shang, H.F. Large C9orf72 repeat expansions are seen in Chinese patients with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 38, e15–e217.e22. [Google Scholar] [CrossRef]
- Iacoangeli, A.; Al Khleifat, A.; Jones, A.R.; Sproviero, W.; Shatunov, A.; Opie-Martin, S.; Morrison, K.E.; Shaw, P.J.; Shaw, C.E.; Fogh, I.; et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol. Commun. 2019, 7, 115. [Google Scholar] [CrossRef]
- Smeyers, J.; Banchi, E.G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef]
- Fratta, P.; Poulter, M.; Lashley, T.; Rohrer, J.D.; Polke, J.M.; Beck, J.; Ryan, N.; Hensman, D.; Mizielinska, S.; Waite, A.J.; et al. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol. 2013, 126, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.J.; Terpstra, M.L.; Zundel, C.A.C.; Vieira De Sá, R.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Jansen-West, K.; Xu, Y.F.; Gendron, T.F.; Bieniek, K.F.; Lin, W.L.; Sasaguri, H.; Caulfield, T.; Hubbard, J.; Daughrity, L.; et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014, 128, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, M.; Ito, D.; Honda, T.; Kubo, K.I.; Noda, M.; Nakajima, K.; Suzuki, N. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum. Mol. Genet. 2015, 24, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- May, S.; Hornburg, D.; Schludi, M.H.; Arzberger, T.; Rentzsch, K.; Schwenk, B.M.; Grässer, F.A.; Mori, K.; Kremmer, E.; Banzhaf-Strathmann, J.; et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014, 128, 485–503. [Google Scholar] [CrossRef]
- Chew, J.; Gendron, T.F.; Prudencio, M.; Sasaguri, H.; Zhang, Y.J.; Castanedes-Casey, M.; Lee, C.W.; Jansen-West, K.; Kurti, A.; Murray, M.E.; et al. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 2015, 348, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Todd, T.W.; McEachin, Z.T.; Chew, J.; Burch, A.R.; Jansen-West, K.; Tong, J.; Yue, M.; Song, Y.; Castanedes-Casey, M.; Kurti, A.; et al. Hexanucleotide Repeat Expansions in c9FTD/ALS and SCA36 Confer Selective Patterns of Neurodegeneration In Vivo. Cell Rep. 2020, 31, 107616. [Google Scholar] [CrossRef]
- Swinnen, B.; Bento-Abreu, A.; Gendron, T.F.; Boeynaems, S.; Bogaert, E.; Nuyts, R.; Timmers, M.; Scheveneels, W.; Hersmus, N.; Wang, J.; et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018, 135, 427–443. [Google Scholar] [CrossRef]
- Shaw, M.P.; Higginbottom, A.; McGown, A.; Castelli, L.M.; James, E.; Hautbergue, G.M.; Shaw, P.J.; Ramesh, T.M. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 2018, 6, 125. [Google Scholar] [CrossRef]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Mizielinska, S.; Lashley, T.; Norona, F.E.; Clayton, E.L.; Ridler, C.E.; Fratta, P.; Isaacs, A.M. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013, 126, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.B.; Chen, H.J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Štalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA Foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Higginbottom, A.; Stopford, M.J.; Highley, J.R.; Ince, P.G.; Wharton, S.B.; Pickering-Brown, S.; Kirby, J.; Hautbergue, G.M.; Shaw, P.J. Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol. 2015, 130, 63–75. [Google Scholar] [CrossRef]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Arzberger, T.; Grässer, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.M.; Schludi, M.H.; Van Der Zee, J.; Cruts, M.; et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Ash, P.E.A.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.L.; DeJesus-Hernandez, M.; Van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef]
- Mann, D.M.A.; Rollinson, S.; Robinson, A.; Bennion Callister, J.; Thompson, J.C.; Snowden, J.S.; Gendron, T.; Petrucelli, L.; Masuda-Suzukake, M.; Hasegawa, M.; et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 2014, 2, 68. [Google Scholar] [CrossRef]
- Schludi, M.H.; May, S.; Grässer, F.A.; Rentzsch, K.; Kremmer, E.; Küpper, C.; Klopstock, T.; Ceballos-Baumann, A.; Danek, A.; Diehl-Schmid, J.; et al. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 2015, 130, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Cristofani, R.; Crippa, V.; Vezzoli, G.; Rusmini, P.; Galbiati, M.; Cicardi, M.E.; Meroni, M.; Ferrari, V.; Tedesco, B.; Piccolella, M.; et al. The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72-related neurodegenerative diseases. Cell Stress Chaperones 2018, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Lehmer, C.; Martínez-Sánchez, A.; Rudack, T.; Beck, F.; Hartmann, H.; Pérez-Berlanga, M.; Frottin, F.; Hipp, M.S.; Hartl, F.U.; et al. In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment. Cell 2018, 172, 696.e12–705.e12. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, B.; LaClair, K.D.; Riemenschneider, H.; Zhou, Q.; Frottin, F.; Mareljic, N.; Czuppa, M.; Farny, D.; Hartmann, H.; Michaelsen, M.; et al. Cell-to-cell transmission of C9orf72 poly-(Gly-Ala) triggers key features of ALS/FTD. EMBO J. 2020, 39, e102811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Gendron, T.F.; Grima, J.C.; Sasaguri, H.; Jansen-West, K.; Xu, Y.F.; Katzman, R.B.; Gass, J.; Murray, M.E.; Shinohara, M.; et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 2016, 19, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Lan, M.; Mojsilovic-Petrovic, J.; Choi, W.H.; Safren, N.; Barmada, S.; Lee, M.J.; Kalb, R. The proline/arginine dipeptide from hexanucleotide repeat expanded C9ORF72 inhibits the proteasome. eNeuro 2017, 4, ENEURO.0249-16.2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nelson, S.C.K.; Viera Ortiz, A.P.; Lee, E.B.; Fairman, R. C9orf72 proline-arginine dipeptide repeats disrupt the proteasome and perturb proteolytic activities. J. Neuropathol. Exp. Neurol. 2023, 82, 901–910. [Google Scholar] [CrossRef]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.B.; Chen, H.J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef]
- Van Eersel, J.; Ke, Y.D.; Gladbach, A.; Bi, M.; Götz, J.; Kril, J.J.; Ittner, L.M. Cytoplasmic accumulation and aggregation of TDP-43 upon proteasome inhibition in cultured neurons. PLoS ONE 2011, 6, e22850. [Google Scholar] [CrossRef]
- MacKenzie, I.R.; Arzberger, T.; Kremmer, E.; Troost, D.; Lorenzl, S.; Mori, K.; Weng, S.M.; Haass, C.; Kretzschmar, H.A.; Edbauer, D.; et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: Clinico-pathological correlations. Acta Neuropathol. 2013, 126, 859–879. [Google Scholar] [CrossRef]
- Gomez-Deza, J.; Lee, Y.B.; Troakes, C.; Nolan, M.; Al-Sarraj, S.; Gallo, J.M.; Shaw, C.E. Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta Neuropathol. Commun. 2015, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Baborie, A.; Griffiths, T.D.; Jaros, E.; Perry, R.; McKeith, I.G.; Burn, D.J.; Masuda-Suzukake, M.; Hasegawa, M.; Rollinson, S.; Pickering-Brown, S.; et al. Accumulation of dipeptide repeat proteins predates that of TDP-43 in frontotemporal lobar degeneration associated with hexanucleotide repeat expansions in C9ORF72 gene. Neuropathol. Appl. Neurobiol. 2015, 41, 601–612. [Google Scholar] [CrossRef]
- Vatsavayai, S.C.; Yoon, S.J.; Gardner, R.C.; Gendron, T.F.; Vargas, J.N.S.; Trujillo, A.; Pribadi, M.; Phillips, J.J.; Gaus, S.E.; Hixson, J.D.; et al. Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain 2016, 139, 3202–3216. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 21, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 2019, 35, 477–500. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Cunningham, K.M.; Maulding, K.; Ruan, K.; Senturk, M.; Grima, J.C.; Sung, H.; Zuo, Z.; Song, H.; Gao, J.; Dubey, S.; et al. Tfeb/mitf links impaired nuclear import to autophagolysosomal dysfunction in c9-als. eLife 2020, 9, e59419. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Lloyd, T.E. Disrupted endoplasmic reticulum-mediated autophagosomal biogenesis in a Drosophila model of C9-ALS-FTD. Autophagy 2023, 20, 94–113. [Google Scholar] [CrossRef]
- Wang, S.M.; Wu, H.E.; Yasui, Y.; Geva, M.; Hayden, M.; Maurice, T.; Cozzolino, M.; Su, T.P. Nucleoporin POM121 signals TFEB-mediated autophagy via activation of SIGMAR1/sigma-1 receptor chaperone by pridopidine. Autophagy 2023, 19, 126–151. [Google Scholar] [CrossRef]
- Zhang, K.; Grima, J.C.; Rothstein, J.D.; Lloyd, T.E. Nucleocytoplasmic transport in C9orf72-mediated ALS/FTD. Nucleus 2016, 7, 132–137. [Google Scholar] [CrossRef]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.N.; Zaepfel, B.L.; Hayes, L.; Fitchman, B.; Salzberg, Y.; Luo, E.C.; Bowen, K.; Trost, H.; Aigner, S.; Rigo, F.; et al. G4C2 Repeat RNA Initiates a POM121-Mediated Reduction in Specific Nucleoporins in C9orf72 ALS/FTD. Neuron 2020, 107, 1124–1140.e11. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.Y.; Mori, E.; Nizami, Z.F.; Lin, Y.; Kato, M.; Xiang, S.; Wu, L.C.; Ding, M.; Yu, Y.; Gall, J.G.; et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. USA 2017, 114, E1111–E1117. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; del Rosso, G.; Yue, M.; Jiang, P.; Gomes, E.; Tong, J.; et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci. Transl. Med. 2020, 12, eabb3774. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Bouché, V.; Espinosa, A.P.; Leone, L.; Sardiello, M.; Ballabio, A.; Botas, J. Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy 2016, 12, 484–498. [Google Scholar] [CrossRef]
- Tognon, E.; Kobia, F.; Busi, I.; Fumagalli, A.; De Masi, F.; Vaccari, T. Control of lysosomal biogenesis and Notch-dependent tissue patterning by components of the TFEB-V-ATPase axis in Drosophila melanogaster. Autophagy 2016, 12, 499–514. [Google Scholar] [CrossRef]
- Bozič, J.; Motaln, H.; Janez, A.P.; Markič, L.; Tripathi, P.; Yamoah, A.; Aronica, E.; Lee, Y.B.; Heilig, R.; Fischer, R.; et al. Interactome screening of C9orf72 dipeptide repeats reveals VCP sequestration and functional impairment by polyGA. Brain 2022, 145, 684–699. [Google Scholar] [CrossRef]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK 1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef]
- Yang, M.; Chen, L.; Swaminathan, K.; Herrlinger, S.; Lai, F.; Shiekhattar, R.; Chen, J.F. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv. 2016, 2, e1601167. [Google Scholar] [CrossRef]
- Sullivan, P.M.; Zhou, X.; Robins, A.M.; Paushter, D.H.; Kim, D.; Smolka, M.B.; Hu, F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 2016, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Corbier, C.; Sellier, C. C9ORF72 is a GDP/GTP exchange factor for Rab8 and Rab39 and regulates autophagy. Small GTPases 2017, 8, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.P.; Smith, E.F.; Grierson, A.J.; De Vos, K.J. C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases 2018, 9, 399–408. [Google Scholar] [CrossRef]
- Wang, M.; Wang, H.; Tao, Z.; Xia, Q.; Hao, Z.; Prehn, J.H.M.; Zhen, X.; Wang, G.; Ying, Z. C9orf72 associates with inactive Rag GTPases and regulates mTORC1-mediated autophagosomal and lysosomal biogenesis. Aging Cell 2020, 19, e13126. [Google Scholar] [CrossRef] [PubMed]
- Boivin, M.; Pfister, V.; Gaucherot, A.; Ruffenach, F.; Negroni, L.; Sellier, C.; Charlet-Berguerand, N. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 2020, 39, e100574. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Subramanian, V.; Acharya, K.R. C9orf72, a protein associated with amyotrophic lateral sclerosis (ALS) is a guanine nucleotide exchange factor. PeerJ 2018, 2018, e5815. [Google Scholar] [CrossRef] [PubMed]
- Marchi, P.M.; Marrone, L.; Brasseur, L.; Coens, A.; Webster, C.P.; Bousset, L.; Destro, M.; Smith, E.F.; Walther, C.G.; Alfred, V.; et al. C9ORF72-derived poly-GA DPRs undergo endocytic uptake in iAstrocytes and spread to motor neurons. Life Sci. Alliance 2022, 5, e202101276. [Google Scholar] [CrossRef]
- Westergard, T.; Jensen, B.K.; Wen, X.; Cai, J.; Kropf, E.; Iacovitti, L.; Pasinelli, P.; Trotti, D. Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72-ALS/FTD. Cell Rep. 2016, 17, 645–652. [Google Scholar] [CrossRef]
- Chang, Y.J.; Jeng, U.S.; Chiang, Y.L.; Hwang, I.S.; Chen, Y.R. The glycine-alanine dipeptide repeat from C9 or f72 hexanucleotide expansions forms toxic amyloids possessing cell-to-cell transmission properties. J. Biol. Chem. 2016, 291, 4903–4911. [Google Scholar] [CrossRef]
- Zhou, Q.; Lehmer, C.; Michaelsen, M.; Mori, K.; Alterauge, D.; Baumjohann, D.; Schludi, M.H.; Greiling, J.; Farny, D.; Flatley, A.; et al. Antibodies inhibit transmission and aggregation of C9orf72 poly-GA dipeptide repeat proteins. EMBO Mol. Med. 2017, 9, 687–702. [Google Scholar] [CrossRef]
- Matus, S.; Valenzuela, V.; Medinas, D.B.; Hetz, C. ER dysfunction and protein folding stress in ALS. Int. J. Cell Biol. 2013, 2013, 674751. [Google Scholar] [CrossRef]
- Prudencio, M.; Belzil, V.V.; Batra, R.; Ross, C.A.; Gendron, T.F.; Pregent, L.J.; Murray, M.E.; Overstreet, K.K.; Piazza-Johnston, A.E.; Desaro, P.; et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 2015, 18, 1175–1182. [Google Scholar] [CrossRef]
- Gami-Patel, P.; van Dijken, I.; Meeter, L.H.; Melhem, S.; Morrema, T.H.J.; Scheper, W.; van Swieten, J.C.; Rozemuller, A.J.M.; Dijkstra, A.A.; Hoozemans, J.J.M. Unfolded protein response activation in C9orf72 frontotemporal dementia is associated with dipeptide pathology and granulovacuolar degeneration in granule cells. Brain Pathol. 2021, 31, 163–173. [Google Scholar] [CrossRef]
- Pilotto, F.; Schmitz, A.; Maharjan, N.; Diab, R.; Odriozola, A.; Tripathi, P.; Yamoah, A.; Scheidegger, O.; Oestmann, A.; Dennys, C.N.; et al. PolyGA targets the ER stress-adaptive response by impairing GRP75 function at the MAM in C9ORF72-ALS/FTD. Acta Neuropathol. 2022, 144, 939–966. [Google Scholar] [CrossRef] [PubMed]
- Kramer, N.J.; Haney, M.S.; Morgens, D.W.; Jovičić, A.; Couthouis, J.; Li, A.; Ousey, J.; Ma, R.; Bieri, G.; Tsui, C.K.; et al. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet. 2018, 50, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, X.; Hao, Z.; Zhang, S.; Wu, D.; Sun, H.; Mu, C.; Ren, H.; Wang, G. Poly-PR in C9ORF72-Related Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Causes Neurotoxicity by Clathrin-Dependent Endocytosis. Neurosci. Bull. 2019, 35, 889–900. [Google Scholar] [CrossRef]
- Zu, T.; Guo, S.; Bardhi, O.; Ryskamp, D.A.; Li, J.; Tusi, S.K.; Engelbrecht, A.; Klippel, K.; Chakrabarty, P.; Nguyen, L.; et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 18591–18599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Gendron, T.F.; Ebbert, M.T.W.; O’Raw, A.D.; Yue, M.; Jansen-West, K.; Zhang, X.; Prudencio, M.; Chew, J.; Cook, C.N.; et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 2018, 24, 1136–1142. [Google Scholar] [CrossRef]
- Sahana, T.G.; Chase, K.J.; Liu, F.; Lloyd, T.E.; Rossoll, W.; Zhang, K. c-Jun N-Terminal Kinase Promotes Stress Granule Assembly and Neurodegeneration in C9orf72-Mediated ALS and FTD. J. Neurosci. 2023, 43, 3186–3197. [Google Scholar] [CrossRef] [PubMed]
- Green, K.M.; Glineburg, M.R.; Kearse, M.G.; Flores, B.N.; Linsalata, A.E.; Fedak, S.J.; Goldstrohm, A.C.; Barmada, S.J.; Todd, P.K. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat. Commun. 2017, 8, 2005. [Google Scholar] [CrossRef]
- Cheng, W.; Wang, S.; Mestre, A.A.; Fu, C.; Makarem, A.; Xian, F.; Hayes, L.R.; Lopez-Gonzalez, R.; Drenner, K.; Jiang, J.; et al. C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2α phosphorylation. Nat. Commun. 2018, 9, 51. [Google Scholar] [CrossRef]
- Sonobe, Y.; Aburas, J.; Krishnan, G.; Fleming, A.C.; Ghadge, G.; Islam, P.; Warren, E.C.; Gu, Y.; Kankel, M.W.; Brown, A.E.X.; et al. A C. elegans model of C9orf72-associated ALS/FTD uncovers a conserved role for eIF2D in RAN translation. Nat. Commun. 2021, 12, 6025. [Google Scholar] [CrossRef]
- Sonobe, Y.; Ghadge, G.; Masaki, K.; Sendoel, A.; Fuchs, E.; Roos, R.P. Translation of dipeptide repeat proteins from the C9ORF72 expanded repeat is associated with cellular stress. Neurobiol. Dis. 2018, 116, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Westergard, T.; McAvoy, K.; Russell, K.; Wen, X.; Pang, Y.; Morris, B.; Pasinelli, P.; Trotti, D.; Haeusler, A. Repeat-associated non- AUG translation in C9orf72- ALS/FTD is driven by neuronal excitation and stress. EMBO Mol. Med. 2019, 11, e9423. [Google Scholar] [CrossRef] [PubMed]
- Tusi, S.K.; Nguyen, L.; Thangaraju, K.; Li, J.; Cleary, J.D.; Zu, T.; Ranum, L.P.W. The alternative initiation factor eIF2A plays key role in RAN translation of myotonic dystrophy type 2 CCUG•CAGG repeats. Hum. Mol. Genet. 2021, 30, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Wang, K.; Wu, Y.; Yan, G.; Zhang, C.; Li, Z.; Wang, L.; Chen, S. C9orf72 regulates the unfolded protein response and stress granule formation by interacting with eIF2α. Theranostics 2022, 12, 7289–7306. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Yagi, T.; Cammack, A.J.; Mahadevan, J.; Kuroda, M.; Harms, M.B.; Miller, T.M.; Urano, F. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum. Mol. Genet. 2016, 25, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Moens, T.G.; Niccoli, T.; Wilson, K.M.; Atilano, M.L.; Birsa, N.; Gittings, L.M.; Holbling, B.V.; Dyson, M.C.; Thoeng, A.; Neeves, J.; et al. C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 2019, 137, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Loveland, A.B.; Svidritskiy, E.; Susorov, D.; Lee, S.; Park, A.; Zvornicanin, S.; Demo, G.; Gao, F.B.; Korostelev, A.A. Ribosome inhibition by C9ORF72-ALS/FTD-associated poly-PR and poly-GR proteins revealed by cryo-EM. Nat. Commun. 2022, 13, 2776. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic Stress Granules: The Ins and Outs of Translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Nicchitta, C.V. An emerging role for the endoplasmic reticulum in stress granule biogenesis. Semin. Cell Dev. Biol. 2022, 156, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e17. [Google Scholar] [CrossRef]
- Lin, Y.; Mori, E.; Kato, M.; Xiang, S.; Wu, L.; Kwon, I.; McKnight, S.L. Toxic PR Poly-Dipeptides Encoded by the C9orf72 Repeat Expansion Target LC Domain Polymers. Cell 2016, 167, 789.e12–802.e12. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Kovacs, D.; Konijnenberg, A.; Timmerman, E.; Volkov, A.; Guharoy, M.; De Decker, M.; Jaspers, T.; Ryan, V.H.; et al. Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Mol. Cell 2017, 65, 1044–1055.e5. [Google Scholar] [CrossRef] [PubMed]
- Chew, J.; Cook, C.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Daughrity, L.M.; Castanedes-Casey, M.; Kurti, A.; Stankowski, J.N.; Disney, M.D.; et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol. Neurodegener. 2019, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.L.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef]
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef]
- Hartmann, H.; Hornburg, D.; Czuppa, M.; Bader, J.; Michaelsen, M.; Farny, D.; Arzberger, T.; Mann, M.; Meissner, F.; Edbauer, D. Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci. Alliance 2018, 1, e201800070. [Google Scholar] [CrossRef]
- Baradaran-Heravi, Y.; Van Broeckhoven, C.; van der Zee, J. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol. Dis. 2020, 134, 104639. [Google Scholar] [CrossRef]
- Yang, P.; Mathieu, C.; Kolaitis, R.M.; Zhang, P.; Messing, J.; Yurtsever, U.; Yang, Z.; Wu, J.; Li, Y.; Pan, Q.; et al. G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell 2020, 181, 325–345.e28. [Google Scholar] [CrossRef] [PubMed]
- Guillén-Boixet, J.; Kopach, A.; Holehouse, A.S.; Wittmann, S.; Jahnel, M.; Schlüßler, R.; Kim, K.; Trussina, I.R.E.A.; Wang, J.; Mateju, D.; et al. RNA-Induced Conformational Switching and Clustering of G3BP Drive Stress Granule Assembly by Condensation. Cell 2020, 181, 346.e17–361.e17. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kedersha, N.; Lee, D.S.W.; Strom, A.R.; Drake, V.; Riback, J.A.; Bracha, D.; Eeftens, J.M.; Iwanicki, A.; Wang, A.; et al. Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell 2020, 181, 306.e28–324.e28. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wu, Y.; Shao, W.; Gendron, T.F.; van der Spek, S.J.F.; Sultanakhmetov, G.; Basu, A.; Castellanos Otero, P.; Jones, C.J.; Jansen-West, K.; et al. Poly(GR) interacts with key stress granule factors promoting its assembly into cytoplasmic inclusions. Cell Rep. 2023, 42, 112822. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808.e9–816.e9. [Google Scholar] [CrossRef]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef]
- Ash, P.E.A.; Lei, S.; Shattuck, J.; Boudeau, S.; Carlomagno, Y.; Medalla, M.; Mashimo, B.L.; Socorro, G.; Al-Mohanna, L.F.A.; Jiang, L.; et al. TIA1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc. Natl. Acad. Sci. USA 2021, 118, e2014188118. [Google Scholar] [CrossRef]
- Xinmei, W.; Wenzhi, T.; Thomas, W.; Karthik, K.; Shamamandri, M.S.; Yingxiao, S.; Shaoyu, L.; Lin, S.; Shneider, N.A.; John, M.; et al. Antisense Proline-Arginine RAN dipeptides linked to C9ORF72- ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar]
- Schludi, M.H.; Becker, L.; Garrett, L.; Gendron, T.F.; Zhou, Q.; Schreiber, F.; Popper, B.; Dimou, L.; Strom, T.M.; Winkelmann, J.; et al. Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol. 2017, 134, 241–254. [Google Scholar] [CrossRef] [PubMed]
- LaClair, K.D.; Zhou, Q.; Michaelsen, M.; Wefers, B.; Brill, M.S.; Janjic, A.; Rathkolb, B.; Farny, D.; Cygan, M.; de Angelis, M.H.; et al. Congenic expression of poly-GA but not poly-PR in mice triggers selective neuron loss and interferon responses found in C9orf72 ALS. Acta Neuropathol. 2020, 140, 121–142. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Frick, P.; Grässer, F.A.; Gendron, T.F.; Petrucelli, L.; Cashman, N.R.; Edbauer, D.; Kremmer, E.; Prudlo, J.; Troost, D.; et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015, 130, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Davidson, Y.S.; Barker, H.; Robinson, A.C.; Thompson, J.C.; Harris, J.; Troakes, C.; Smith, B.; Al-Saraj, S.; Shaw, C.; Rollinson, S.; et al. Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 2014, 2, 70. [Google Scholar] [CrossRef] [PubMed]
- Sakae, N.; Bieniek, K.F.; Zhang, Y.J.; Ross, K.; Gendron, T.F.; Murray, M.E.; Rademakers, R.; Petrucelli, L.; Dickson, D.W. Poly-GR dipeptide repeat polymers correlate with neurodegeneration and Clinicopathological subtypes in C9ORF72-related brain disease. Acta Neuropathol. Commun. 2018, 6, 63. [Google Scholar] [CrossRef]
- Saberi, S.; Stauffer, J.E.; Jiang, J.; Garcia, S.D.; Taylor, A.E.; Schulte, D.; Ohkubo, T.; Schloffman, C.L.; Maldonado, M.; Baughn, M.; et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 459–474. [Google Scholar] [CrossRef]
- Morón-Oset, J.; Fischer, L.K.S.; Carcolé, M.; Giblin, A.; Zhang, P.; Isaacs, A.M.; Grönke, S.; Partridge, L. Toxicity of C9orf72-associated dipeptide repeat peptides is modified by commonly used protein tags. Life Sci. Alliance 2023, 6, e202201739. [Google Scholar] [CrossRef]
- Lee, Y.B.; Baskaran, P.; Gomez-Deza, J.; Chen, H.J.; Nishimura, A.L.; Smith, B.N.; Troakes, C.; Adachi, Y.; Stepto, A.; Petrucelli, L.; et al. C9orf72 poly GA RAN-translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum. Mol. Genet. 2017, 26, 4765–4777. [Google Scholar] [CrossRef]
- Yang, D.; Abdallah, A.; Li, Z.; Lu, Y.; Almeida, S.; Gao, F.B. FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol. 2015, 130, 525–535. [Google Scholar] [CrossRef]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Wek, R.C. Phosphorylation of the α-subunit of the eukaryotic initiation factor-2 (eIF2α) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J. Biol. Chem. 2005, 280, 14189–14202. [Google Scholar] [CrossRef] [PubMed]
- Seguin, S.J.; Morelli, F.F.; Vinet, J.; Amore, D.; De Biasi, S.; Poletti, A.; Rubinsztein, D.C.; Carra, S. Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell Death Differ. 2014, 21, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Mazroui, R.; Di Marco, S.; Kaufman, R.J.; Gallouzi, I.E. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 2007, 18, 2603–2618. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. XEukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, Z.; Kang, Y.; Yi, Q.; Wang, T.; Bai, Y.; Liu, Y. Stress granule homeostasis is modulated by TRIM21-mediated ubiquitination of G3BP1 and autophagy-dependent elimination of stress granules. Autophagy 2023, 19, 1934–1951. [Google Scholar] [CrossRef]
- Zhang, K.; Daigle, J.G.; Cunningham, K.M.; Coyne, A.N.; Ruan, K.; Grima, J.C.; Bowen, K.E.; Wadhwa, H.; Yang, P.; Rigo, F.; et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell 2018, 173, 958.e17–971.e17. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).