

Chromosome Transplantation: Opportunities and Limitations
 , , , ,
, , , ,
Abstract
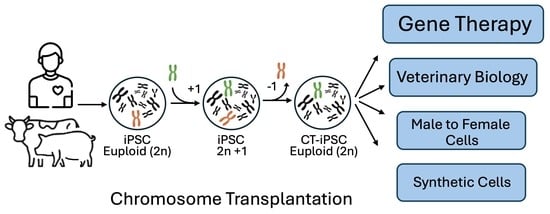
1. Introduction
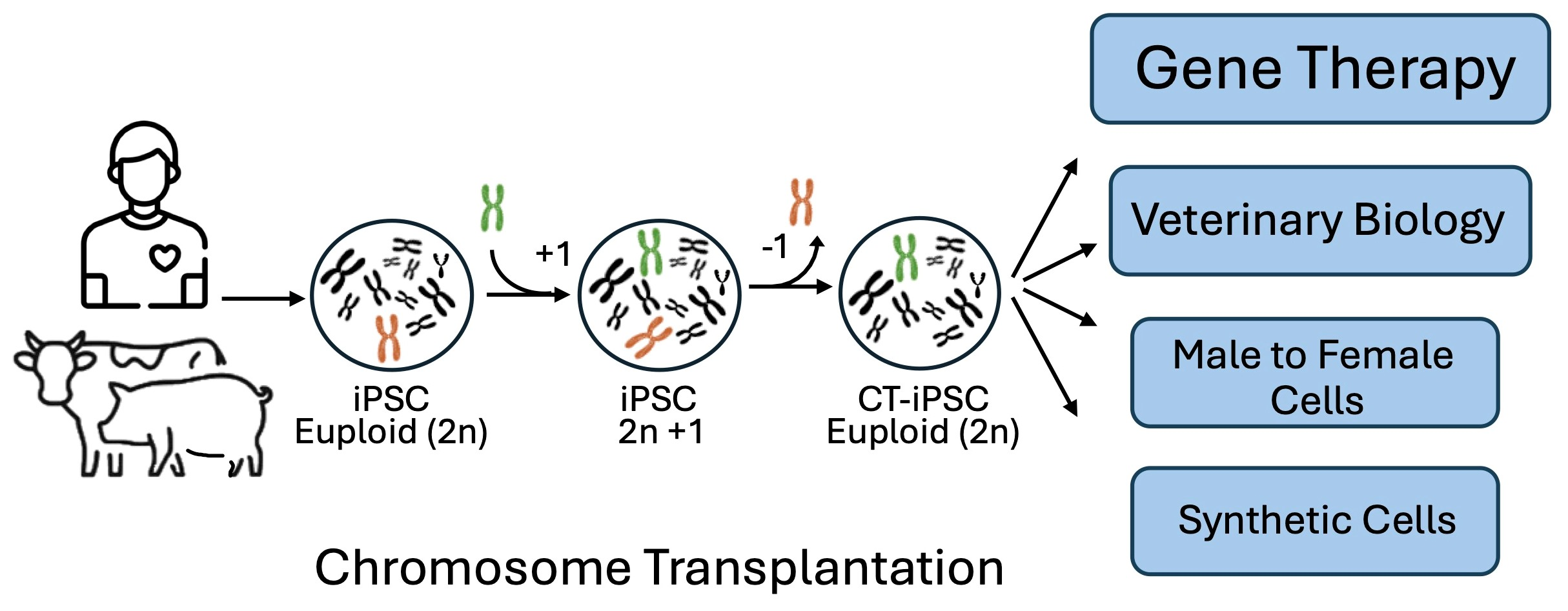
2. The Technique of CT

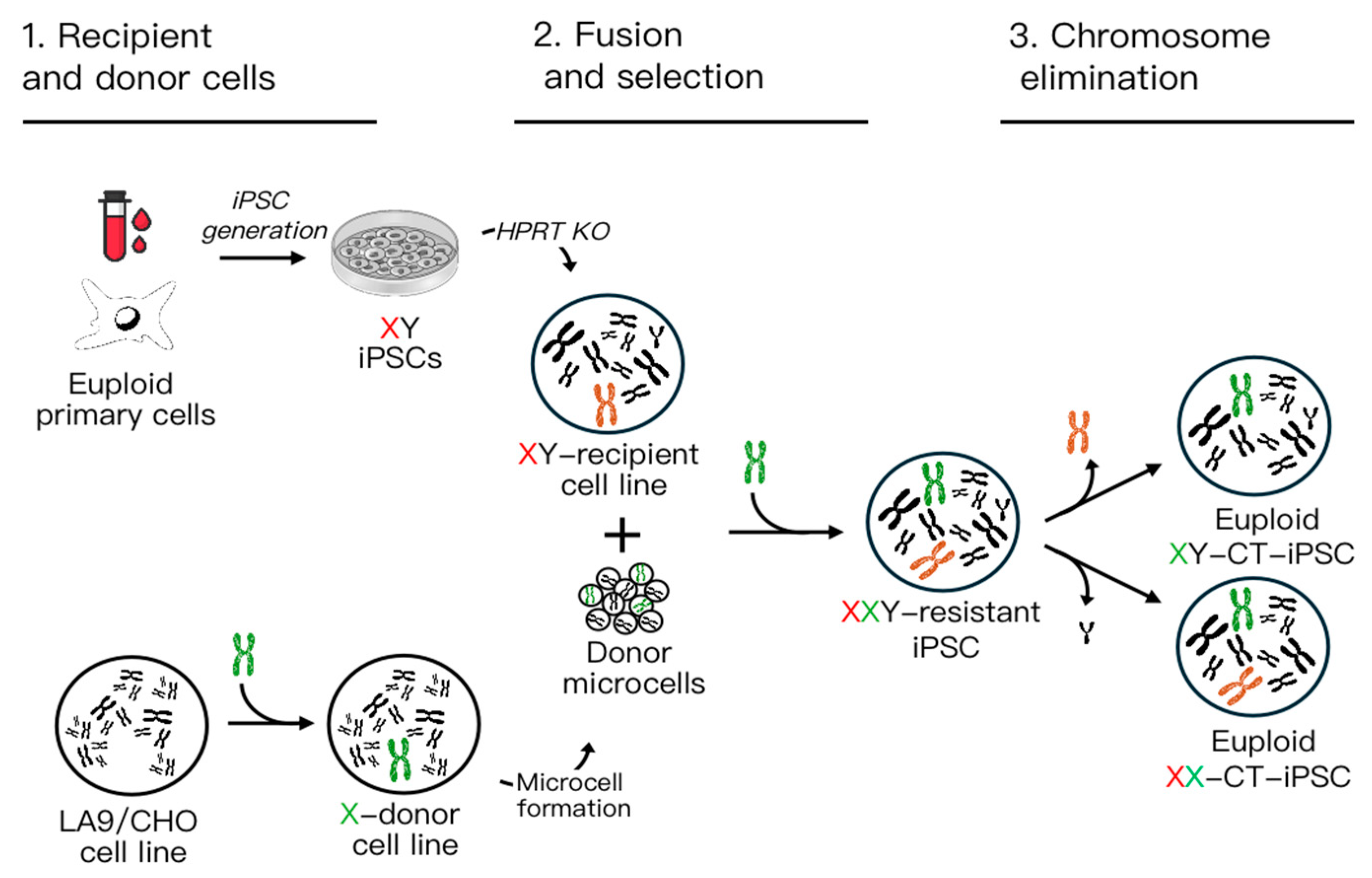
2.1. Step 1: Donor Cell Line and Micronucleation
2.2. Step 2: Fusion and Selection
2.3. Step 3: Chromosome Elimination
3. Results Achieved So Far
4. CT for Autosomes
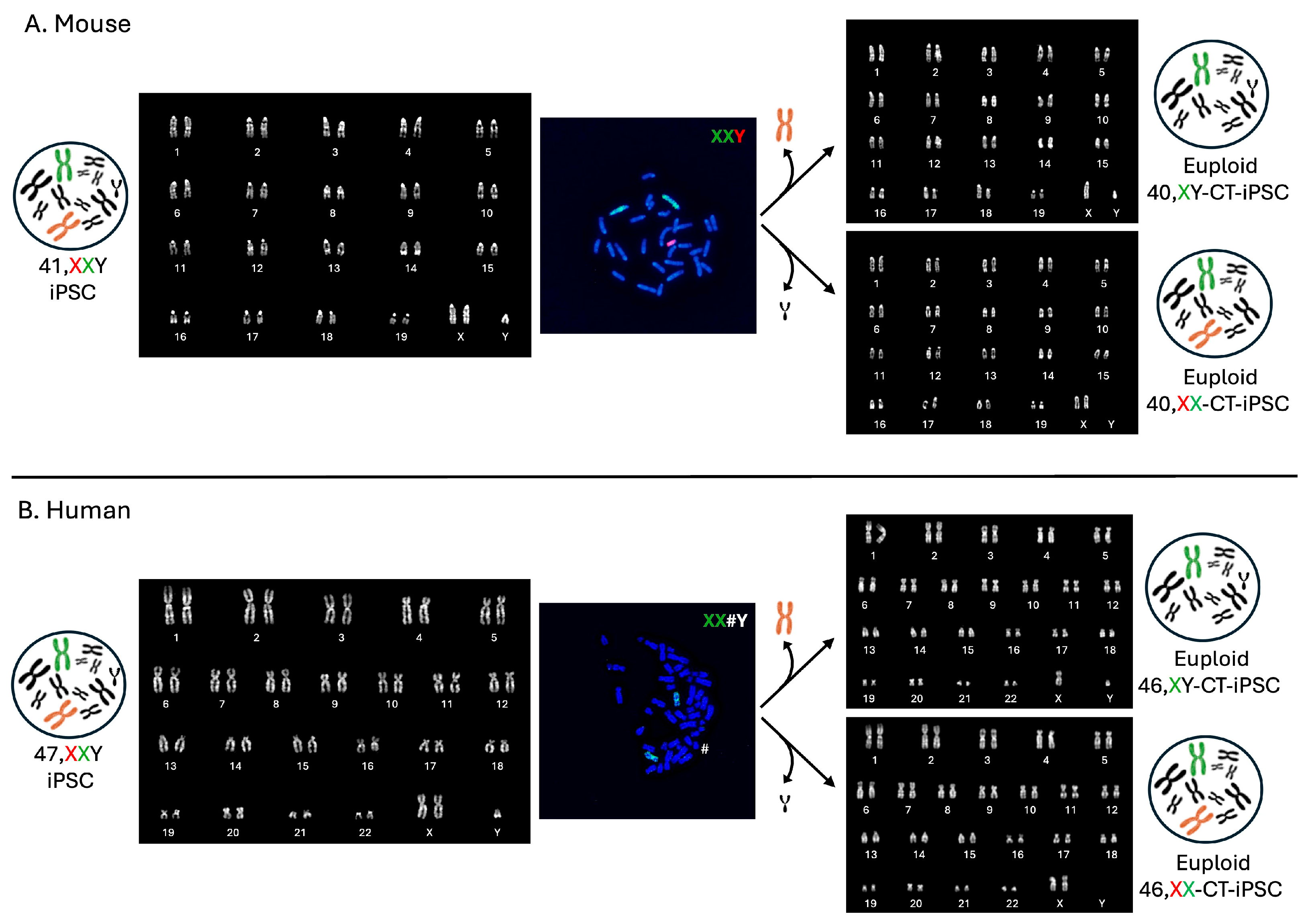
5. Male to Female
6. Large Animals
7. Limitations
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ferrari, S.; Valeri, E.; Conti, A.; Scala, S.; Aprile, A.; Di Micco, R.; Kajaste-Rudnitski, A.; Montini, E.; Ferrari, G.; Aiuti, A.; et al. Genetic engineering meets hematopoietic stem cell biology for next-generation gene therapy. Cell Stem Cell 2024, 30, 549–570. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A. Gene therapy for inborn errors of immunity: Past, present and future. Nat. Rev. Immunol. 2023, 23, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998, 14, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Ege, T.; Ringertz, N.R. Preparation of microcells by enucleation of micronucleate cells. Exp. Cell Res. 1974, 87, 378–382. [Google Scholar] [CrossRef]
- Fournier, R.E.; Ruddle, F.H. Microcell-mediated transfer of murine chromosomes into mouse, Chinese hamster, and human somatic cells. Proc. Natl. Acad. Sci. USA 1977, 74, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kazuki, Y.; Hara, T.; Oshimura, M. Current advances in microcell-mediated chromosome transfer technology and its applications. Exp. Cell Res. 2020, 390, 111915. [Google Scholar] [CrossRef] [PubMed]
- Sinenko, S.A.; Ponomartsev, S.V.; Tomilin, A.N. Pluripotent stem cell-based gene therapy approach: Human de novo synthesized chromosomes. Cell. Mol. Life Sci. 2021, 78, 1207–1220. [Google Scholar] [CrossRef]
- Kazuki, Y.; Uno, N.; Abe, S.; Kajitani, N.; Kazuki, K.; Yakura, Y.; Sawada, C.; Takata, S.; Sugawara, M.; Nagashima, Y.; et al. Engineering of human induced pluripotent stem cells via human artificial chromosome vectors for cell therapy and disease modeling. Mol. Ther. Nucleic Acids 2020, 23, 629–639. [Google Scholar] [CrossRef]
- Farr, C.J.; Bayne, R.A.; Kipling, D.; Mills, W.; Critcher, R.; Cooke, H.J. Generation of a human X-derived minichromosome using telomere-associated chromosome fragmentation. EMBO J. 1995, 14, 5444–5554. [Google Scholar] [CrossRef]
- Heller, R.; Brown, K.E.; Burgtorf, C.; Brown, W.R. Mini-chromosomes derived from the human Y chromosome by telomere directed chromosome breakage. Proc. Natl. Acad. Sci. USA 1996, 93, 7125–7130. [Google Scholar] [CrossRef]
- Raimondi, E.; Balzaretti, M.; Moralli, D.; Vagnarelli, P.; Tredici, F.; Bensi, M.; De Carli, L. Gene targeting to the centromeric DNA of a human minichromosome. Hum. Gene Ther. 1996, 7, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Harrington, J.J.; Bokkelen, G.V.; Mays, R.W.; Gustashaw, K.; Willard, H.F. Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat. Genet. 1997, 15, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Kouprina, N.; Petrov, N.; Molina, O.; Liskovykh, M.; Pesenti, E.; Ohzekiv, J.I.; Masumoto, H.; Earnshaw, W.C.; Larionov, V. Human artificial chromosome with regulated centromere: A tool for genome and cancer studies. ACS Synth. Biol. 2018, 7, 1974–1989. [Google Scholar] [CrossRef] [PubMed]
- Sinenko, S.A.; Skvortsova, E.V.; Liskovykh, M.A.; Ponomartsev, S.V.; Kuzmin, A.A.; Khudiakov, A.A.; Malashicheva, A.B.; Alenina, N.; Larionov, V.N.; Kouprina, N.; et al. Transfer of synthetic human chromosome into human induced Pluripotent Stem cells for Biomedical applications. Cells 2018, 7, 261. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Gambogi, C.W.; Liskovykh, M.A.; Barrey, E.J.; Larionov, V.; Miga, K.H.; Heun, P.; Black, B.E. Human artificial chromosomes that Bypass centromeric DNA. Cell 2019, 178, 624–639. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lampson, M.; Black, B. Centromere identity and function put to use: Construction and transfer of mammalian artificial chromosomes to animal models. Essays Biochem. 2020, 64, 185–192. [Google Scholar] [PubMed]
- Lee, N.C.O.; Petrov, N.S.; Larionov, V.; Kouprina, N. Assembly of multiple full-size genes or genomic DNA fragments on human artificial chromosomes using the iterative integration system. Curr. Protoc. 2021, 1, e316. [Google Scholar]
- Molina, O.; Kouprina, N.; Masumoto, H.; Larionov, V.; Earnshaw, W.C. Using human artificial chromosomes to study centromere assembly and function. Chromosoma 2017, 126, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, T.; Abe, S.; Oshimura, M.; Kazuki, Y. Transchromosomic technology for genomically humanized animals. Exp. Cell Res. 2020, 390, 111914. [Google Scholar] [CrossRef]
- Moralli, D.; Monaco, Z.L. Gene expressing human artificial chromosome vectors: Advantages and challenges for gene therapy. Exp. Cell Res. 2020, 390, 111931. [Google Scholar] [CrossRef]
- Nakayama, Y.; Uno, N.; Uno, K.; Mizoguchi, Y.; Komoto, S.; Kazuki, Y.; Nanba, E.; Inoue, T.; Oshimura, M. Recurrent micronucleation through cell cycle progression in the presence of microtubule inhibitors. Cell Struct. Funct. 2015, 40, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Paulis, M.; Susani, L.; Castelli, A.; Suzuki, T.; Hara, T.; Straniero, L.; Duga, S.; Strina, D.; Mantero, S.; Caldana, E.; et al. Chromosome Transplantation: A Possible Approach to Treat Human X-linked Disorders. Mol. Ther. Methods Clin. Dev. 2020, 17, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Uno, N.; Satofuka, H.; Miyamoto, H.; Honma, K.; Suzuki, T.; Yamazaki, K.; Ito, R.; Moriwaki, T.; Hamamichi, S.; Tomizuka, K.; et al. Treatment of CHO cells with Taxol and reversine improves micronucleation and microcell-mediated chromosome transfer efficiency. Mol. Ther. Nucleic Acids 2023, 33, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Liskovykh, M.; Lee, N.C.; Larionov, V.; Kouprina, N. Moving toward a higher efficiency of microcell-mediated chromosome transfer. Mol. Ther. Methods Clin. Dev. 2016, 3, 16043. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Shen, M.H. Polyethylene glycol-mediated cell fusion. Methods Mol. Biol. 2006, 325, 59–66. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Ren, X.; Katoh, M.; Miyata, M.; Fukushima, H.; Inoue, T.; Oshimura, M. A new method of microcell-mediated transfer of human artificial chromosomes using a hemagglutinating virus of Japan envelope. Chromosome Sci. 2006, 9, 65–73. [Google Scholar]
- Katoh, M.; Kazuki, Y.; Kazuki, K.; Kajitani, N.; Takiguchi, M.; Nakayama, Y.; Nakamura, T.; Oshimura, M. Exploitation of the interaction of measles virus fusogenic envelope proteins with the surface receptor CD46 on human cells for microcell-mediated chromosome transfer. BMC Biotechnol. 2010, 10, 37. [Google Scholar] [CrossRef]
- Suzuki, T.; Kazuki, Y.; Oshimura, M.; Hara, T. Highly Efficient Transfer of Chromosomes to a Broad Range of Target Cells Using Chinese Hamster Ovary Cells Expressing Murine Leukemia Virus-Derived Envelope Proteins. PLoS ONE 2016, 11, e0157187. [Google Scholar] [CrossRef]
- Tomita, A.; Sasanuma, H.; Owa, T.; Nakazawa, Y.; Shimada, M.; Fukuoka, T.; Ogi, T.; Nakada, S. Inducing multiple nicks promotes interhomolog homologous recombination to correct heterozygous mutations in somatic cells. Nat. Commun. 2023, 14, 5607. [Google Scholar] [CrossRef]
- Paulis, M.; Castelli, A.; Susani, L.; Lizier, M.; Lagutina, I.; Focarelli, M.L.; Recordati, C.; Uva, P.; Faggioli, F.; Neri, T.; et al. Chromosome transplantation as a novel approach for correcting complex genomic disorders. Oncotarget 2015, 6, 35218–35230. [Google Scholar] [CrossRef]
- Castelli, A.; Susani, L.; Menale, C.; Muggeo, S.; Caldana, E.; Strina, D.; Cassani, B.; Recordati, C.; Scanziani, E.; Ficara, F.; et al. Chromosome Transplantation: Correction of the Chronic Granulomatous Disease Defect in Mouse Induced Pluripotent Stem Cells. Stem Cells 2019, 37, 876–887. [Google Scholar] [CrossRef]
- Murakami, K.; Hamazaki, N.; Hamada, N.; Nagamatsu, G.; Okamoto, I.; Ohta, H.; Nosaka, Y.; Ishikura, Y.; Kitajima, T.S.; Semba, Y.; et al. Generation of functional oocytes from male mice in vitro. Nature 2023, 615, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, S.N.; Miyamoto, T.; Oba, D.; Tomioka, K.; Ochiai, H.; Ohashi, H.; Matsuura, S. iPSC reprogramming-mediated aneuploidy correction in autosomal trisomy syndromes. PLoS ONE 2022, 17, e0264965. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.R.; Capecchi, M.R. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 1987, 51, 503–512. [Google Scholar] [CrossRef]
- Sato, H.; Kato, H.; Yamaza, H.; Masuda, K.; Nguyen, H.T.; Pham, T.T.; Han, X.; Hirofuji, Y.; Nonaka, K. Engineering of Systematic Elimination of a Targeted Chromosome in Human Cells. BioMed Res. Int. 2017, 2017, 6037159. [Google Scholar] [CrossRef] [PubMed]
- Li, L.B.; Chang, K.H.; Wang, P.R.; Hirata, R.K.; Papayannopoulou, T.; Russell, D.W. Trisomy correction in Down syndrome induced pluripotent stem cells. Cell Stem Cell 2012, 11, 615–619. [Google Scholar] [CrossRef]
- Matsumura, H.; Tada, M.; Otsuji, T.; Yasuchika, K.; Nakatsuji, N.; Surani, A.; Tada, T. Targeted chromosome elimination from ES-somatic hybrid cells. Nat. Methods 2007, 4, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Lewandoski, M.; Martin, G.R. Cre-mediated chromosome loss in mice. Nat. Genet. 1997, 17, 223–225. [Google Scholar] [CrossRef]
- Zuo, E.; Huo, X.; Yao, X.; Hu, X.; Sun, Y.; Yin, J.; He, B.; Wang, X.; Shi, L.; Ping, J.; et al. CRISPR/Cas9-mediated targeted chromosome elimination. Genome Biol. 2017, 18, 224. [Google Scholar] [CrossRef]
- Adikusuma, F.; Williams, N.; Grutzner, F.; Hughes, J.; Thomas, P. Targeted Deletion of an Entire Chromosome Using CRISPR/Cas9. Mol. Ther. 2017, 25, 1736–1738. [Google Scholar] [CrossRef]
- Bosco, N.; Goldberg, A.; Zhao, X.; Mays, J.C.; Cheng, P.; Johnson, A.F.; Bianchi, J.J.; Toscani, C.; Di Tommaso, E.; Katsnelson, L.; et al. KaryoCreate: A CRISPR-based technology to study chromosome-specific aneuploidy by targeting human centromeres. Cell 2023, 186, 1985–2001.e19. [Google Scholar] [CrossRef] [PubMed]
- Tovini, L.; Johnson, S.C.; Guscott, M.A.; Andersen, A.M.; Spierings, D.C.J.; Wardenaar, R.; Foijer, F.; McClelland, S.E. Targeted assembly of ectopic kinetochores to induce chromosome-specific segmental aneuploidies. EMBO J. 2023, 42, e111587. [Google Scholar] [CrossRef] [PubMed]
- Truong, M.A.; Cané-Gasull, P.; Lens, S.M.A. Modeling specific aneuploidies: From karyotype manipulations to biological insights. Chromosome Res. 2023, 31, 25. [Google Scholar] [CrossRef] [PubMed]
- Girish, V.; Lakhani, A.A.; Thompson, S.L.; Scaduto, C.M.; Brown, L.M.; Hagenson, R.A.; Sausville, E.L.; Mendelson, B.E.; Kandikuppa, P.K.; Lukow, D.A.; et al. Oncogene-like addiction to aneuploidy in human cancers. Science 2023, 381, eadg4521. [Google Scholar] [CrossRef]
- Maclean, G.A.; Menne, T.F.; Guo, G.; Sanchez, D.J.; Park, I.H.; Daley, G.Q.; Orkin, S.H. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc. Natl. Acad. Sci. USA 2012, 109, 17567–17572. [Google Scholar] [CrossRef] [PubMed]
- Bershteyn, M.; Hayashi, Y.; Desachy, G.; Hsiao, E.C.; Sami, S.; Tsang, K.M.; Weiss, L.A.; Kriegstein, A.R.; Yamanaka, S.; Wynshaw-Boris, A. Cell-autonomous correction of ring chromosomes in human induced pluripotent stem cells. Nature 2014, 507, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Plona, K.; Wynshaw-Boris, A. A novel system for correcting large-scale chromosomal aberrations: Ring chromosome correction via reprogramming into induced pluripotent stem cell (iPSC). Chromosoma 2017, 126, 457–463. [Google Scholar] [CrossRef]
- Royer-Pokora, B.; Kunkel, L.M.; Monaco, A.P.; Goff, S.C.; Newburger, P.E.; Baehner, R.L.; Cole, F.S.; Curnutte, J.T.; Orkin, S.H. Cloning the gene for an inherited human disorder—Chronic granulomatous disease—On the basis of its chromosomal location. Nature 1986, 322, 32–38. [Google Scholar] [CrossRef]
- Chiriaco, M.; Di Matteo, G.; Sinibaldi, C.; Giardina, E.; Nardone, A.M.; Folgori, L.; D’Argenio, P.; Rossi, P.; Finocchi, A. Identification of deletion carriers in X-linked chronic granulomatous disease by real-time PCR. Genet. Test. Mol. Biomark. 2009, 13, 785–789. [Google Scholar] [CrossRef]
- Marciano, B.E.; Allen, E.S.; Cantilena, C.C.; Kristosturyan, E.; Klein, H.G.; Fleisher, T.A.; Holland, S.M.; Malech, H.L.; Rosenzweig, S.D. Granulocyte transfusions in patients with chronic granulomatous disease and refractory infections: The NIH experience. J. Allergy Clin. Immunol. 2017, 140, 622–625. [Google Scholar] [CrossRef]
- Tedesco, F.S. Human artificial chromosomes for Duchenne muscular dystrophy and beyond: Challenges and hopes. Chromosome Res. 2015, 23, 135–141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tedesco, F.S.; Hoshiya, H.; D’Antona, G.; Gerli, M.F.; Messina, G.; Antonini, S.; Tonlorenzi, R.; Benedetti, S.; Berghella, L.; Torrente, Y.; et al. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci. Transl. Med. 2011, 3, 96ra78. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Miyamoto, H.; Okamoto, K.; Nakano, K.; Matsunari, H.; Kazuki, K.; Hasegawa, K.; Uchikura, A.; Takayanagi, S.; Umeyama, K.; et al. Phenotypic features of dystrophin gene knockout pigs harboring a human artificial chromosome containing the entire dystrophin gene. Mol. Ther. Nucleic Acids 2023, 33, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.L.; Bassel-Duby, R.; Olson, E.N. CRISPR Correction of Duchenne Muscular Dystrophy. Annu. Rev. Med. 2019, 70, 239–255. [Google Scholar] [CrossRef]
- Saad, F.A.; Siciliano, G.; Angelini, C. Advances in Dystrophinopathy Diagnosis and Therapy. Biomolecules 2023, 13, 1319. [Google Scholar] [CrossRef] [PubMed]
- Waldhorn, I.; Turetsky, T.; Steiner, D.; Gil, Y.; Benyamini, H.; Gropp, M.; Reubinoff, B.E. Modeling sex differences in humans using isogenic induced pluripotent stem cells. Stem Cell Rep. 2022, 17, 2732–2744. [Google Scholar] [CrossRef]
- Eggan, K.; Rode, A.; Jentsch, I.; Samuel, C.; Hennek, T.; Tintrup, H.; Zevnik, B.; Erwin, J.; Loring, J.; Jackson-Grusby, L.; et al. Male and female mice derived from the same embryonic stem cell clone by tetraploid embryo complementation. Nat. Biotechnol. 2002, 20, 455–459. [Google Scholar] [CrossRef]
- Deng, J.M.; Satoh, K.; Wang, H.; Chang, H.; Zhang, Z.; Stewart, M.D.; Cooney, A.J.; Behringer, R.R. Generation of viable male and female mice from two fathers. Biol. Reprod. 2011, 84, 613–618. [Google Scholar] [CrossRef]
- Sparrow, R. Human enhancement through the lens of sex selection. In The Routledge Handbook of the Ethics of Human Enhancement; Jotterand, F., Ienca, M., Eds.; Taylor & Francis Group: London, UK, 2023; Chapter 7. [Google Scholar]
- Suvá, M.; Arnold, V.H.; Wiedenmann, E.A.; Jordan, R.; Galvagno, E.; Martínez, M.; Vichera, G.D. First sex modification case in equine cloning. PLoS ONE 2023, 18, e0279869. [Google Scholar] [CrossRef] [PubMed]
- Horer, S.; Feichtinger, M.; Rosner, M.; Hengstschläger, M. Pluripotent Stem Cell-Derived In Vitro Gametogenesis and Synthetic Embryos-It Is Never Too Early for an Ethical Debate. Stem Cells Transl. Med. 2023, 12, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Schnieke, A. How genome editing changed the world of large animal research. Front. Genome Ed. 2023, 5, 1272687. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Reinoso, M.A.; Aponte, P.M.; Garcia-Herreros, M. Genomic Analysis, Progress and Future Perspectives in Dairy Cattle Selection: A Review. Animals 2021, 11, 599. [Google Scholar] [CrossRef]
- Cartwright, S.L.; Schmied, J.; Karrow, N.; Mallard, B.A. Impact of heat stress on dairy cattle and selection strategies for thermotolerance: A review. Front. Vet. Sci. 2023, 10, 1198697. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Achievements | Limitations | |
|---|---|---|
| Donor cell line (CHO 1/HSA 2) | Single-chromosome universal donor cell line for the treatment of the disorder mapping to the specific chromosome. Cell line able to form microcells. | Demonstrated only for X-linked disorders (CHO/HSAX). |
| Recipient cell line (iPSCs 3) | Cells with a normal diploid genome, indefinite growth, pluripotency and capacity to differentiate into various tissues. | Long term in vitro culturing could accumulate mutations. |
| Fusion (retro-MMCT 4) | Retro-MMCT is based on murine leukemia virus envelope protein (Env) mediated fusion: less cytotoxic and more efficient compared to classic PEG 5 based MMCT fusion. | It is necessary to infect the donor cells with a lentivirus carrying the Env gene. |
| Selection (drug selection) | Use of an endogenous selectable gene (i.e., HPRT 6) or classical drug selection. | Endogenous selection system limited for a few chromosomes. |
| Chromosome loss (spontaneous loss) | Spontaneous loss of trisomic chromosomes without genomic manipulation. | Alternatively, more complex approaches could be used to drive the chromosome loss. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Grua, A.; Rao, I.; Susani, L.; Lucchini, F.; Raimondi, E.; Vezzoni, P.; Paulis, M. Chromosome Transplantation: Opportunities and Limitations. Cells 2024, 13, 666. https://doi.org/10.3390/cells13080666
La Grua A, Rao I, Susani L, Lucchini F, Raimondi E, Vezzoni P, Paulis M. Chromosome Transplantation: Opportunities and Limitations. Cells. 2024; 13(8):666. https://doi.org/10.3390/cells13080666
Chicago/Turabian StyleLa Grua, Angela, Ilaria Rao, Lucia Susani, Franco Lucchini, Elena Raimondi, Paolo Vezzoni, and Marianna Paulis. 2024. "Chromosome Transplantation: Opportunities and Limitations" Cells 13, no. 8: 666. https://doi.org/10.3390/cells13080666
APA StyleLa Grua, A., Rao, I., Susani, L., Lucchini, F., Raimondi, E., Vezzoni, P., & Paulis, M. (2024). Chromosome Transplantation: Opportunities and Limitations. Cells, 13(8), 666. https://doi.org/10.3390/cells13080666