Abstract
The tumor microenvironment (TME) plays an important role in the process of tumorigenesis, regulating the growth, metabolism, proliferation, and invasion of cancer cells, as well as contributing to tumor resistance to the conventional chemoradiotherapies. Several types of cells with relatively stable phenotypes have been identified within the TME, including cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), neutrophils, and natural killer (NK) cells, which have been shown to modulate cancer cell proliferation, metastasis, and interaction with the immune system, thus promoting tumor heterogeneity. Growing evidence suggests that tumor-cell-derived extracellular vesicles (EVs), via the transfer of various molecules (e.g., RNA, proteins, peptides, and lipids), play a pivotal role in the transformation of normal cells in the TME into their tumor-associated protumorigenic counterparts. This review article focuses on the functions of EVs in the modulation of the TME with a view to how exosomes contribute to the transformation of normal cells, as well as their importance for cancer diagnosis and therapy.
1. Introduction
Extracellular vesicles (EVs) are lipid-membrane-bound secretory structures that play a crucial role in cancer biology, mediating intercellular communication between tumor and normal cells within the tumor microenvironment (TME) [1]. According to the International Society for Extracellular Vesicles (and MISEV2024 update), EVs are classified according to various criteria, as follows: (a) biogenesis pathway—endosome-origin “exosomes” or plasma-membrane-derived “ectosomes” (i.e., microparticles/microvesicles); (b) size—small (<100 nm) or large (>200 nm); (c) biochemical composition (CD63+/CD81+-EVs, annexin-A5-stained EVs, etc.); and (d) according to the cell of origin or physiological processes (podocyte EVs, hypoxic EVs, large oncosomes, apoptotic bodies, etc.) [2]. The most common classification is related to the pathway of EV biogenesis: exomeres, exosomes, ectosomes (i.e., microvesicles), migrasomes, apoptotic bodies, and oncosomes (Figure 1) [3]. The constitutive contents of exosomes include various RNAs (e.g., miRNA, lncRNA, and circRNA), DNA fragments, transcription factors, signaling proteins, and many other molecules [4]. EVs have a number of advantages as mediators. Firstly, because of their lipid membrane bilayer, they preserve the contents from external influences and allow them to be activated only inside the recipient cell. Secondly, surface markers enable the targeted delivery of the exosomal content [5]. The uptake of extracellular vesicles by recipient cells is mediated by several mechanisms including phagocytosis, clathrin- and caveolin-mediated endocytosis, macropinocytosis, and membrane fusion [6].
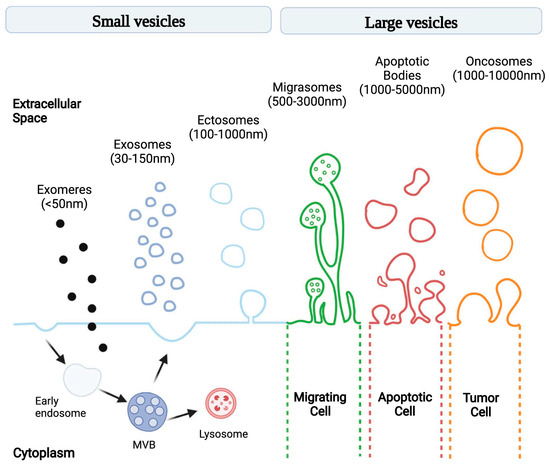
Figure 1.
Schematic illustration of EV subtypes according to their different size and genesis models [3].
Tumor cells under stressful conditions are characterized by an increased level of exosome release [7]. Within a tumor, exosomes are involved in modulating the tumor microenvironment. Presumably, tumor-derived EVs ensure the heterogeneity of normal cell populations through the delivery of physiologically active cargo. The heterogeneity of cancer cells has long been documented [8]. However, in addition to tumor heterogeneity, there is also heterogeneity in other cell groups [9]. The cohabitation of different types of cells within the TME under harsh conditions creates several types of interactions. Merlo et al. identified five types of interactions: competition, predation, parasitism, mutualism, and commensalism [10]. Transformation of normal cells in this case can be considered a means of creating the most favorable conditions for the tumor and, accordingly, changing the interaction toward a symbiotic relationship. Considering that stable populations of transformed normal cells, such as cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs) [11], tumor-associated neutrophils (TANs), and others, are found within the solid tumor, one could assume that during tumor-driven selection, they have some advantage over others (Table 1).

Table 1.
Properties of tumor microenvironment cells associated with protumorigenic effect.
However, there are no data on further modulation of already-transformed normal cells. Thus, fibroblasts transformed into CAFs have no well-defined phenotype that can be used to describe this cellular subpopulation. This tumor cell–CAF interaction is the only documented example of mutualism in a tumor beneficial for both cell types [33]. Accordingly, it has been shown that exosomal LncRNA LINC00659 of CAF enhances colorectal cancer cell progression via the miR-342-3p/ANXA2 axis [34]. On the other hand, the heterogeneous TME is influenced by the antitumor responses of the organism, mainly associated with the immune system [35]. Thus, the formation of cellular subpopulations of transformed normal cells within the TME occurs under the influence of various factors derived from the tumor itself and the organism (Figure 2). The question of the direction of these factors remains unclear. Additionally, it is worth considering that the release of EVs by tumor cells also depends on external conditions. Thus, hypoxic conditions increase the release of EVs by tumor cells which results in increased angiogenesis and migration [36]. The molecular mechanisms of the influence of EV cargo on the heterogeneity of specific cell types are discussed in the following sections.
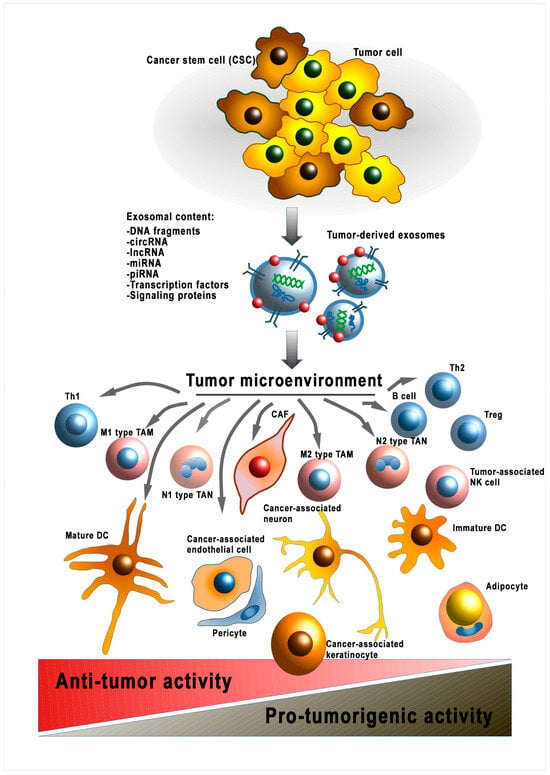
Figure 2.
Modulation of the tumor microenvironment by cancer-cell-derived EVs. sRNA—ribonucleic acid; miRNA—micro RNA; lncPRN—long noncoding RNA; circRNA—circular RNA; piRNA—piwi-interacting RNA; DNA—deoxyribonucleic acid; Th1—T-helper 1; TAM—tumor-associated macrophage; Treg—T regulatory cell; TAN—tumor-associated neutrophil; CAF—cancer-associated fibroblast; DC—dendritic cell; Th2—T-helper 2; NK cell—natural killer cell.
In addition to the exosomal cross-talk mechanism, other mechanisms of cancer-associated cell transformation have been described, including close intercellular contacts, paracrine signaling pathways, growth factors (BMP2 and BMP4), and stress-related conditions (hypoxia, low pH, etc.). Kyuno et al. described a significant impact of the cell–cell junction on the epithelial-to-mesenchymal transition of cancer cells [37]. The main molecular player among the soluble factors is considered to be TGF-β [38]. It is secreted by different cells of the TME and induces significant modulation of the tumor environment, bringing about a cascade of changes to tumorigenesis [39]. Although various stress-related conditions have been identified, including hypoxia, oxidative stress, and other factors, it is still quite difficult to estimate the input of each factor for TME transformation, as these factors are usually simultaneously present in the tumor and have a synergistic effect on the infiltrating cells [40,41,42,43].
In this review, we focus on tumor-derived EVs and their role in the modulation of normal cells infiltrating the sites of solid tumors, as well as on the processes of transformation of a normal microenvironment into a tumor-associated microenvironment mediated by EV crosstalk. We attempt to describe the most significant events in the acquisition of a relatively stable tumor-associated phenotype TME by EVs.
2. Cancer-Derived EVs Transform Normal Cells into Cancer Cells
The ability of cancer cell secretions, including EVs, to induce the carcinogenic transformation of normal cells into cancer cells was first experimentally demonstrated, in 1999, by Olmo and colleagues [44]. Subsequently, this hypothesis of a cell-free circulation factor inducing distant tumorigenic transformation was termed “genometastasis”. Early evidence suggested a key role for the horizontal transfer of cell-free DNA into recipient cells [45]. Subsequently, this hypothesis was further elaborated and expanded in [46,47], and tumor-derived EVs have been shown to be the key players in this process [48]. Currently, not all components of EVs have been identified and, more importantly, the mechanism of their malignant effect on normal cells has not been described. Another problem in the studies to date is the difficulty they faced in determining the exact moment of the transformation of normal cells into cancer cells. A change in the phenotype in favor of highly proliferative and apoptosis-resistant cells does not always indicate the ability to develop tumors in vivo. The preferred criteria for identifying cell malignancy could, therefore, relate to in vivo tumor formation upon cell transplantation. The consequence of a transformation of normal cells into cancer cells is an increase in tumor heterogeneity and, accordingly, a decrease in the tumoral response to treatment. Ernesto Yagu et al. showed that the acquired multidrug resistance (MDR) can occur before the malignant transformation stage. The minimal set of changes necessary to obtain pretumorigenic drug-resistant cells includes the expression of telomerase and inactivation of p53 and pRb [49]. A similar alteration might emerge in a normal cell within tumorigenic processes. Indeed, MCF10A normal breast cells transformed with exosomes derived from MCF10A.NeuT tumor cells showed increased resistance to radiation therapy compared to normal cells [50]. A major contribution to the development of tumor EV research was made, in 2008, when Janus Rak’s group introduced the term “oncosomes” and showed their participation in the spread of a mutant form of the epidermal growth factor receptor, termed EGFRvIII, from cancer cells to normal cells [51]. Oncosomes are defined as large vesicles of ectosome pathway biogenesis that facilitate the export of specific oncogenic cargo [52]. Despite the importance of determining the EV subtype, in practice this is a rather difficult task; therefore, most studies are related to common secretory EVs.
One of the most studied components of tumor EVs is RNA. Noncoding circRNA EVs of cancer cells can induce uncontrolled proliferation of normal cells with subsequent transformation into tumor cells [53]. Xiangyu Dai showed that arsenic-induced cancer cells produce exosomes containing circRNA_100284 [54]. When integrated into hepatic epithelial (L-02) cells, circRNA_100284 acts as a sponge of microRNA-217 [55], hybridizes with miRNAs, and inhibits EZH2/cyclin-D1 gene silencing. In another study, it was demonstrated that exosomal hsa_circ_0000069 secreted by SW1990 pancreatic cancer cells promoted the proliferation, migration, and cell cycle progression of human pancreatic duct epithelial cells (HPDEs). These experiments indicate the critical role of exosome-delivered circRNA in tumor progression. Moreover, the malignant changes were reversed through downregulation of hsa_-circ_0000069. When determining the mechanism of action, it was proposed that hsa_circ_0000069 acts as a sponge for miR-144 by increasing the expression of the STIL gene by the main regulator of the mitotic centrosome [56]. In addition to circulating RNAs, it has been shown that gastric cancer cells secrete exosome-packaged long noncoding RNA (lncRNA) SND1-IT1 [57]. Internalization of this lncRNA by human gastric mucosa epithelial GES-1 cells caused cell malignant transformation by enhancing the expression of ubiquitin-specific protease 3 (USP3), inhibiting miR-1245b-5p, and simultaneously recruiting DEAD-box helicase 54 (DDX54). USP3 mediates the deubiquitination of snail family transcriptional repressor 1 (SNAIL1) by activating tumor development and cell transformation [58]. The participation of miRNA packed in EVs in the modulation of the phenotype of recipient cells has been shown in numerous studies, including those on the malignant transformation of normal cells [59,60]. Thus, exosomal miR-224-5p secreted by human colorectal cancer SW620 cells induces cancerous transformation of human normal colon epithelial CCD 841 CoN cells by inhibiting the oncosuppressor CMTM4 [61]. Different types of RNAs are, indeed, powerful tools for modulating cellular processes and creating a heterogeneous population. However, they are not capable of directly causing tumor mutations in the genetic apparatus, which are critical in cell malignancy. We assume that the occurrence of these mutations can be, in part, mediated by indirect mechanisms such as aberrations of DNA repair due to the effects of different types of RNAs.
Our hypothesis is supported by a study of exosomal delivery of annexin A1 (ANXA1) by medullary thyroid carcinoma SW579 cells, which caused the malignant transformation of Nthy ori3 1 thyroid follicular epithelial cell lines that, subsequently, resulted in an increased levels of proliferation, invasion, and epithelial–mesenchymal transition [62]. Recently, Stefanius et al. proposed a two-stage scheme for the involvement of cancer exosomes in the transformation of normal cells into cancer cells (Figure 3) [63].
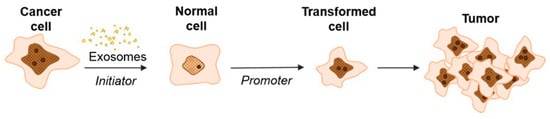
Figure 3.
Schematic model of exosome-mediated transformation [63].
According to this hypothesis, exosomes secreted by cancer cells, upon being internalized into normal cells, initiate the development of mutations but are not yet able to cause complete transformation into a tumor cell. In this case, exosomes replaced the genotoxic carcinogen 3-MCA (3-methylcholanthrene) that was used as a control and induced random genetic change. During the second stage, the influence of TPA (12-O-tetradecanoylphorbol 13-acetate) enhanced the proliferation in the initiated cells selectively, thus driving malignant transformation of the cells [37,64]. These data are consistent with the studies of Abdour and colleagues, who showed that oncosuppressor genes can work as protectors from the absorption of cancer EVs and, accordingly, from subsequent malignant transformation [64]. In their study, BRCA1 knockout fibroblasts were more susceptible to exosomes isolated from the serum of patients with various tumor locations (colorectal cancer [CRC], hepatocellular carcinoma [HCC], pancreatic cancer [PC], and ovarian cancer) [37]. The treatment led to the malignant transformation of fibroblasts into tumors with the feature of highly proliferative adenocarcinoma. Moreover, the transformed fibroblasts showed signs of a primary tumor: the expression of epithelial markers typical of colorectal adenocarcinoma (CK7, positive for CEA, CK20, CDX-2, and AE1/AE3), markers reflecting early differentiation into pancreatic cancer (positive for cytokeratin CK19 and AE1/AE3, CK7 focal positive patches), and ovarian cancer markers positive for WT-1 and EMA. Interestingly, fibroblasts with wild-type BRCA1 did not transform into cancer cells.
Summarizing the described data, the primary interaction of normal cells with tumor EVs leads to the creation of a heterogeneous population of activated normal cells which, through selection and modification, become either tumor-bearing or cancer-associated or are destroyed by immune surveillance (Figure 4).
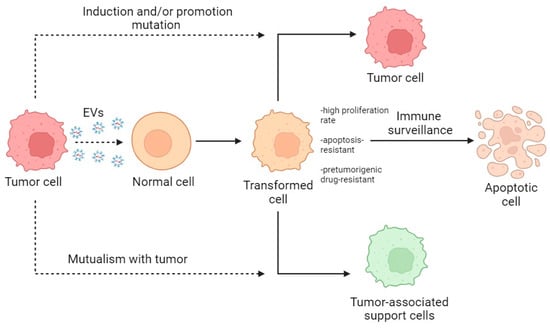
Figure 4.
Scheme of the transformation of normal cells within the TME. Dotted lines indicate the direction of action; solid lines indicate the direction of transformation.
In conclusion, cancer-derived EVs can be both initiators and promoters of the malignant transformation of normal cells. In the case of initiatory participation, they introduce nondirectional changes and create conditions for the competitive selection of subpopulations. With promoter involvement, susceptibility to EVs allows for a more directed change in the cell phenotype toward a tumor type. In both cases, there is an accumulation of modifications leading to a malignancy of normal cells. In 2022, Douglas Hanahan updated the list of the main hallmarks of cancer [65]. From this point of view, cancer-derived EVs components are responsible for ensuring the acquisition of these characteristics (Table 2). Unfortunately, not all tumor hallmarks were described for transformed normal cells, but data can be extrapolated from other cells’ types of tumor microenvironment. At the same time, a certain portion of altered normal cells might transform into cancer-associated support cells.
Table 2.
Influence of cancer-derived EVs on the acquisition of tumor hallmarks in transformed normal cells.
3. Cancer-Derived EVs Transform Cancer Cells into Cancer Stem Cells
Cancer stem cells (CSCs), discovered in 1994, consistently attract the attention of researchers [74]. The phenotype of stem cells allows them to self-renew and differentiate into different cell subpopulations. Moreover, the plasticity of CSCs allows them to dedifferentiate back under various stimuli, including those provided by EVs [75]. In addition to the CSC phenotype, because of the tumor heterogeneity, other subpopulations of tumor cells can be detected within the TME [76]. Extracellular vesicles play a significant role in the transfer of an aggressive tumor phenotype to nonaggressive tumor cells, as a result of which the latter acquire the stem cell-like properties [77,78,79].
3.1. Support for Cancer Stem Cell Niche by EVs
The features of cancer stem cells require the existence of a stem cell niche that in normal tissues plays an essential role in maintaining stem cells or preventing tumorigenesis by providing primarily inhibitory signals for both proliferation and differentiation. CSC niche characterization, therefore, arises from an intrinsic mutation, leading to self-sufficient cell proliferation, and/or it may also involve the deregulation or alteration of the niche by dominant proliferation-promoting signals. Moreover, tumor development may recruit mechanisms of the niche of normal stem cells for invasion and metastasis [49]. TME cells, particularly cancer-associated fibroblasts, produce EVs that support the population of CSCs and promote the development of tumor chemoresistance. This could be attributed to the enhancement of the Wnt signaling pathway [80,81,82]. Another cell type, glioma-associated human mesenchymal stem cells (GA-hMSCs), has been shown to produce exosomes containing miR-1587, which is known as a tumor-suppressive nuclear receptor corepressor, NCOR1, and leads to increases in the proliferation and clonogenicity of GA-hMSCs according to in vitro experiments [83]. miRNAs also play a significant role in maintaining the stemness of cancer cells [84]. Thus, Yanxia Zhan et al. showed that hypoxic CAF-derived exosomes modulate breast cancer cell stemness through exonic circHIF1A by miR-580-5p cells in breast cancer [85]. In this case, the development of the CSC population can be considered to be a way of adapting the tumor to hypoxic conditions. In general terms, the properties of the cancer stem cell niche are similar to conventional stem cell niches.
3.2. EMT-Mediated Transformation of Non-CSCs into CSCs
The induction of non-CSC-to-CSC transformation may be mediated by the epithelial–mesenchymal transition (EMT) [86]. Among various cytokines, it has been demonstrated that TGF-b1 plays an important role in the EMT process. Researchers showed that EVs secreted by chronic myeloid leukemia cells containing TGF-b1 on the membrane trigger the corresponding cascade of events in acceptor cancer cells and promote both the acquisition of the stem cell phenotype and EMT [87]. In addition to the activation of the canonical transcription factor SMAD, signal transduction and activation of the MAPK PI3K/Akt proliferation and survival pathways, like (TGF)-β, PI3K/AKT, and MAPK, were also observed [88]. In addition to activating cancer cell reprogramming of signaling pathways through the activation of receptors, vesicles directly alter the cell transcriptome via the contained transcription factors [89]. In general terms, the reprogramming of cancer cells into cancer stem cells through the EMT has similarities with technology that creates induced stem cells using Yamanaka factors, which include cell pretreatment with TGF-b [90,91].
3.3. Role of EV piRNAs in Transformation of Non-CSCs into CSCs
P-element-induced wimpy testis (PIWI) and PIWI-interacting RNA (piRNA) proteins are specific markers for the germline state. They are responsible for maintaining cell stemness [38]. The involvement of piRNAs in the functioning of cancer stem cells was first demonstrated on a testicular germ-cell tumor, where the overexpression of one of the four PIWI protein genes, HIWI, was detected [92]. Since then, the overexpression of piRNAs and PIWI has been found in several other cancers, including breast carcinoma [93,94]. Ross et al., relying on data of increased expression of mobile genetic elements in tumor cells [95], linked the activities of piRNAs and PIWI with the compensatory mechanism of genetic stability. Thus, preadipocyte-derived exosomes have been shown to enhance the growth and survival of breast cancer cells. The exosomal components miR-140/SOX2 and SOX9 were shown to contribute to the maintenance of tumor stemness in that study [96]. Senescent neutrophil exosomes can transfer chemoresistance and EMT characteristics to recipient breast cancer cells through cell-to-cell transfer of piR-17560. Moreover, exosomal piR-17560 promotes the EMT by regulating ZEB1 of breast cancer cells through FTO-dependent m6A demethylation. These findings indicate the critical role of senescent neutrophils in the regulation of the EMT and, consequently, the acquisition of the CSC phenotype [97]. piR-823 also had different activities in various types of cancer. For example, piR-823 promotes cell proliferation and tumorigenesis by enhancing HSF1 phosphorylation and transcriptional activity in colorectal cancer [98]. In multiple myeloma, piR-823 promotes tumorigenesis by regulating de novo DNA methylation [99]. However, piR-823 has been shown to inhibit gastric carcinogenesis [100] and induces the expression of stem cell markers (including OCT4, SOX2, KLF4, NANOG, and hTERT) and increases the formation of mammospheres, as has been demonstrated on breast cancer MCF-7 and T-47D cells. The authors attribute this to the activation of the Wnt signaling pathway through increased expression of DNA methyltransferase (DNMT) and methylation of the adenomatous polyposis coli (APC) gene [101]. The induction of the overexpression of piR-823 has not yet been studied. It has recently been shown that piR-823 can be secreted by cancer cells via exosomes, and it was found in biopsies from patients with colorectal cancer [102]. Extrapolating from these data, piR-823 secreted by CSC during tumor development may be involved in the autocrine and paracrine maintenance of the stem phenotype in the cancer population. It also creates a favorable environment, as shown in a multiple myeloma model, whereby exosomal piR-823 induced the transformation of EA.hy926 endothelial cells by enhancing the expression of VEGF, IL-6, and ICAM-1, and attenuating apoptosis [49].
3.4. Role of Other EV Cargoes in the Transformation of Non-CSCs into CSCs
Another important molecule for CSC maintenance is Wip1 (wild-type p53-inducible phosphatase-1) [103]. Indeed, the inhibitions of p53 and p38 MAPK pathway activities have been found to result in an extended MSC life span and their increased differential potential in normal mesenchymal stem cells [104]. A similar role for Wip1 has been identified in cancer stem cells, in which it is able to directly dephosphorylate p53 at Ser15 and dephosphorylate MDM2, an E3 p53 ubiquitin ligase, leading to the destabilization of the p53 protein. Wip1 also dephosphorylates and inactivates ATM, Chk1, and Chk2, which are upstream activators of p53 kinase [105,106]. The inhibition of the p38 MAPK pathway is also exerted via the Wip1 phosphatase activity [107]. So far, there are no data on the exosomal secretion of Wip1 by cancer cells, although it is known that changes in Wip1 expression in cells can occur under the influence of exosomal circSHKBP1 secreted by cancer cells [108].
In general terms, the transformation of cancer cells into CSCs and maintenance of the stem phenotype by EVs are similar to the creation of a stem niche [109]. The questions of why a CSC population is formed in a tumor and why it is so relatively small still remain open. The accumulated data clearly indicate that CSCs increase tumor resistance to therapy [110]. Indeed, the CSC population is heterogeneous and, during treatment-related selection, relapses quickly because of high proliferative activity and plasticity [111]. As was shown by Bao et al., glioma stem cells (defined by CD133 expression) contributed to tumor radioresistance through the activation of the DNA damage checkpoint response, which was reversed by the application of specific inhibitors of checkpoint kinases (Chk1 and Chk2) [112]. Another study showed that the BMP pathway in glioma stem cells is related to cell resistance to not only radiotherapy but also to chemotherapy with temozolomide [113].
4. Cancer-Derived EVs Transform Stromal Microenvironment Cells into Cancer-Associated Support Cells
The cellular composition of the TME, on the one hand, is quite well studied, but on the other hand, in practice, determining the cell type is a rather difficult task due to high heterogeneity and necrotic processes [105]. Most data are available regarding cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), and tumor-infiltrated/-associated natural cells (TINKs/TANKs) (Table 3). The remaining cellular components of the TME, however, have been studied to a lesser extent. The transformative influence of cancer-derived EVs on the formation of cancer-associated support cells with certain phenotypes is shown in Table 3.
Table 3.
Participation of some EV cargo components in the transformation into cancer-associated support cells with certain phenotypes.
4.1. Transformation of Fibroblasts into Cancer-Associated Fibroblasts (CAFs)
Fibroblasts are connective tissue cells of mesenchymal origin [131]. They are often identified by their morphology, location, and absence of clonal markers of endothelial cells, epithelial cells, and leukocytes. However, their unique molecular markers have not yet been identified. Vimentin and platelet-derived growth factor receptor-α (PDGFRα) can be used as auxiliary markers [12]. Cancer-associated fibroblasts functionally differ from normal ones by their more pronounced tumor-modulating effect, expressed in the induction of angiogenesis, inflammation, and remodulation of the extracellular matrix (ECM) [132]. The identification of CAFs has typically been carried out on the basis of the expressions of various “CAF markers”, such as fibroblast activation protein alpha (FAP) and alpha smooth muscle actin (αSMA). However, αSMA can also be highly expressed in normal fibroblasts when wound healing occurs [13]. Moreover, αSMA is constitutively expressed in smooth muscle cells, as is FAP in adipocytes and osteocytes [133,134]. The high heterogeneity of CAFs is reflected by their various subpopulations within a tumor [135]. It has been found that subpopulations of inflammatory fibroblasts (iCAFs) and myofibroblasts (myCAFs) in PDACs are spatially separated. myCAFs are located in the periglandular region, whereas iCAFs (characterized by the secretion of IL6, IL11, and LIF and a stimulated STAT pathway) are distantly located from cancer cells and myCAFs [136]. The main distinguishing feature and boundary of the transition between CAFs and tumor cells is the absence of genetic mutations. The identification of CAFs is usually made on the basis of many traits, including molecular markers, morphologies, and genotypes [132]. Depending on the expression of CAF markers (i.e., FAP, αSMA, and integrin β1 [CD29]), various CAF subsets were previously identified for ovarian and breast cancers [137,138,139]. The myCAF subsets (FAPHigh SMAHigh CD29Med-High and FAPNeg SMAHigh CD29High) have been shown to be correlated with a poor prognosis [140,141]. The process by which fibroblasts are converted to CAFs is not fully understood. However, various molecules and biochemical processes, including inflammatory cytokines (IL-1, IL-6, TNF, and TGFβ), RTK ligands (PDGF and FGF), physiological stress (e.g., ROS and disrupted metabolism), and DNA damage, have been shown to play a role in CAF formation [142,143,144,145]. The stiffness and composition of the ECM, as well as contact signals (i.e., Notch and Eph/ephrins), are also important for fibroblasts’ transformation [146]. The particular role of EVs in this process of CAF formation is rather difficult to estimate because of the additive effect of the multiple factors listed above. However, one cannot underestimate the significance of cancer-derived EVs in the transformation of fibroblasts into CAFs. CSCs play an important role in this process through their involvement in modulating the microenvironment [147]. The main effect of CSCs produced by EVs is to enhance fibroblasts’ proliferation, migration, and invasion [148,149,150]. Thus, it has been shown that Piwil2-induced cancer stem cells (Piwil2-iCSCs) increase the expressions of MMP2 and MMP9, which are responsible for enhanced invasiveness and migration through exosomes [114]. It has been shown in melanoma models that exosomes secreted by B16F0 cells induce reprogramming of NIH/3T3 fibroblast cells to CAFs, as evidenced by increased expressions of CAF-related markers (α-SMA and FAP) and facilitation of cell migration. EVs secreted by B16F0 melanoma cells deliver Gm26809 to NIH/3T3 cells, where Gm26809 (long noncoding RNA [lncRNA]) mediates the reprogramming of normal fibroblasts. As expected, knockdown of the Gm26809 gene disrupts exosome-induced transformation [115]. Other miRNAs within cancer-derived exosomes can also induce transformation [144]. Thus, exosomes of breast cancer cells containing miR-146a inhibited the expression of the TXNIP gene in fibroblast cells and activated the transition to CAFs via activation of the Wnt signaling pathway [116]. Another agent, miR-27a, found in abundance in exosomes of gastric cancer, is able to directly target the oncosuppressor CSRP2 by binding to its 3’-UTR, thereby triggering Rac1 activation and inductions of the ERK and PAK/LIMK/cortactin signaling cascades [117,118]. Gutkin et al. determined that hTERT mRNA, a transcript of the telomerase enzyme, is transferred from cancer cells through exosomes to telomerase-negative fibroblasts, where it is translated into a fully active enzyme. Subsequently, these telomerase-positive cells represent a new cell type—nontumor cells with enhanced telomerase activity [119]. A more recent study reported that exosomal telomerase may play a role in modifying normal fibroblasts into cancer-associated fibroblasts by upregulating αSMA and vimentin [68]. As previously mentioned, the interaction of tumor cells with CAFs is mutualistic. In addition to the obvious benefits for the tumor in the form of increased angiogenesis, extracellular matrix remodeling, and activation of the inflammatory processes, it has been shown that CAFs with exosomal lncRNA POU3F3 induced tumor resistance to cisplatin due to the secretion of inflammatory cytokines [151]. Accordingly, they cause an increase in tumor resistance to therapy [152]. However, the involvement of CAFs in tumor development shows a dual role. In a study by Daniela et al., two subpopulations were identified, as follows: CAF-N with a transcriptome and secretome similar to normal fibroblasts, and tumor-promoting CAF-D with an expression pattern that differed from normal fibroblasts and CAF-N [153]. In addition, CAFs expressing FSP1 have been shown to inhibit tumor development by encapsulating carcinogenesis. When the carcinogen methylcholanthrene (MCA) was administered subcutaneously, a concentration of FSP1-positive fibroblasts around the lesion and increased secretion of collagens were observed [154]. Another marker of antitumor CAFs is the glycosylphosphatidylinositol-anchored protein Meflin, the increased expression of which induces an improvement in chemosensitivity in PDAC [155]. Complete depletion of αSM-positive CAFs in the tumor PDAC model in a transgene murine model resulted in reduced desmoplasia and stromal stiffness and increased tumor invasiveness and metastatic activity [156]. The idea of the existence of protumorogenic and antitumorogenic types of CAFs emerged after a series of failures associated with CAF depletion [157]. Today, it is considered that the most promising anticancer strategy mediated by CAFs is to switch CAFs to a quiescent state [158]. Silke Haubeiss et al. determined the therapeutic role of dasatinib mediated through the transformation of CAFs into normal resting fibroblasts; moreover, the incubation of tumor cells with conditioned medium from CAFs preincubated with dasatinib significantly reduced tumor cell proliferation [159].
Unfortunately, there are no data on the dynamic changes in CAFs during cancer progression. According to the previously reported studies, CAF senescence occurs over several passages in vitro and depends on the age of the tissue [131]. When drawing analogies between the processes of transformation of fibroblasts into CAFs and wound healing (in both cases, activation of fibroblasts and acquisition of the myofibroblast phenotype are observed), the role of tumor EVs consists in the maintenance of a chronic activated state of CAFs [160]. Unfortunately, there are no experimental data on the dynamics of the changes in the CAF phenotype and reversible tumor effects. Targeted uptake of EVs has been documented as therapeutic [161]. Furthermore, it has been previously shown that tumor EVs can carry PDGF on their surfaces [162], and PDGFRα/β is one of the markers of CAFs [12]. This suggests the existence of the targeted secretion of EVs by tumor cells for CAFs. However, the question of whether this is the case remains open and requires in-depth study.
4.2. Transformation of Nerve Cells into Cancer-Associated Nerve Cells
Tumor innervation, along with angiogenesis, is a necessary process for disease progression [163]. Phenotypic transformations of nerve cells under the influence of tumors can be divided into the following three aspects of innervation: axonogenesis, reprogramming of nerve cells, and neurogenesis (Figure 5) [164]. Nerve cells with a phenotype that changed under the influence of tumor cells do not have a specific classification, unlike fibroblasts or macrophages. Identification is mainly provided by histological staining and morphology. As candidate molecular markers, high expressions of Eph receptors as the main stimuli of innervation could potentially be used in the future [165]. The main task solved by these aspects of innervation is to increase the supply of the tumor with various neurotransmitters. In a prostate cancer model, it has been shown that norepinephrine (NE) delivered from nerve terminals acts on b2- and b3-adrenergic receptors (Adrb2, Adrb) expressed on stromal cells, promoting the survival of cancer cells and the initial development of the tumor. Nerve fibers from the parasympathetic nervous system (PNS) also invade tumors, delivering acetylcholine (Ach), which promotes tumor cell proliferation and egress to lymph nodes and distant organs through the type 1 muscarinic receptor (Chrm1) expressed on stromal cells [166].
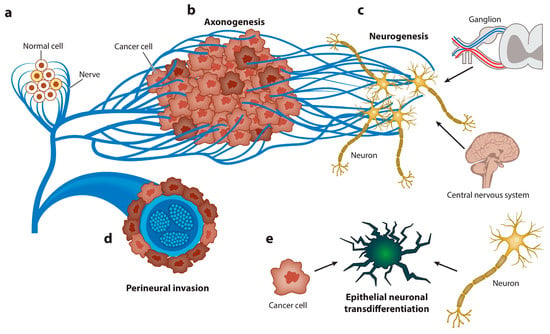
Figure 5.
Graphical representation of the spectrum of neuroepithelial interactions. Cancer recapitulates the biology of the neural regulation of epithelial tissues. (a) Nerves regulate the homeostasis and energetic metabolism of normal epithelial cells. (b) Axonogenesis from pre-existing nerves takes place to supply the new malignant epithelial growth. Cancer cells rarely develop in denervated organs, and denervation affects tumorigenesis in vivo and in humans. (c) Neurogenesis occurs later, first in ganglia around organs or the spinal column and, subsequently, through recruitment of neuroblasts from the central nervous system. The hallmark of this stage is the regulation of homeostasis and energetic metabolism. (d) Perineural invasion is the most efficient interaction between cancer cells and nerves. The hallmarks of this stage are increased proliferation and decreased apoptosis. (e) Finally, carcinoma cells transdifferentiate into a neuronal profile to gain neural independence [167].
Axonogenesis is among the aspects of tumor innervation [27]. The intensity of this process correlates with a poor treatment prognosis and the rate of tumor progression. It is not completely known what mechanisms underlie the connectivity of these processes. It is known that the extracellular release of neurotrophic factors, such as nerve growth factor [168], by tumor cells can promote cancer development [169]. Similarly, NGF is synthesized in tumor cells in the form of proNGF. Following intracellular internalization with subsequent furin protease cleavage or after extracellular procession by metalloproteases, proNGF is transformed to its matured form [170]. One of the main roles in the induction of tumor-associated axonogenesis is exerted by cancer-derived EVs. Madeo et al. determined that exosamal EphrinB1 induces and activates axonal outgrowth in tumors, enhancing tumor proliferation [28]. In addition, plasma-derived exosomal EphrinB1 isolated from human head and neck cancer specimens and mouse oropharyngeal squamous cell carcinoma caused increased neurite outgrowth in rat pheochromocytoma PC12 cells compared to exosomes derived from control plasma of healthy donors [171]. The involvement of exosomal miRNAs was shown in a study by Amit et al., in which the authors showed that genetically aberrant, p53-knockout or -mutant (p53C176F and p53A161S) oral squamous cell carcinoma (OCSCC) cells release EVs that promote neuritogenesis in the posterior ganglion roots. The effect was attributed to the miR-34a, miR-21, and miR-324 contained in exosomes [172].
Neurogenesis, as a process of de novo formation of neurons, is extremely difficult to study, especially within the framework of tumorigenesis. However, Mauffrey and colleagues expounded on this hypothesis, demonstrating that neural progenitor cells expressing the neural stem cell marker doublecortin (DCXþ) migrate from neurogenic regions of the brain’s subventricular zone (SVZ) to tumorous and metastatic niches via the bloodstream, differentiating into noradrenergic and mature neuronal phenotypes [173]. In addition, the acquisition of a neuron-like phenotype by cancer stem cells has also been demonstrated. These cells expressed autonomic nerve markers such as VAChT (a marker of parasympathetic neurons) or TH (tyrosine hydroxylase, which is characteristic of sympathetic neurons) [174].
To a particular extent, the transformation of nerve cells under the influence of tumors is relevant for the human brain environment, which is distinguished by a wide variety of cells, including astrocytes, macrophages, vascular endothelial cells, microglia, macrophages, and fibroblasts [175]. It is known that the development of a tumor near the nerve pathways is mediated by the growth in cancer cells around the peripheral nerves and, as a result, by intrusion into those nerves; this process is termed perineural invasion (PNI). In the case of tumors of the central nervous system, such a variant of development is unavoidable. Research in this area received further impetus with the discovery of close interactions between tumor cells and brain cells mediated by long membranous protrusions termed tumor microtubes (TMs) [176]. The most obvious factor in the transformation of neurons in CNS tumors is solid stress developed during the promotion of cell density, which can cause nonreversible and reversible nerve dysfunction [177]. The role of EVs in these processes is reduced to the formation of a pro-oncogenic microenvironment, often mediated by neuroinflammation. EVs released by glioblastoma cells induce migration and secretion of growth and pro-inflammatory factors in astrocytes. In addition, the regulation of the transcription factors TP53 and MYC by EVs induced the appearance of a senescence-associated secretory phenotype (SASP) in astrocytes [178].
4.3. Transformation of Immune Cells into Cancer-Associated Types
Immunological surveillance is one of the first barriers to tumor occurrence. However, with further tumor progression, immune cells can become supportive of tumor growth. A prime example is macrophages, in which the tumor-associated macrophage (TAM) phenotype has been identified [179]. TAMs are identified as protumor members of the microenvironment through immunohistochemical staining of neoplastic tissue for CD68 [180]. However, it has also been determined that the TAM population is heterogeneous and includes both M1- and M2-activated macrophages, exhibiting either protumorigenic or antitumorigenic effects, respectively. The overall effect of the TAM environment on the tumor is considered in the “macrophage balance hypothesis” paradigm [181]. Four approaches are commonly used to characterize macrophage subpopulations: analysis of cell surface marker expression, expression of transcription factors, production of cytokines, and production of specific enzymes [182]. The role of each population is still not defined, but they all been found to have a close relationship with the tumor cells and their microenvironment [183].
M2 polarization of macrophages is the main event associated with the formation of TAM and is characterized by the secretion of anti-inflammatory factors and pro-oncogenic activity [184]. It has been demonstrated that this process is mediated by EVs that contain certain cytokines (IL-6, TGF-b, IFN-a, and others) in various types of tumors [185]. In addition, M2 polarization is mediated by a vast ensemble of different RNAs contained in EVs [168]. M1 polarization is often associated with the secretion of pro-inflammatory factors and, accordingly, antitumor activity. The EVs of such macrophages enhance the therapeutic effect of, for example, paclitaxel [186]. The reprogramming of M1 macrophages into M2 and, accordingly, into TAMs can be carried out by EVs [168]. This process is similar to the natural processes that occur with macrophages during wound healing [187]. Reverse transformation of M2 macrophages into M1 is a promising strategy to enhance anticancer therapy [188]. Nonetheless, an increase in the TAM population in a tumor cannot only be facilitated by the EVs of tumor cells. For example, it has been shown that EVs secreted by human and mouse MSCs accelerated breast cancer progression by inducing differentiation of monocytic myeloid-derived suppressor cells (MDSCs) into highly immunosuppressive M2-polarized macrophages. The EVs of MSCs, in contrast to the EVs of tumor cells, contained TGF-β, C1q, and semaphorins, which induce the overexpression of PD-L1 in both immature myelo-monocytic precursors and committed CD206+ macrophages and, thereby, increase overall immune tolerogenicity [189].
The tumor-associated neutrophil (TAN) is another immune participant in the tumor microenvironment. In analogy with macrophages, a subpopulation of transformed neutrophils with protumor functions is termed N2 TAN [16]. Researchers attribute a central role in this process to TGF-b, the expression of which is controlled not only by the cytokine components of exosomes but also by miRNA [190]. In addition to this, an important exosomal component of this process is high-mobility group box-1 (HMGB1), which activates the NF-κB pathway through interaction with TLR4, resulting in an increased autophagic response in neutrophils [124]. The main antitumor effects of the N1 TAN population are associated with the activation of T cells, induction of apoptosis of tumor cells (via TRAIL), and production and secretion of ROS, as well as participation in ADCC [16]. Using a model of TAN transformation, it has been shown that the acquisition of a cancer-associated phenotype not only supports the tumor progression but also prolongs the life cycle of neutrophils through the induction of resistance to apoptosis [191].
Natural killer (NK) cells are another type of leukocyte capable of infiltrating tumors and acquiring a tumor-associated phenotype (TANKs) found in peripheral blood [18]. Like other immune cells, they exhibit a dual role in the TME [192]. They have been shown to play a role in enhancing angiogenesis and tumor invasion. It has also been demonstrated that a high content of TINKs in the TME in primary head and neck squamous cell carcinoma (HNSCC) correlates with a good prognosis [11,193]. TINKs have been characterized as CD56 bright, and they overexpress NKG2A, as well as lower levels of KIRs and leukocyte immunoglobulin-like receptor subfamily B member 1 (LILRB1) [19]. Tumor EVs primarily have an immunosuppressive effect on NK cells [194,195]. The main exosomal component responsible for reducing the cytotoxicity of natural killer cells is TGF-β1, which causes a decrease in the expression of activating receptors, including NKG2D, NKP30, NKP44, NPK46, and NKG2C [126]. It is noteworthy that the tumor mechanisms of the inhibition of NK activity through the release of NKG2DL are similar to the processes of embryonic development, when the placenta secretes NKG2DL to protect the fetus from the cytotoxic effects of NK [127,196]
Immune cells often assemble into tertiary lymphoid structures (TLSs) [197]. TLSs are organized aggregates of immune cells that form in nonlymphoid tissues associated with autoimmune reaction, chronic infection, cancer, and other diseases in the postnatal period characterized by an inner zone of CD20+ B cells that is surrounded by CD3+ T cells [198]. The molecular mechanisms by which tumor-derived EVs influence TLS formation remain unknown [199]. Thus, EVs are involved in the formation of TLSs at the initiation and maturation stages, and apoptotic bodies play a key role in these processes [200]. The function of TLSs in tumor progression remains unclear (Figure 6). On the one hand, TLSs duplicate the role of SLO and are involved in enhancing the immune response to cancer cells [201]. It has been shown that in primary melanoma, a high density of DC-Lamp+, a mature DC found within lymphoid aggregates, is associated with a strong infiltration of activated T cells and significantly higher rates of disease-free survival [202]. The anti- or protumorigenic role of DC is defined by its cell maturity status [203]. Immature DCs that have infiltrated into the tumor display low expression of costimulatory molecules (CD80 and CD86) and high expression of inhibitory molecules (PD-L1 and CTLA-4), and, therefore, create immunotolerogenic conditions within the TME [204]. Meanwhile, mature DCs in the TME display antitumor immune responses through the release of IFN-λ1, which stimulates Th1 differentiation and activation and effector CD8+ T-cell activation together with enhanced IFN-γ production through IL-12p70 production, which, as a result, increases overall survival [205]. On the other hand, a correlation has been found between late-stage progression of breast, bladder, or stomach cancer and the number of TLSs formed [206,207,208]. Clearly, the participation of TLSs in tumor progression depends, first of all, on cellular components [209] such as subpopulations of dendritic cells [210], T lymphocytes [211], and B lymphocytes [30,31]. Whether the effect of cancer EVs on TLS formation is inhibitory or enhancing is unknown. Despite this, TLSs remain promising prognostic markers and a potential target for therapy [197].
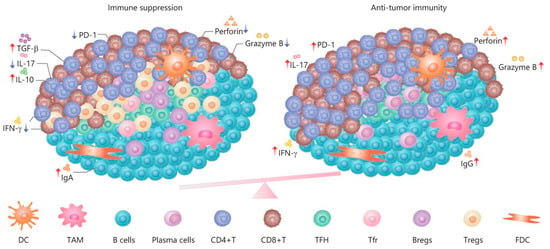
Figure 6.
The dual role and immune regulation in TLSs [212].
The remaining fractions of immune cells are much less studied and phenotypically identified in the light of tumor transformation, which is due to the lower contents of these cells in the tumor microenvironment [213].
4.4. Cancer-Derived EVs for Transformation of Other Cell Types
In addition to the previously described cells, the tumor microenvironment includes a number of other tumor-supporting cell types, including endothelial cells, adipocytes, and keratinocytes [214].
Endotheliocytes are the main cells of blood vessels and one of the main participants in angiogenesis, which, in turn, is targeted by many secreted factors including EVs [215]. Tumor interactions with endothelial cells induce their transformation into tumor endothelial cells (TECs) characterized morphologically by irregular surfaces, excessively fenestrated cell walls, and loose intercellular junctions to adjacent cells. They exhibit a stem-cell-like origin, thus playing a key role in tumor neo-angiogenesis [22]. The activation of VEGF signals released by tumor cells and the TME plays a central role in this process [216]. In addition, tumors have been shown to release exosomes that inhibit the mechanosensitive ion channel transient receptor vanilloid 4 (TRPV4), whose expression and activity is significantly reduced in tumor endothelial cells (TECs). The activation of TRPV4 has been shown to normalize the tumor vasculature and improved the efficacy of anticancer therapy [129].
Adipocytes are cells involved in energy metabolism. They are found in large numbers in the mammary glands and are therefore active participants in the TME in breast cancer [217]. A recently described phenotype of cancer-associated adipocytes (CAAs) (characterized by the production of CCL5, CCL2, IL-6, and TNF-α) has been shown to promote the proliferation and invasion of tumor cells, as well as neovascularization [25]. One of its key roles in CAA activation is attributed to exosomal miR-1304, which regulates GATA2 gene expression and enhances lipid release from adipocytes [130]. Subsequently, Munteanu et al. proposed treatment in other tumor models based on the downregulation of CAAs [218].
Keratinocytes, cells that are mainly located in the epidermis of the skin, are constitutive participants in the TME in various forms of melanoma [219]. In one study, Danella et al. reported that cancer-associated keratinocytes in a head and neck squamous cell carcinoma (HNSCC) model secreted TGFβ and TNFα, which enhanced the invasion of HNSCC cells [26].
Pericytes, together with smooth muscle cells, are mural cells serving to support and stabilize the endothelium. This explains their significant role in the processes of neoangiogenesis [220]. In competition with pericytes, cancer cells replace them and line blood vessels. In this case, detached pericytes undergo pericyte–fibroblast transition (PFT), which facilitates vascular penetration and metastasis of cancer cells [221]. Xiaofei Ning et al. showed that an exosome from gastric cancer induced the transformation of pericytes via the activation of the PI3K/AKT and MEK/ERK signaling pathways. On the contrary, the inhibition of the BMP pathway reversed cancer exosome-induced CAF transition [222].
Lymphatic endothelial cells (LECs) constitute the main cell type in lymph nodes and vessels. A major breakthrough in the study of lymphatic endothelial cells occurred with the discovery of their specific markers: 5’-nucleotidase, lymphatic vessel endothelial receptor-1, vascular endothelial growth factor receptor-3, podoplanin, and Prox-1 [223]. It was subsequently demonstrated that LECs are an integral part of the TME involved in processes of lymph metastasis. Phenotypic changes in LECs within the tumor supported the identification of structures called tumor-draining lymph nodes [32]. This resulted in the identification of the following processes: (1) lymphangiogenesis and the expansionof lymphatic sinuses, (2) dilation and dedifferentiation of high endothelial venules (HEVs), and (3) remodeling of fibroblastic reticular cells (FRCs) [224].
5. Therapeutic Applications of EVs Involved in TME Modulation
Close interactions of TME cellular components can be utilized in tumor diagnostics and therapy. Indeed EVs, as carriers of various molecules (mRNA, functional proteins, etc.), were shown to provide a real-time valuable biomarker platform. From what was described earlier, it is also clear that the role of EVs within TME interaction is considerable. The use of artificial target exosomes for tumor therapy is a well-studied strategy [225]. Such therapeutic exosomes have a number of advantages, such as biocompatibility and biodegradation, low toxicity, high stability, high permeability through cellular barriers, high specificity, accumulation in tumor tissues, and the potential for design [5]. Engineered EVs often target tumor cells directly, but the targeting of other TME cells for reprogramming has recently received a new impetus (Table 4).
Table 4.
Exosome-based TME reprogramming therapy.
As previously described, this strategy of cell reprogramming, compared with complete depletion using the example of CAFs, shows great promise [156]. Targeted reprogramming of individual TME cell types makes it possible to alter the composition of a solid tumor [229]. This, in turn, allows greater tumor susceptibility to chemo- and radiotherapy, immunotherapy, and other treatment strategies [233]. Unfortunately, EVs have currently only been developed for certain types of TME cells. Additionally, the arsenal of therapeutic agents is quite limited. We hope that increased understanding of cellular cross-talk within the TME will help to expand this tumor treatment strategy.
The application of EVs in TME modulation is not, however, limited to the depletion and reprogramming of cellular components. Modified exosomes with multivalent antibodies on the surface specific to T-cell CD3 and cancer-cell-associated EGFR redirect and activate cytotoxic T cells toward cancer cells for killing [234]. In addition, therapeutic exosomes aimed at metabolic reprogramming of the TME deserve special attention [235]. Furthermore, strategies are being developed in parallel to inhibit either the release of EVs or their uptake by TME cells [236]. The development of exosome-based vaccines deserves special attention, as reviewed in [237,238]. The clinical application of EVs in the treatment of tumors is becoming an increasingly promising tool. Many synthetic, natural, and combined EVs are undergoing clinical trials (Table 5). We believe that the development of exosome-based tumor therapy will be associated with the construction of more complex networks of interactions between TME components and more targeted effects. There have already been studies linking tumor gene therapy with subsequent TME remodeling induced by primary exposure [239].
Table 5.
EVs-based tumor therapy drugs.
6. Conclusions
Although great strides have been made in recent decades in the study of the transformation of the tumor microenvironment under the influence of cancer cells (mediated by EVs), specific mechanisms still remain poorly understood. The scientific research carried out to date has mainly focused on the study of a binary system, in which, on the one hand, there is a cancer cell, and on the other, a normal cell of the body, which undergoes certain changes under the action of tumor molecules. However, this system does not take into account the influences of various transformed cells of the tumor microenvironment (for example, endotheliocytes, CAFs, pericytes, and TAMs) on each other, which undoubtedly determine the progression of the tumor, its heterogeneity, and resistance to therapy. It is presumably these complex intercellular interactions that determine the occurrence of metastatic niches and the formation of microemboli (conglomerates of normal and cancer cells) [240]. Accordingly, such metastatic niche formation was shown to be mediated by EVs during the processes of apoptosis and senescence of CAFs due to the abundant release of inflammatory and growth factors [241,242]. In these aspects, TME may be regarded as an ecosystem, in which EVs play a significant role in the systemic tumor–host interplay, modulating interactions among cancer cells and their local microenvironment, distant organ niches, and nervous, endocrine, and immune systems [243,244].
It is also worth noting that cancer cells successfully hijack various physiological processes (wound healing and stem niche formation) in the body for cell recruitment and remodeling [245]. This may be due to the imitation of other pathological processes by the TME. Further study of the tumor-derived EVs may help to elucidate the complex underlying pathways of normal cell recruitment to the tumor site and subsequent transformation. The presence of many tumor-associated phenotypes of normal cells in a tumor site is associated with a complex biocenotic system. At the same time, EV crosstalk is one of the pathways of transformation of normal competitive cells into supporting tumor-associated cells.
Tumor-derived EVs have shown potential to be employed not only as detectable biomarkers for early diagnosis but also as a promising tool for targeted therapeutic strategies. The accumulated data clearly indicate that novel therapeutic strategies should include targeting the TME, which, in turn, may sever tumor resistance mechanisms and increase the efficacy of applied therapies.
Author Contributions
Conceptualization, A.T. and M.S.; methodology, A.T. and M.S.; software, A.T.; validation, A.T. and M.S.; formal analysis, A.T. and M.S.; investigation, A.T. and M.S.; resources, M.S.; data curation, A.T. and M.S.; writing—original draft preparation, A.T., V.F., V.K., H.G. and M.S.; writing—review and editing, A.T., V.F., V.K., H.G. and M.S.; visualization, A.T. and M.S.; supervision, M.S.; project administration, M.S.; funding acquisition, M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Ministry of Science and Higher Education of the Russian Federation (project # FZNS-2023-0017, chapters 1,2,3) and Russian academic leadership program Priority 2030, chapters 4,5,6).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings are available from the corresponding author.
Acknowledgments
The authors are grateful for Nan-Jong Lee for preparing Figure 1.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Li, I.; Nabet, B.Y. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 2019, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Goberdhan, D.C.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.; Erdbrügger, U. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef] [PubMed]
- Di Daniele, A.; Antonucci, Y.; Campello, S. Migrasomes, new vescicles as Hansel and Gretel white pebbles? Biol. Direct 2022, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K.J.M.c. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zheng, Y.; Zhao, M. Exosome-based cancer therapy: Implication for targeting cancer stem cells. Front. Pharmacol. 2017, 7, 533. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef]
- Prakash, J. The Tumor Stroma: Biology and Therapeutics; Jenny Stanford Publishing: Dubai, United Arab Emirates, 2022. [Google Scholar]
- Merlo, L.M.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef]
- Weil, S.; Memmer, S.; Lechner, A.; Huppert, V.; Giannattasio, A.; Becker, T.; Müller-Runte, A.; Lampe, K.; Beutner, D.; Quaas, A.; et al. Natural killer group 2D ligand depletion reconstitutes natural killer cell immunosurveillance of head and neck squamous cell carcinoma. Front. Immunol. 2017, 8, 387. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Weymouth, N.; Shi, Z. Smooth muscle α actin (Acta2) and myofibroblast function during hepatic wound healing. PLoS ONE 2013, 8, e77166. [Google Scholar] [CrossRef] [PubMed]
- Cortese, N.; Carriero, R.; Laghi, L.; Mantovani, A.; Marchesi, F. Prognostic significance of tumor-associated macrophages: Past, present and future. Semin. Immunol. 2020, 48, 101408. [Google Scholar] [CrossRef] [PubMed]
- Donadon, M.; Torzilli, G.; Cortese, N.; Soldani, C.; Di Tommaso, L.; Franceschini, B.; Carriero, R.; Barbagallo, M.; Rigamonti, A.; Anselmo, A.; et al. Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J. Exp. Med. 2020, 217, e20191847. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Albelda, S.M. Tumor-associated neutrophils: Friend or foe? Carcinogenesis 2012, 33, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhai, J.; Zhang, T.; Han, S.; Zhang, Y.; Yao, X.; Shen, L. Tumor-associated neutrophils can predict lymph node metastasis in early gastric cancer. Front. Oncol. 2020, 10, 570113. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Ferlazzo, G.; Albini, A.; Noonan, D. A think tank of TINK/TANKs: Tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J. Natl. Cancer Inst. 2014, 106, dju200. [Google Scholar] [CrossRef] [PubMed]
- Habif, G.; Crinier, A.; André, P.; Vivier, E.; Narni-Mancinelli, E. Targeting natural killer cells in solid tumors. Cell. Mol. Immunol. 2019, 16, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Ruffell, B. Dendritic cells and cancer immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef]
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36. [Google Scholar] [CrossRef]
- Nagl, L.; Horvath, L.; Pircher, A.; Wolf, D. Tumor endothelial cells (TECs) as potential immune directors of the tumor microenvironment–new findings and future perspectives. Front. Cell Dev. Biol. 2020, 8, 766. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Ohga, N.; Akiyama, K.; Maishi, N.; Hida, Y. Heterogeneity of tumor endothelial cells. Cancer Sci. 2013, 104, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Takehara, M.; Sato, Y.; Kimura, T.; Noda, K.; Miyamoto, H.; Fujino, Y.; Miyoshi, J.; Nakamura, F.; Wada, H.; Bando, Y.; et al. Cancer-associated adipocytes promote pancreatic cancer progression through SAA1 expression. Cancer Sci. 2020, 111, 2883–2894. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Danella, E.B.; Costa De Medeiros, M.; D’Silva, N.J. Cancer-associated keratinocytes: New members of the microenvironment in head and neck cancer. Mol. Cell. Oncol. 2021, 8, 1933329. [Google Scholar] [CrossRef] [PubMed]
- Entschladen, F.; Palm, D.; Niggemann, B.; Zaenker, K.S. The cancer’s nervous tooth: Considering the neuronal crosstalk within tumors. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2008; pp. 171–175. [Google Scholar]
- Madeo, M.; Colbert, P.L.; Vermeer, D.W.; Lucido, C.T.; Cain, J.T.; Vichaya, E.G.; Grossberg, A.J.; Muirhead, D.; Rickel, A.P.; Hong, Z.; et al. Cancer exosomes induce tumor innervation. Nat. Commun. 2018, 9, 4284. [Google Scholar] [CrossRef]
- Rahir, G.; Moser, M. Tumor microenvironment and lymphocyte infiltration. Cancer Immunol. Immunother. 2012, 61, 751–759. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef]
- Tsou, P.; Katayama, H.; Ostrin, E.J.; Hanash, S.M. The emerging role of B cells in tumor immunity. Cancer Res. 2016, 76, 5597–5601. [Google Scholar] [CrossRef]
- He, M.; He, Q.; Cai, X.; Chen, Z.; Lao, S.; Deng, H.; Liu, X.; Zheng, Y.; Liu, X.; Liu, J.; et al. Role of lymphatic endothelial cells in the tumor microenvironment—A narrative review of recent advances. Transl. Lung Cancer Res. 2021, 10, 2252. [Google Scholar] [CrossRef]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef]
- Zhou, L.; Li, J.; Tang, Y.; Yang, M. Exosomal LncRNA LINC00659 transferred from cancer-associated fibroblasts promotes colorectal cancer cell progression via miR-342-3p/ANXA2 axis. J. Transl. Med. 2021, 19, 8. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S. Immune surveillance: A balance between protumor and antitumor immunity. Curr. Opin. Genet. Dev. 2008, 18, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Hao, Y.; He, C.; Li, L.; Zhu, G. Exosome-orchestrated hypoxic tumor microenvironment. Mol. Cancer 2019, 18, 57. [Google Scholar] [CrossRef]
- Kyuno, D.; Takasawa, A.; Kikuchi, S.; Takemasa, I.; Osanai, M.; Kojima, T. Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells. Biochim. Biophys. Acta (BBA)-Biomembr. 2021, 1863, 183503. [Google Scholar] [CrossRef]
- Stover, D.G.; Bierie, B.; Moses, H.L. A delicate balance: TGF-β and the tumor microenvironment. J. Cell. Biochem. 2007, 101, 851–861. [Google Scholar] [CrossRef]
- Joshi, A.; Cao, D. TGF-β signaling, tumor microenvironment and tumor progression: The butterfly effect. Front. Biosci. 2010, 15, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Pavlović, N.; Heindryckx, F. Targeting ER stress in the hepatic tumor microenvironment. FEBS J. 2022, 289, 7163–7176. [Google Scholar] [CrossRef]
- Cook, J.A.; Gius, D.; Wink, D.A.; Krishna, M.C.; Russo, A.; Mitchell, J.B. Oxidative stress, redox, and the tumor microenvironment. In Seminars in Radiation Oncology; WB Saunders: Philadelphia, PA, USA, 2004; pp. 259–266. [Google Scholar]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef]
- Khawar, I.A.; Kim, J.H.; Kuh, H.-J. Improving drug delivery to solid tumors: Priming the tumor microenvironment. J. Control. Release 2015, 201, 78–89. [Google Scholar] [CrossRef]
- Olmo, D.G.; Olmo, D.G.; Ontanon, J.; Martinez, E.; Vallejo, M. Tumor DNA circulating in the plasma might play a role in metastasis. The hypothesis of the genometastasis. Histol. Histopathol. 1999, 14, 1159–1164. [Google Scholar]
- García-Olmo, D.; García-Olmo, D.C.; Ontañón, J.; Martinez, E. Horizontal transfer of DNA and the “genometastasis hypothesis”. Blood J. Am. Soc. Hematol. 2000, 95, 724–725. [Google Scholar] [CrossRef]
- García-Olmo, D.; García-Olmo, D.C. Functionality of circulating DNA: The hypothesis of genometastasis. Ann. N. Y. Acad. Sci. 2001, 945, 265–275. [Google Scholar] [CrossRef] [PubMed]
- García-Casas, A.; García-Olmo, D.C.; García-Olmo, D. Further the liquid biopsy: Gathering pieces of the puzzle of genometastasis theory. World J. Clin. Oncol. 2017, 8, 378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ji, Q.; Yang, Y.; Li, Q.; Wang, Z. Exosome: Function and role in cancer metastasis and drug resistance. Technol. Cancer Res. Treat. 2018, 17, 1533033818763450. [Google Scholar] [CrossRef] [PubMed]
- Yagüe, E.; Arance, A.; Kubitza, L.; O’Hare, M.; Jat, P.; Ogilvie, C.M.; Hart, I.R.; Higgins, C.F.; Raguz, S. Ability to acquire drug resistance arises early during the tumorigenesis process. Cancer Res. 2007, 67, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, J.; Wu, H.; Lu, Q.; Tai, Y.; Liu, Q.; Wang, C. Oncogenic transformation of normal breast epithelial cells co-cultured with cancer cells. Cell Cycle 2018, 17, 2027–2040. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2015; pp. 41–51. [Google Scholar]
- Zhou, Y.; Ren, H.; Dai, B.; Li, J.; Shang, L.; Huang, J.; Shi, X. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J. Exp. Clin. Cancer Res. 2018, 37, 324. [Google Scholar] [CrossRef]
- Dai, X.; Chen, C.; Yang, Q.; Xue, J.; Chen, X.; Sun, B.; Luo, F.; Liu, X.; Xiao, T.; Xu, H.; et al. Exosomal circRNA_100284 from arsenite-transformed cells, via microRNA-217 regulation of EZH2, is involved in the malignant transformation of human hepatic cells by accelerating the cell cycle and promoting cell proliferation. Cell Death Dis. 2018, 9, 454. [Google Scholar] [CrossRef]
- Zhang, M.; Li, M.; Li, N.; Zhang, Z.; Liu, N.; Han, X.; Liu, Q.; Liao, C. miR-217 suppresses proliferation, migration, and invasion promoting apoptosis via targeting MTDH in hepatocellular carcinoma. Oncol. Rep. 2017, 37, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhu, Z.; Xie, J.; Feng, Z.; Li, Y.; Xu, X.; Li, W.; Chen, W. Hsa_circ_0000069 knockdown inhibits tumorigenesis and exosomes with downregulated hsa_circ_0000069 suppress malignant transformation via inhibition of STIL in pancreatic cancer. Int. J. Nanomed. 2020, 15, 9859–9873. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sun, F.; Jin, J.; Xu, W.; Qian, H. Exosomal LncRNAs in Gastrointestinal Cancer: Biological Functions and Emerging Clinical Applications. Cancers 2023, 15, 959. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Zhang, J.; Cao, T.; Chen, B.; Tian, Y.; Shi, Y. Exosome-mediated lncRNA SND1-IT1 from gastric cancer cells enhances malignant transformation of gastric mucosa cells via up-regulating SNAIL1. J. Transl. Med. 2022, 20, 284. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Lu, J.; Mercer, K.L.; Golub, T.R.; Jacks, T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 2007, 39, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Gu, H.; Xiong, X.; Ao, H.; Cao, J.; Lin, W.; Yu, M.; Lin, J.; Cui, Q. MicroRNAs involved in carcinogenesis, prognosis, therapeutic resistance, and applications in human triple-negative breast cancer. Cells 2019, 8, 1492. [Google Scholar] [CrossRef]
- Wu, F.; Yang, J.; Shang, G.; Zhang, Z.; Niu, S.; Liu, Y.; Liu, H.; Jing, J.; Fang, Y. Exosomal miR-224-5p from colorectal cancer cells promotes malignant transformation of human normal colon epithelial cells by promoting cell proliferation through downregulation of CMTM4. Oxid. Med. Cell. Longev. 2022, 2022, 5983629. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, W.; Wang, Z.; Wang, C.; Ai, Z. Exosomal ANXA1 derived from thyroid cancer cells is associated with malignant transformation of human thyroid follicular epithelial cells by promoting cell proliferation. Int. J. Oncol. 2021, 59, 104. [Google Scholar] [CrossRef] [PubMed]
- Stefanius, K.; Servage, K.; de Souza Santos, M.; Gray, H.F.; Toombs, J.E.; Chimalapati, S.; Kim, M.S.; Malladi, V.S.; Brekken, R.; Orth, K. Human pancreatic cancer cell exosomes, but not human normal cell exosomes, act as an initiator in cell transformation. Elife 2019, 8, e40226. [Google Scholar] [CrossRef]
- Abdouh, M.; Hamam, D.; Gao, Z.-H.; Arena, V.; Arena, M.; Arena, G.O. Exosomes isolated from cancer patients’ sera transfer malignant traits and confer the same phenotype of primary tumors to oncosuppressor-mutated cells. J. Exp. Clin. Cancer Res. 2017, 36, 113. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Abd Elmageed, Z.Y.; Yang, Y.; Thomas, R.; Ranjan, M.; Mondal, D.; Moroz, K.; Fang, Z.; Rezk, B.M.; Moparty, K.; Sikka, S.C. Neoplastic reprogramming of patient-derived adipose stem cells by prostate cancer cell-associated exosomes. Stem Cells 2014, 32, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Gyukity-Sebestyén, E.; Harmati, M.; Dobra, G.; Németh, I.B.; Mihály, J.; Zvara, Á.; Hunyadi-Gulyás, É.; Katona, R.; Nagy, I.; Horváth, P. Melanoma-derived exosomes induce PD-1 overexpression and tumor progression via mesenchymal stem cell oncogenic reprogramming. Front. Immunol. 2019, 10, 461312. [Google Scholar] [CrossRef] [PubMed]
- Likonen, D.; Pinchasi, M.; Beery, E.; Sarsor, Z.; Signorini, L.F.; Gervits, A.; Sharan, R.; Lahav, M.; Raanani, P.; Uziel, O. Exosomal telomerase transcripts reprogram the microRNA transcriptome profile of fibroblasts and partially contribute to CAF formation. Sci. Rep. 2022, 12, 16415. [Google Scholar] [CrossRef] [PubMed]
- Strnadová, K.; Pfeiferová, L.; Přikryl, P.; Dvořánková, B.; Vlčák, E.; Frýdlová, J.; Vokurka, M.; Novotný, J.; Šáchová, J.; Hradilová, M. Exosomes produced by melanoma cells significantly influence the biological properties of normal and cancer-associated fibroblasts. Histochem. Cell Biol. 2022, 157, 153–172. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Qian, J.; Tan, S.; Li, W.; Liu, P.; Zhao, J.; Zeng, Y.; Xu, L.; Wang, Z.; Cai, J. Tumor cell-derived exosomes deliver TIE2 protein to macrophages to promote angiogenesis in cervical cancer. Cancer Lett. 2022, 529, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Yang, Y.; Yang, W.; Han, Y.; Huang, L.; Yang, R.; Hu, Z.; Tao, Y.; Liu, L.; Li, Y.; et al. Exosomal cargos-mediated metabolic reprogramming in tumor microenvironment. J. Exp. Clin. Cancer Res. 2023, 42, 59. [Google Scholar] [CrossRef]
- Yan, W.; Wu, X.; Zhou, W.; Fong, M.Y.; Cao, M.; Liu, J.; Liu, X.; Chen, C.-H.; Fadare, O.; Pizzo, D.P. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat. Cell Biol. 2018, 20, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, B.; Rodríguez, M.; San Millán, C.; García, V.; Fernandez-Perianez, R.; Gil-Calderon, B.; Martín, P.; García-Grande, A.; Silva, J.; Bonilla, F. Tumor-derived exosomes are enriched in ΔNp73, which promotes oncogenic potential in acceptor cells and correlates with patient survival. Hum. Mol. Genet. 2014, 23, 467–478. [Google Scholar] [CrossRef]
- Shimada, H.; Chatten, J.; Newton, W.A.; Sachs, N.; Hamoudi, A.B.; Chiba, T.; Marsden, H.B.; Misugi, K. Histopathologic prognostic factors in neuroblastic tumors: Definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. JNCI J. Natl. Cancer Inst. 1984, 73, 405–416. [Google Scholar] [CrossRef]
- Hatina, J. The dynamics of cancer stem cells. Neoplasma 2012, 59, 700. [Google Scholar] [CrossRef] [PubMed]
- Safri, F.; Nguyen, R.; Zerehpooshnesfchi, S.; George, J.; Qiao, L. Heterogeneity of hepatocellular carcinoma: From mechanisms to clinical implications. Cancer Gene Ther. 2024, 1–8. [Google Scholar] [CrossRef]
- Das, K.; Paul, S.; Singh, A.; Ghosh, A.; Roy, A.; Ansari, S.A.; Prasad, R.; Mukherjee, A.; Sen, P. Triple-negative breast cancer-derived microvesicles transfer microRNA221 to the recipient cells and thereby promote epithelial-to-mesenchymal transition. J. Biol. Chem. 2019, 294, 13681–13696. [Google Scholar] [CrossRef]
- Das, K.; Prasad, R.; Singh, A.; Bhattacharya, A.; Roy, A.; Mallik, S.; Mukherjee, A.; Sen, P. Protease-activated receptor 2 promotes actomyosin dependent transforming microvesicles generation from human breast cancer. Mol. Carcinog. 2018, 57, 1707–1722. [Google Scholar] [CrossRef]
- Das, K.; Prasad, R.; Roy, S.; Mukherjee, A.; Sen, P. The protease activated receptor2 promotes Rab5a mediated generation of pro-metastatic microvesicles. Sci. Rep. 2018, 8, 7357. [Google Scholar] [CrossRef]
- Zhang, X.; Lou, Y.; Wang, H.; Zheng, X.; Dong, Q.; Sun, J.; Han, B. Wnt signaling regulates the stemness of lung cancer stem cells and its inhibitors exert anticancer effect on lung cancer SPC-A1 cells. Med. Oncol. 2015, 32, 95. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Wang, S.; Zhao, R.C. Exosomes from human adipose-derived mesenchymal stem cells promote migration through Wnt signaling pathway in a breast cancer cell model. Mol. Cell. Biochem. 2013, 383, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef]
- Figueroa, J.; Phillips, L.M.; Shahar, T.; Hossain, A.; Gumin, J.; Kim, H.; Bean, A.J.; Calin, G.A.; Fueyo, J.; Walters, E.T. Exosomes from Glioma-Associated Mesenchymal Stem Cells Increase the Tumorigenicity of Glioma Stem-like Cells via Transfer of miR-1587GA-hMSCs Regulate GSCs via Exosomal miRNA. Cancer Res. 2017, 77, 5808–5819. [Google Scholar] [CrossRef]
- Yoshida, K.; Yamamoto, Y.; Ochiya, T. miRNA signaling networks in cancer stem cells. Regen. Ther. 2021, 17, 1–7. [Google Scholar] [CrossRef]
- Zhan, Y.; Du, J.; Min, Z.; Ma, L.; Zhang, W.; Zhu, W.; Liu, Y. Carcinoma-associated fibroblasts derived exosomes modulate breast cancer cell stemness through exonic circHIF1A by miR-580-5p in hypoxic stress. Cell Death Discov. 2021, 7, 141. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef]
- Raimondo, S.; Saieva, L.; Corrado, C.; Fontana, S.; Flugy, A.; Rizzo, A.; De Leo, G.; Alessandro, R. Chronic myeloid leukemia-derived exosomes promote tumor growth through an autocrine mechanism. Cell Commun. Signal. 2015, 13, 8. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; ten Dijke, P. Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 2013, 38, 612–620. [Google Scholar] [CrossRef]
- Ung, T.H.; Madsen, H.J.; Hellwinkel, J.E.; Lencioni, A.M.; Graner, M.W. Exosome proteomics reveals transcriptional regulator proteins with potential to mediate downstream pathways. Cancer Sci. 2014, 105, 1384–1392. [Google Scholar] [CrossRef]
- Liu, X.; Sun, H.; Qi, J.; Wang, L.; He, S.; Liu, J.; Feng, C.; Chen, C.; Li, W.; Guo, Y. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT–MET mechanism for optimal reprogramming. Nat. Cell Biol. 2013, 15, 829–838. [Google Scholar] [CrossRef]
- Chen, J.; Han, Q.; Pei, D. EMT and MET as paradigms for cell fate switching. J. Mol. Cell Biol. 2012, 4, 66–69. [Google Scholar] [CrossRef]
- Qiao, D.; Zeeman, A.-M.; Deng, W.; Looijenga, L.H.; Lin, H. Molecular characterization of hiwi, a human member of the piwi gene family whose overexpression is correlated to seminomas. Oncogene 2002, 21, 3988–3999. [Google Scholar] [CrossRef]
- Liu, X.; Sun, Y.; Guo, J.; Ma, H.; Li, J.; Dong, B.; Jin, G.; Zhang, J.; Wu, J.; Meng, L. Expression of hiwi gene in human gastric cancer was associated with proliferation of cancer cells. Int. J. Cancer 2006, 118, 1922–1929. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Schütte, D.; Wulf, G.; Füzesi, L.; Radzun, H.-J.; Schweyer, S.; Engel, W.; Nayernia, K. Stem-cell protein Piwil2 is widely expressed in tumors and inhibits apoptosis through activation of Stat3/Bcl-XL pathway. Hum. Mol. Genet. 2006, 15, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette III, L.J.; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K. Landscape of somatic retrotransposition in human cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef]
- Gernapudi, R.; Yao, Y.; Zhang, Y.; Wolfson, B.; Roy, S.; Duru, N.; Eades, G.; Yang, P.; Zhou, Q. Targeting exosomes from preadipocytes inhibits preadipocyte to cancer stem cell signaling in early-stage breast cancer. Breast Cancer Res. Treat. 2015, 150, 685–695. [Google Scholar] [CrossRef]
- Ou, B.; Liu, Y.; Gao, Z.; Xu, J.; Yan, Y.; Li, Y.; Zhang, J. Senescent neutrophils-derived exosomal piRNA-17560 promotes chemoresistance and EMT of breast cancer via FTO-mediated m6A demethylation. Cell Death Dis. 2022, 13, 905. [Google Scholar] [CrossRef]
- Yin, J.; Jiang, X.Y.; Qi, W.; Ji, C.G.; Xie, X.L.; Zhang, D.X.; Cui, Z.J.; Wang, C.K.; Bai, Y.; Wang, J. piR-823 contributes to colorectal tumorigenesis by enhancing the transcriptional activity of HSF 1. Cancer Sci. 2017, 108, 1746–1756. [Google Scholar] [CrossRef]
- Ai, L.; Mu, S.; Sun, C.; Fan, F.; Yan, H.; Qin, Y.; Cui, G.; Wang, Y.; Guo, T.; Mei, H. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol. Cancer 2019, 18, 88. [Google Scholar] [CrossRef]
- Cheng, J.; Deng, H.; Xiao, B.; Zhou, H.; Zhou, F.; Shen, Z.; Guo, J. piR-823, a novel non-coding small RNA, demonstrates in vitro and in vivo tumor suppressive activity in human gastric cancer cells. Cancer Lett. 2012, 315, 12–17. [Google Scholar] [CrossRef]
- Ding, X.; Li, Y.; Lü, J.; Zhao, Q.; Guo, Y.; Lu, Z.; Ma, W.; Liu, P.; Pestell, R.G.; Liang, C. piRNA-823 is involved in cancer stem cell regulation through altering DNA methylation in association with luminal breast cancer. Front. Cell Dev. Biol. 2021, 9, 641052. [Google Scholar] [CrossRef]
- Tong, Y.; Guan, B.; Sun, Z.; Dong, X.; Chen, Y.; Li, Y.; Jiang, Y.; Li, J. Ratiometric fluorescent detection of exosomal piRNA-823 based on Au NCs/UiO-66-NH2 and target-triggered rolling circle amplification. Talanta 2023, 257, 124307. [Google Scholar] [CrossRef]
- Zhu, Y.H.; Bulavin, D.V. Wip1-dependent signaling pathways in health and diseases. Prog. Mol. Biol. Transl. Sci. 2012, 106, 307–325. [Google Scholar] [PubMed]
- Lee, J.-S.; Lee, M.-O.; Moon, B.-H.; Shim, S.H.; Fornace, A.J., Jr.; Cha, H.-J. Senescent growth arrest in mesenchymal stem cells is bypassed by Wip1-mediated downregulation of intrinsic stress signaling pathways. Stem Cells 2009, 27, 1963–1975. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Tumor microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef]
- Lu, X.; Nguyen, T.-A.; Moon, S.-H.; Darlington, Y.; Sommer, M.; Donehower, L.A. The type 2C phosphatase Wip1: An oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008, 27, 123–135. [Google Scholar] [CrossRef]
- Deng, K.; Liu, L.; Tan, X.; Zhang, Z.; Li, J.; Ou, Y.; Wang, X.; Yang, S.; Xiang, R.; Sun, P. WIP1 promotes cancer stem cell properties by inhibiting p38 MAPK in NSCLC. Signal Transduct. Target. Ther. 2020, 5, 36. [Google Scholar] [CrossRef]
- Chen, W.; Tang, D.; Lin, J.; Huang, X.; Lin, S.; Shen, G.; Dai, Y. Exosomal circSHKBP1 participates in non-small cell lung cancer progression through PKM2-mediated glycolysis. Mol. Ther.–Oncolytics 2022, 24, 470–485. [Google Scholar] [CrossRef]
- Ju, F.; Atyah, M.M.; Horstmann, N.; Gul, S.; Vago, R.; Bruns, C.J.; Zhao, Y.; Dong, Q.-Z.; Ren, N. Characteristics of the cancer stem cell niche and therapeutic strategies. Stem Cell Res. Ther. 2022, 13, 233. [Google Scholar] [CrossRef]
- Rezayatmand, H.; Razmkhah, M.; Razeghian-Jahromi, I. Drug resistance in cancer therapy: The Pandora’s Box of cancer stem cells. Stem Cell Res. Ther. 2022, 13, 181. [Google Scholar] [CrossRef]
- Morrison, R.; Schleicher, S.M.; Sun, Y.; Niermann, K.J.; Kim, S.; Spratt, D.E.; Chung, C.H.; Lu, B. Targeting the mechanisms of resistance to chemotherapy and radiotherapy with the cancer stem cell hypothesis. J. Oncol. 2011, 2011, 941876. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Sachdeva, R.; Wu, M.; Johnson, K.; Kim, H.; Celebre, A.; Shahzad, U.; Graham, M.S.; Kessler, J.A.; Chuang, J.H.; Karamchandani, J. BMP signaling mediates glioma stem cell quiescence and confers treatment resistance in glioblastoma. Sci. Rep. 2019, 9, 14569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, D.; Shen, L.; Hu, D.; Tang, B.; Guo, W.; Wang, Z.; Zhang, Z.; Wei, G.; He, D. Exosomes derived from Piwil2-induced cancer stem cells transform fibroblasts into cancer-associated fibroblasts Corrigendum in/10.3892/or. 2022.8273. Oncol. Rep. 2020, 43, 1125–1132. [Google Scholar] [PubMed]
- Hu, T.; Hu, J. Melanoma-derived exosomes induce reprogramming fibroblasts into cancer-associated fibroblasts via Gm26809 delivery. Cell Cycle 2019, 18, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-S.; Ma, S.; Dou, H.; Liu, F.; Zhang, S.-Y.; Jiang, C.; Xiao, M.; Huang, Y.-X. Breast cancer-derived exosomes regulate cell invasion and metastasis in breast cancer via miR-146a to activate cancer associated fibroblasts in tumor microenvironment. Exp. Cell Res. 2020, 391, 111983. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guan, X.; Zhang, Y.; Ge, S.; Zhang, L.; Li, H.; Wang, X.; Liu, R.; Ning, T.; Deng, T. Exosomal miR-27a derived from gastric cancer cells regulates the transformation of fibroblasts into cancer-associated fibroblasts. Cell. Physiol. Biochem. 2018, 49, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Long, X.; Duan, S.; Liu, X.; Chen, J.; Lan, J.; Liu, X.; Huang, W.; Geng, J.; Zhou, J. CSRP2 suppresses colorectal cancer progression via p130Cas/Rac1 axis-meditated ERK, PAK, and HIPPO signaling pathways. Theranostics 2020, 10, 11063. [Google Scholar] [CrossRef] [PubMed]
- Gutkin, A.; Uziel, O.; Beery, E.; Nordenberg, J.; Pinchasi, M.; Goldvaser, H.; Henick, S.; Goldberg, M.; Lahav, M. Tumor cells derived exosomes contain hTERT mRNA and transform nonmalignant fibroblasts into telomerase positive cells. Oncotarget 2016, 7, 59173. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-L.; Hung, J.-Y.; Chang, W.-A.; Jian, S.-F.; Lin, Y.-S.; Pan, Y.-C.; Wu, C.-Y.; Kuo, P.-L. Hypoxic lung-cancer-derived extracellular vesicle microRNA-103a increases the oncogenic effects of macrophages by targeting PTEN. Mol. Ther. 2018, 26, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Qiao, B.; Gao, N.; Lin, N.; He, W. Oral squamous cell carcinoma-derived exosomes promote M2 subtype macrophage polarization mediated by exosome-enclosed miR-29a-3p. Am. J. Physiol.-Cell Physiol. 2019, 316, C731–C740. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Chen, T.; Zheng, X.; Yang, S.; Xu, K.; Chen, X.; Xu, F.; Wang, L.; Shen, Y.; Wang, T. Colorectal cancer-derived small extracellular vesicles establish an inflammatory premetastatic niche in liver metastasis. Carcinogenesis 2018, 39, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Shang, A.; Gu, C.; Liu, W.; Wang, X.; Zeng, B.; Chen, C.; Chang, W.; Ping, Y.; Sun, J.; Ji, P. Exosomal KRAS mutation promotes promotion of colorectal cancer by the formation of tumor-associated neutrophil extracellular traps. Electronic 2019, 10, 2617. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Yuan, X.; Jiang, P.; Qian, H.; Xu, W. Tumor-derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration. Mol. Cancer 2018, 17, 146. [Google Scholar] [CrossRef]
- Wang, L.; Shan, Y.; Zheng, S.; Li, J.; Zhu, A. Exosomal circ-CTNNB1 derived from colorectal cancer cells induces N2 polarization of neutrophils to promote colorectal cancer cell growth and immune escape. Biomed. Signal Process. Control 2023, 85, 104960. [Google Scholar] [CrossRef]
- Wang, L.; Sun, Z.; Wang, H. Extracellular vesicles and the regulation of tumor immunity: Current progress and future directions. J. Cell. Biochem. 2021, 122, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [PubMed]
- Briand, J.; Garnier, D.; Nadaradjane, A.; Clément-Colmou, K.; Potiron, V.; Supiot, S.; Bougras-Cartron, G.; Frenel, J.-S.; Heymann, D.; Vallette, F.M. Radiotherapy-induced overexpression of exosomal miRNA-378a-3p in cancer cells limits natural killer cells cytotoxicity. Epigenomics 2020, 12, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Guarino, B.D.; Adapala, R.K.; Kanugula, A.K.; Lenkey, N.M.; Dougherty, J.A.; Paruchuri, S.; Khan, M.; Thodeti, C.K. Extracellular vesicles from pathological microenvironment induce endothelial cell transformation and abnormal angiogenesis via modulation of TRPV4 channels. Front. Cell Dev. Biol. 2019, 7, 344. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Wu, K.; Sharma, S.; Xing, F.; Wu, S.-Y.; Tyagi, A.; Deshpande, R.; Singh, R.; Wabitsch, M.; Mo, Y.-Y.; et al. Exosomal miR-1304-3p promotes breast cancer progression in African Americans by activating cancer-associated adipocytes. Nat. Commun. 2022, 13, 7734. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and biology of cancer-associated fibroblasts. Physiol. Rev. 2020, 101, 147–176. [Google Scholar] [CrossRef]
- Mhaidly, R.; Mechta-Grigoriou, F. Fibroblast heterogeneity in tumor micro-environment: Role in immunosuppression and new therapies. Semin. Immunol. 2020, 48, 101417. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [PubMed]
- Givel, A.-M.; Kieffer, Y.; Scholer-Dahirel, A.; Sirven, P.; Cardon, M.; Pelon, F.; Magagna, I.; Gentric, G.; Costa, A.; Bonneau, C. miR200-regulated CXCL12β promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat. Commun. 2018, 9, 1056. [Google Scholar] [CrossRef] [PubMed]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.-M.; Attieh, Y.; Barbazan, J. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020, 11, 404. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M. Inter-and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Tauriello, D.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef]
- Yang, F.; Ning, Z.; Ma, L.; Liu, W.; Shao, C.; Shu, Y.; Shen, H. Exosomal miRNAs and miRNA dysregulation in cancer-associated fibroblasts. Mol. Cancer 2017, 16, 148. [Google Scholar] [CrossRef]
- Hassan, M.S.; Cwidak, N.; Awasthi, N.; von Holzen, U. Cytokine interaction with cancer-associated fibroblasts in esophageal cancer. Cancer Control 2022, 29, 10732748221078470. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Hu, C.; Liu, P.; Lu, M. Tumor-derived exosomes in cancer metastasis risk diagnosis and metastasis therapy. Clin. Transl. Oncol. 2019, 21, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, X.; Zhang, B.; Shi, H.; Yuan, X.; Sun, Y.; Pan, Z.; Qian, H.; Xu, W. Exosomes derived from gastric cancer cells activate NF-κB pathway in macrophages to promote cancer progression. Tumor Biol. 2016, 37, 12169–12180. [Google Scholar] [CrossRef] [PubMed]
- Gangoda, L.; Liem, M.; Ang, C.S.; Keerthikumar, S.; Adda, C.G.; Parker, B.S.; Mathivanan, S. Proteomic profiling of exosomes secreted by breast cancer cells with varying metastatic potential. Proteomics 2017, 17, 1600370. [Google Scholar] [CrossRef] [PubMed]
- Sakha, S.; Muramatsu, T.; Ueda, K.; Inazawa, J. Exosomal microRNA miR-1246 induces cell motility and invasion through the regulation of DENND2D in oral squamous cell carcinoma. Sci. Rep. 2016, 6, 38750. [Google Scholar] [CrossRef]
- Tong, Y.; Yang, L.; Yu, C.; Zhu, W.; Zhou, X.; Xiong, Y.; Wang, W.; Ji, F.; He, D.; Cao, X. Tumor-secreted exosomal lncRNA POU3F3 promotes cisplatin resistance in ESCC by inducing fibroblast differentiation into CAFs. Mol. Ther.-Oncolytics 2020, 18, 1–13. [Google Scholar] [CrossRef]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef]
- Costea, D.E.; Hills, A.; Osman, A.H.; Thurlow, J.; Kalna, G.; Huang, X.; Pena Murillo, C.; Parajuli, H.; Suliman, S.; Kulasekara, K.K. Identification of two distinct carcinoma-associated fibroblast subtypes with differential tumor-promoting abilities in oral squamous cell carcinoma. Cancer Res. 2013, 73, 3888–3901. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.; Liu, X.; Kammertoens, T.; Blankenstein, T.; Qin, Z. Fibroblast-specific protein 1/S100A4–positive cells prevent carcinoma through collagen production and encapsulation of carcinogens. Cancer Res. 2013, 73, 2770–2781. [Google Scholar] [CrossRef]
- Iida, T.; Mizutani, Y.; Esaki, N.; Ponik, S.M.; Burkel, B.M.; Weng, L.; Kuwata, K.; Masamune, A.; Ishihara, S.; Haga, H. Pharmacologic conversion of cancer-associated fibroblasts from a protumor phenotype to an antitumor phenotype improves the sensitivity of pancreatic cancer to chemotherapeutics. Oncogene 2022, 41, 2764–2777. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef]
- Rimal, R.; Desai, P.; Daware, R.; Hosseinnejad, A.; Prakash, J.; Lammers, T.; Singh, S. Cancer-associated fibroblasts: Origin, function, imaging, and therapeutic targeting. Adv. Drug Deliv. Rev. 2022, 189, 114504. [Google Scholar] [CrossRef] [PubMed]
- Haubeiss, S.; Schmid, J.O.; Mürdter, T.E.; Sonnenberg, M.; Friedel, G.; van der Kuip, H.; Aulitzky, W.E. Dasatinib reverses cancer-associated fibroblasts (CAFs) from primary lung carcinomas to a phenotype comparable to that of normal fibroblasts. Mol. Cancer 2010, 9, 168. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, E.; Wu, N.C.; Cadavid, J.L.; McGuigan, A.P. The life cycle of cancer-associated fibroblasts within the tumour stroma and its importance in disease outcome. Br. J. Cancer 2020, 122, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Corbett, A.L.; Taatizadeh, E.; Tasnim, N.; Little, J.P.; Garnis, C.; Daugaard, M.; Guns, E.; Hoorfar, M.; Li, I.T. Challenges and opportunities in exosome research—Perspectives from biology, engineering, and cancer therapy. APL Bioeng. 2019, 3, 011503. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Yang, F.; Zhang, Y.; Liu, L.; Zhou, Y.; Wang, F.; Zhang, G.-J. ExoAPP: Exosome-oriented, aptamer nanoprobe-enabled surface proteins profiling and detection. Anal. Chem. 2018, 90, 14402–14411. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, Q.; Lu, Z.; Wang, L.; Ding, L.; Xia, L.; Zhang, H.; Wang, M.; Chen, Y.; Li, G. Role of the nervous system in cancers: A review. Cell Death Discov. 2021, 7, 76. [Google Scholar] [CrossRef]
- Silverman, D.A.; Martinez, V.K.; Dougherty, P.M.; Myers, J.N.; Calin, G.A.; Amit, M. Cancer-Associated Neurogenesis and Nerve-Cancer Cross-talkNerve-Cancer cross-talk. Cancer Res. 2021, 81, 1431–1440. [Google Scholar] [CrossRef]
- Zhou, C.Q.; Lee, J.; Henkemeyer, M.J.; Lee, K.H. Disruption of ephrin B/Eph B interaction results in abnormal cochlear innervation patterns. Laryngoscope 2011, 121, 1541–1547. [Google Scholar] [CrossRef]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G. Neuroepithelial Interactions in Cancer. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 493–514. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.S.; Roy, A.; Rajpoot, S.; Liu, D.; Savai, R.; Banerjee, S.; Kawada, M.; Faisal, S.M.; Saluja, R.; Saqib, U. Tumor-derived exosomes in the regulation of macrophage polarization. Inflamm. Res. 2020, 69, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Demir, I.E.; D’Haese, J.G.; Tieftrunk, E.; Kujundzic, K.; Schorn, S.; Xing, B.; Kehl, T.; Friess, H.; Ceyhan, G.O. The neurotrophic factor neurturin contributes toward an aggressive cancer cell phenotype, neuropathic pain and neuronal plasticity in pancreatic cancer. Carcinogenesis 2014, 35, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of cell survival by secreted proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Hyenne, V.; Labouesse, M.; Goetz, J.G. The Small GTPase Ral orchestrates MVB biogenesis and exosome secretion. Small GTPases 2018, 9, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Amit, M.; Takahashi, H.; Dragomir, M.P.; Lindemann, A.; Gleber-Netto, F.O.; Pickering, C.R.; Anfossi, S.; Osman, A.A.; Cai, Y.; Wang, R. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 2020, 578, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Bemelmans, A.-P.; Firlej, V.; Allory, Y.; Romeo, P.-H.; Magnon, C. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 2019, 569, 672–678. [Google Scholar] [CrossRef]
- Lu, R.; Fan, C.; Shangguan, W.; Liu, Y.; Li, Y.; Shang, Y.; Yin, D.; Zhang, S.; Huang, Q.; Li, X.; et al. Neurons generated from carcinoma stem cells support cancer progression. Signal Transduct. Target. Ther. 2017, 2, 16036. [Google Scholar] [CrossRef]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2011, 59, 1169–1180. [Google Scholar] [CrossRef]
- Yang, Y.; Schubert, M.C.; Kuner, T.; Wick, W.; Winkler, F.; Venkataramani, V. Brain tumor networks in diffuse glioma. Neurotherapeutics 2022, 19, 1832–1843. [Google Scholar] [CrossRef]
- Seano, G.; Nia, H.T.; Emblem, K.E.; Datta, M.; Ren, J.; Krishnan, S.; Kloepper, J.; Pinho, M.C.; Ho, W.W.; Ghosh, M.; et al. Solid stress in brain tumours causes neuronal loss and neurological dysfunction and can be reversed by lithium. Nat. Biomed. Eng. 2019, 3, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular vesicles released by glioblastoma cells stimulate normal astrocytes to acquire a tumor-supportive phenotype via p53 and MYC signaling pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.; Kuttan, G. Role of macrophages in tumour progression. Immunol. Lett. 2009, 123, 97–102. [Google Scholar] [CrossRef]
- Heusinkveld, M.; van Der Burg, S.H. Identification and manipulation of tumor associated macrophages in human cancers. J. Transl. Med. 2011, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, A.T.; Hanjani, N.A.; Movahed, E.; Doroudian, M.J.C.; Letters, M.B. The role of macrophage subtypes and exosomes in immunomodulation. Cell. Mol. Biol. Lett. 2022, 27, 83. [Google Scholar] [CrossRef]
- Kerneur, C.; Cano, C.E.; Olive, D. Major pathways involved in macrophage polarization in cancer. Front. Immunol. 2022, 13, 1026954. [Google Scholar] [CrossRef]
- Kovaleva, O.; Sorokin, M.; Egorova, A.; Petrenko, A.; Shelekhova, K.; Gratchev, A. Macrophage–tumor cell interaction beyond cytokines. Front. Oncol. 2023, 13, 1078029. [Google Scholar] [CrossRef]
- Wanderley, C.W.; Colon, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef]
- Kim, H.; Wang, S.Y.; Kwak, G.; Yang, Y.; Kwon, I.C.; Kim, S.H. Exosome-guided phenotypic switch of M1 to M2 macrophages for cutaneous wound healing. Adv. Sci. 2019, 6, 1900513. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, Y.; Li, S.; Guan, X.; Jiang, X. In Situ Reprogramming of Tumor-Associated Macrophages with Internally and Externally Engineered Exosomes. Angew. Chem. 2023, 135, e202217089. [Google Scholar] [CrossRef]
- Biswas, S.; Mandal, G.; Roy Chowdhury, S.; Purohit, S.; Payne, K.K.; Anadon, C.; Gupta, A.; Swanson, P.; Yu, X.; Conejo-Garcia, J.R. Exosomes produced by mesenchymal stem cells drive differentiation of myeloid cells into immunosuppressive M2-polarized macrophages in breast cancer. J. Immunol. 2019, 203, 3447–3460. [Google Scholar] [CrossRef]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-β signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 2022, 15, 135. [Google Scholar] [CrossRef]
- Guimarães-Bastos, D.; Frony, A.C.; Barja-Fidalgo, C.; Moraes, J.A. Melanoma-derived extracellular vesicles skew neutrophils into a pro-tumor phenotype. J. Leukoc. Biol. 2022, 111, 585–596. [Google Scholar] [CrossRef]
- Cózar, B.; Greppi, M.; Carpentier, S.; Narni-Mancinelli, E.; Chiossone, L.; Vivier, E. Tumor-infiltrating natural killer cells. Cancer Discov. 2021, 11, 34–44. [Google Scholar] [CrossRef]
- Concha-Benavente, F.; Kansy, B.; Moskovitz, J.; Moy, J.; Chandran, U.; Ferris, R.L. PD-L1 mediates dysfunction in activated PD-1+ NK cells in head and neck cancer patients. Cancer Immunol. Res. 2018, 6, 1548–1560. [Google Scholar] [CrossRef]
- Bennit, H.R.F.; Gonda, A.; Oppegard, L.J.; Chi, D.P.; Khan, S.; Wall, N.R. Uptake of lymphoma-derived exosomes by peripheral blood leukocytes. Blood Lymphat. Cancer Targets Ther. 2017, 7, 9–23. [Google Scholar] [CrossRef]
- Morrissey, S.M.; Yan, J. Exosomal PD-L1: Roles in tumor progression and immunotherapy. Trends Cancer 2020, 6, 550–558. [Google Scholar] [CrossRef]
- Hedlund, M.; Stenqvist, A.-C.; Nagaeva, O.; Kjellberg, L.; Wulff, M.; Baranov, V.; Mincheva-Nilsson, L. Human placenta expresses and secretes NKG2D ligands via exosomes that down-modulate the cognate receptor expression: Evidence for immunosuppressive function. J. Immunol. 2009, 183, 340–351. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Thommen, D.S. Tertiary lymphoid structures in cancer. Science 2022, 375, eabf9419. [Google Scholar] [CrossRef]
- Sautès-Fridman, C.; Lawand, M.; Giraldo, N.A.; Kaplon, H.; Germain, C.; Fridman, W.H.; Dieu-Nosjean, M.-C. Tertiary lymphoid structures in cancers: Prognostic value, regulation, and manipulation for therapeutic intervention. Front. Immunol. 2016, 7, 407. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Larsen, M.S.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Dieudé, M.; Kaci, I.; Hébert, M.-J. The impact of programmed cell death on the formation of tertiary lymphoid structures. Front. Immunol. 2021, 12, 696311. [Google Scholar] [CrossRef]
- Junt, T.; Scandella, E.; Ludewig, B. Form follows function: Lymphoid tissue microarchitecture in antimicrobial immune defence. Nat. Rev. Immunol. 2008, 8, 764–775. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Li, H.; Shi, B. Tolerogenic dendritic cells and their applications in transplantation. Cell. Mol. Immunol. 2015, 12, 24–30. [Google Scholar] [CrossRef]
- Kushwah, R.; Wu, J.; Oliver, J.R.; Jiang, G.; Zhang, J.; Siminovitch, K.A.; Hu, J. Uptake of apoptotic DC converts immature DC into tolerogenic DC that induce differentiation of Foxp3+ Treg. Eur. J. Immunol. 2010, 40, 1022–1035. [Google Scholar] [CrossRef]
- Hubert, M.; Gobbini, E.; Couillault, C.; Manh, T.-P.V.; Doffin, A.-C.; Berthet, J.; Rodriguez, C.; Ollion, V.; Kielbassa, J.; Sajous, C. IFN-III is selectively produced by cDC1 and predicts good clinical outcome in breast cancer. Sci. Immunol. 2020, 5, eaav3942. [Google Scholar] [CrossRef]
- Hill, D.G.; Yu, L.; Gao, H.; Balic, J.J.; West, A.; Oshima, H.; McLeod, L.; Oshima, M.; Gallimore, A.; D’Costa, K. Hyperactive gp130/STAT3-driven gastric tumourigenesis promotes submucosal tertiary lymphoid structure development. Int. J. Cancer 2018, 143, 167–178. [Google Scholar] [CrossRef]
- Koti, M.; Xu, A.S.; Ren, K.Y.M.; Visram, K.; Ren, R.; Berman, D.M.; Siemens, D.R. Tertiary lymphoid structures associate with tumour stage in urothelial bladder cancer. Bladder Cancer 2017, 3, 259–267. [Google Scholar] [CrossRef]
- Figenschau, S.L.; Fismen, S.; Fenton, K.A.; Fenton, C.; Mortensen, E.S. Tertiary lymphoid structures are associated with higher tumor grade in primary operable breast cancer patients. BMC Cancer 2015, 15, 101. [Google Scholar] [CrossRef]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef]
- Gerhard, G.M.; Bill, R.; Messemaker, M.; Klein, A.M.; Pittet, M.J. Tumor-infiltrating dendritic cell states are conserved across solid human cancers. J. Exp. Med. 2021, 218, e20200264. [Google Scholar] [CrossRef]
- Oh, D.Y.; Fong, L.; Newell, E.W.; Turk, M.J.; Chi, H.; Chang, H.Y.; Satpathy, A.T.; Fairfax, B.; Silva-Santos, B.; Lantz, O. Toward a better understanding of T cells in cancer. Cancer Cell 2021, 39, 1549–1552. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, H.; Zhou, Q.; Ren, X. Insights into tertiary lymphoid structures in the solid tumor microenvironment: Anti-tumor mechanism, functional regulation, and immunotherapeutic strategies. Cancer Biol. Med. 2021, 18, 981. [Google Scholar] [CrossRef]
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M. The tumor microenvironment. Tumor Microenviron 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N.; Torii, C.; Hida, Y. Tumor angiogenesis—Characteristics of tumor endothelial cells. Int. J. Clin. Oncol. 2016, 21, 206–212. [Google Scholar] [CrossRef]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.-M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)-key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. J. Clin. Investig. 2017, 2, e87489. [Google Scholar] [CrossRef]
- Munteanu, R.; Onaciu, A.; Moldovan, C.; Zimta, A.-A.; Gulei, D.; Paradiso, A.V.; Lazar, V.; Berindan-Neagoe, I. Adipocyte-based cell therapy in oncology: The role of cancer-associated adipocytes and their reinterpretation as delivery platforms. Pharmaceutics 2020, 12, 402. [Google Scholar] [CrossRef]
- Wang, J.X.; Fukunaga-Kalabis, M.; Herlyn, M. Crosstalk in skin: Melanocytes, keratinocytes, stem cells, and melanoma. J. Cell Commun. Signal. 2016, 10, 191–196. [Google Scholar] [CrossRef]
- Barlow, K.D.; Sanders, A.M.; Soker, S.; Ergun, S.; Metheny-Barlow, L.J. Pericytes on the tumor vasculature: Jekyll or hyde? Cancer Microenviron. 2013, 6, 1–17. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, J.; Li, L.; Liao, S.; He, J.; Zhou, S.; Zhou, Y. Pericytes in the tumor microenvironment. Cancer Lett. 2023, 556, 216074. [Google Scholar] [CrossRef]
- Ning, X.; Zhang, H.; Wang, C.; Song, X. Exosomes released by gastric cancer cells induce transition of pericytes into cancer-associated fibroblasts. Med Sci. Monit. 2018, 24, 2350. [Google Scholar] [CrossRef]
- Kato, S.; Shimoda, H.; Ji, R.C.; Miura, M. Lymphangiogenesis and expression of specific molecules as lymphatic endothelial cell markers. Anat. Sci. Int. 2006, 81, 71–83. [Google Scholar] [CrossRef]
- du Bois, H.; Heim, T.A.; Lund, A.W. Tumor-draining lymph nodes: At the crossroads of metastasis and immunity. Sci. Immunol. 2021, 6, eabg3551. [Google Scholar] [CrossRef]
- Von Schulze, A.; Deng, F. A review on exosome-based cancer therapy. J. Cancer Metastasis Treat. 2020, 6, 42. [Google Scholar] [CrossRef]
- Lin, S.-L.; Chang, D.C.; Chang-Lin, S.; Lin, C.-H.; Wu, D.T.; Chen, D.T.; Ying, S.-Y. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. Rna 2008, 14, 2115–2124. [Google Scholar] [CrossRef]
- Zhou, S.; Abdouh, M.; Arena, V.; Arena, M.; Arena, G.O. Reprogramming malignant cancer cells toward a benign phenotype following exposure to human embryonic stem cell microenvironment. PLoS ONE 2017, 12, e0169899. [Google Scholar] [CrossRef]
- Lee, K.S.; Choi, J.S.; Cho, Y.W. Reprogramming of cancer stem cells into non-tumorigenic cells using stem cell exosomes for cancer therapy. Biochem. Biophys. Res. Commun. 2019, 512, 511–516. [Google Scholar] [CrossRef]
- Kamerkar, S.; Leng, C.; Burenkova, O.; Jang, S.C.; McCoy, C.; Zhang, K.; Dooley, K.; Kasera, S.; Zi, T.; Sisó, S. Exosome-mediated genetic reprogramming of tumor-associated macrophages by exoASO-STAT6 leads to potent monotherapy antitumor activity. Sci. Adv. 2022, 8, eabj7002. [Google Scholar] [CrossRef]
- Kim, H.; Park, H.-J.; Chang, H.W.; Back, J.H.; Lee, S.J.; Park, Y.E.; Kim, E.H.; Hong, Y.; Kwak, G.; Kwon, I.C. Exosome-guided direct reprogramming of tumor-associated macrophages from protumorigenic to antitumorigenic to fight cancer. Bioact. Mater. 2023, 25, 527–540. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, Y.; Chen, X.; Ning, T.; Chen, H.; Guo, Q.; Zhang, Y.; Liu, P.; Zhang, Y.; Li, C. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials 2021, 268, 120546. [Google Scholar] [CrossRef]
- Sherman, M.H.; Ruth, T.Y.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Gong, L.; Yan, Q.; Zhang, Y.; Fang, X.; Liu, B.; Guan, X. Cancer cell reprogramming: A promising therapy converting malignancy to benignity. Cancer Commun. 2019, 39, 48. [Google Scholar] [CrossRef]
- Cheng, Q.; Shi, X.; Han, M.; Smbatyan, G.; Lenz, H.-J.; Zhang, Y. Reprogramming exosomes as nanoscale controllers of cellular immunity. J. Am. Chem. Soc. 2018, 140, 16413–16417. [Google Scholar] [CrossRef]
- Yang, E.; Wang, X.; Gong, Z.; Yu, M.; Wu, H.; Zhang, D. Exosome-mediated metabolic reprogramming: The emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct. Target. Ther. 2020, 5, 242. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Roy, S.; Saha, P.; Chatterjee, N.; Bishayee, A. Trends in research on exosomes in cancer progression and anticancer therapy. Cancers 2021, 13, 326. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zeng, S.; Gong, Z.; Yan, Y. Exosome-based immunotherapy: A promising approach for cancer treatment. Mol. Cancer 2020, 19, 160. [Google Scholar] [CrossRef] [PubMed]
- Dhar, R.; Bhattacharya, B.; Mandal, D.; Devi, A.; Thorat, N.D. Exosome-based cancer vaccine: A cutting-edge approach–Correspondence. Int. J. Surg. 2022, 108, 106993. [Google Scholar] [CrossRef] [PubMed]
- Su, M.-J.; Aldawsari, H.; Amiji, M. Pancreatic cancer cell exosome-mediated macrophage reprogramming and the role of microRNAs 155 and 125b2 transfection using nanoparticle delivery systems. Sci. Rep. 2016, 6, 30110. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Zhu, L.; Yakoub, M.; Reißfelder, C.; Loges, S.; Schölch, S. Cell–cell interactions drive metastasis of circulating tumor microemboli. Cancer Res. 2022, 82, 2661–2671. [Google Scholar] [CrossRef]
- Yasuda, T.; Koiwa, M.; Yonemura, A.; Miyake, K.; Kariya, R.; Kubota, S.; Yokomizo-Nakano, T.; Yasuda-Yoshihara, N.; Uchihara, T.; Itoyama, R.; et al. Inflammation-driven senescence-associated secretory phenotype in cancer-associated fibroblasts enhances peritoneal dissemination. Cell Rep. 2021, 34, 108779. [Google Scholar] [CrossRef] [PubMed]
- Hassona, Y.; Cirillo, N.; Heesom, K.; Parkinson, E.; Prime, S.S. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br. J. Cancer 2014, 111, 1230–1237. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. The theory of tumor ecosystem. Cancer Commun. 2022, 42, 587–608. [Google Scholar] [CrossRef]
- Mitchell, M.I.; Loudig, O. Communicator Extraordinaire: Extracellular Vesicles in the Tumor Microenvironment Are Essential Local and Long-Distance Mediators of Cancer Metastasis. Biomedicines 2023, 11, 2534. [Google Scholar] [CrossRef]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3, e99911. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).